3.2. Preparation and Characterization of Compounds
3.2.1. Synthesis of Weinreb’s Amides 2
General Procedure. A solution of N,O-dimethylhydroxylamine hydrochloride (5.30 g, 54.0 mmol) in dry dichloromethane (30.0 mL) was stirred at −78 °C for 15 min. Then, a 2.0 M solution of AlMe3 in toluene (27.0 mL, 54.0 mmol) was slowly added over 1 h. The reaction mixture was stirred for 12 h and allowed to warm up until it reached 23 °C. After that, the reaction flask was cooled down at 0 °C, and the corresponding lactone 1 (18.0 mmol) was added. The resulting reaction mixture was stirred for 30 min at the same temperature, and for 2 h at 23 °C. After that, it was hydrolyzed with an aqueous solution (10.0 mL) of Rochelle’s salt (7.0 g). The resulting suspension was filtered through celite and repeatedly washed with dichloromethane (3 × 10 mL). Then, the solvent was removed under vacuum (15 Torr, <30 °C), giving rise to the expected compounds 2, which was used in the next reaction step without the need for further purification.
5-Hydroxy-
N-methoxy-
N-methylpentanamide (
2a) [
31]. Following the general procedure, compound
2a was obtained from δ-valerolactone (
1a) as a colorless oil (2.870 g, 17.82 mmol, 99%): C
7H
15NO
3;
Rf 0.33 (hexane/EtOAc 1:4);
1H NMR (400 MHz, CDCl
3) δ 3.68 (s, 3H), 3.63 (t,
J = 6.3 Hz, 2H), 3.17 (s, 3H), 2.46 (t,
J = 7.2 Hz, 2H), 1.78–1.66 (m, 2H), 1.66–1.52 (m, 2H);
13C NMR (100 MHz, CDCl
3) δ 174.7 (C), 62.0 (CH
2), 61.2 (CH
3), 32.3 (CH
2), 32.2 (CH
3), 31.3 (CH
2), 20.4 (CH
2); LRMS (EI)
m/
z 162 (M
+ + 1, 3%), 144 (3), 101 (91), 83 (51), 61 (99), 57 (27), 55 (100).
4-Hydroxy-
N-methoxy-
N-methylbutanamide (
2b) [
32]. Following the general procedure, compound
2b was obtained from γ-butyrolactone (
1b) as a colorless oil (2.593 g, 17.64 mmol, 98%): C
6H
13NO
3;
Rf 0.36 (hexane/EtOAc 1:4);
1H NMR (400 MHz, CDCl
3) δ 3.68 (s, 3H), 3.67 (t,
J = 6.3 Hz, 3H), 3.17 (s, 3H), 2.57 (t,
J = 7.0 Hz, 2H), 1.94–1.82 (m, 2H);
13C NMR (100 MHz, CDCl
3) δ 174.7 (C), 62.1 (CH
2), 61.2 (CH
3), 32.12 (CH
3), 28.9 (CH
2), 27.3 (CH
2); LRMS (EI)
m/
z 148 (M
+ + 1, 1%), 87 (41), 69 (38), 61 (100).
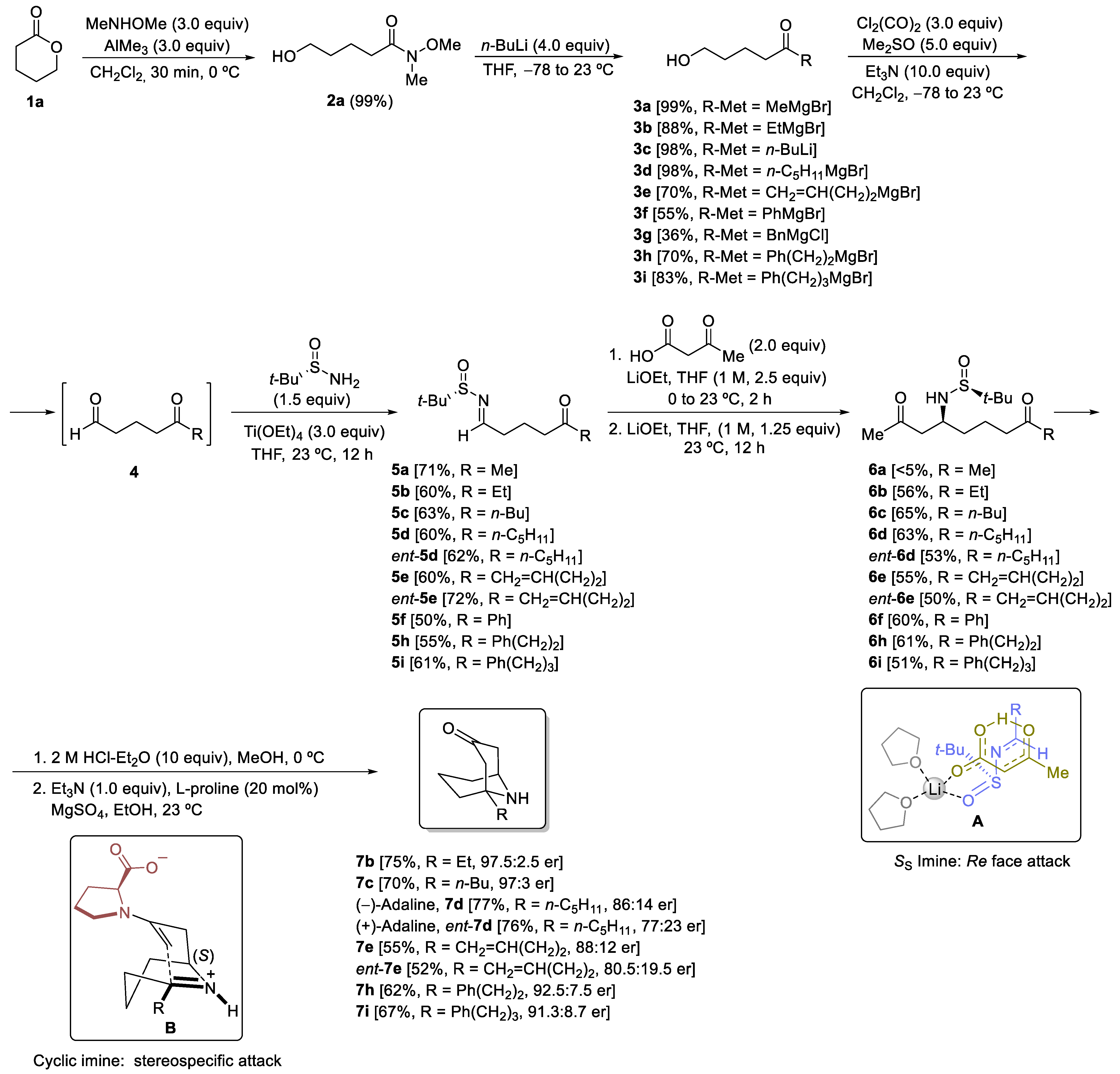
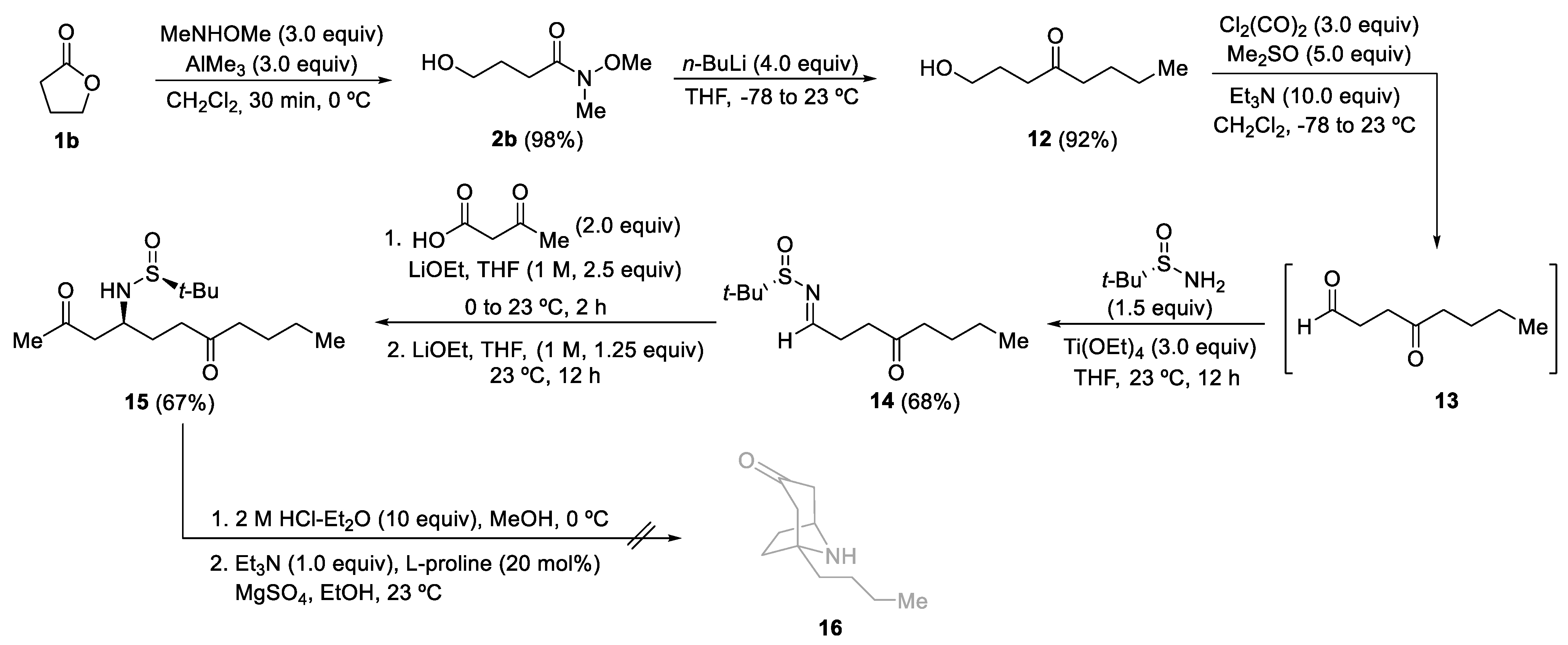
3.2.2. Synthesis of Hydroxy Ketones 3 and 12
General Procedure. A solution of the corresponding Weireb’s amide 2 (6.0 mmol) in dry THF (50.0 mL) was stirred at −78 °C for 15 min. Then, a solution of the corresponding organolithium or Grignard reagent (24.0 mmol) was slowly added. The reaction mixture was stirred and allowed to warm up until it reached 23 °C for 15 h. After that, it was hydrolyzed with water (10 mL), extracted with ethyl acetate (3 × 20 mL), dried over magnesium sulfate, and the solvent was evaporated under a vacuum (15 Torr). The residue was pure enough to be used in the following step for compounds 3a–d and 3i. The residue for compounds 3e–h and 12 was purified by column chromatography (silica gel, hexane/EtOAc) to yield pure products 3 and 12.
6-Hydroxyhexan-2-one (3a). Following the general procedure, compound 3a was obtained from amide 2a as a colorless oil (689.1 mg, 5.94 mmol, 99%): C6H12O2; Rf 0.35 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 3.62 (t, J = 6.3 Hz, 2H), 2.49 (t, J = 7.1 Hz, 2H), 2.15 (s, 3H), 1.74–1.61 (m, 3H), 1.61–1.49 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 209.4 (C), 62.1, 43.2 (CH2), 32.0 (CH2), 29.8 (CH3), 19.8 (CH2); LRMS (EI) m/z 116 (M+, 1%), 98 (99), 83 (54), 58 (47), 56 (41), 55 (100); HRMS (EI-TOF) Calcd for C6H12O2 [M+]: 116.0884, found: 116.0889.
7-Hydroxyheptan-3-one (3b). Following the general procedure, compound 3b was obtained from amide 2a as a colorless oil (686.4 mg, 5.28 mmol, 88%): C7H14O2; Rf 0.40 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 3.61 (t, J = 6.3 Hz, 2H), 2.48–2.40 (m, 4H), 1.70–1.50 (m, 5H), 1.04 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 212.1 (C), 62.1 (CH2), 41.9 (CH2), 35.9 (CH2), 32.1 (CH2), 19.9 (CH2), 7.8 (CH3); LRMS (EI) m/z 112 (M+-H2O, 27%), 101 (22), 83 (40), 57 (100), 55 (70); HRMS (EI-TOF) Calcd for C7H14O2 [M+]: 130.0994, found: 130.0990.
1-Hydroxynonan-5-one (3c). Following the general procedure, compound 3c was obtained from amide 2a as a colorless oil (929.1 mg, 5.88 mmol, 98%): C9H18O2; Rf 0.42 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 3.62 (t, J = 6.2 Hz, 2H), 2.51–2.36 (m, 4H), 1.72–1.62 (m, 3H), 1.62–1.47 (m, 4H), 1.36–1.21 (m, 2H), 0.90 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 211.8 (C), 62.2 (CH2), 42.6 (CH2), 42.3 (CH2), 32.1 (CH2), 26.0 (CH2), 22.35 (CH2), 19.7 (CH2), 13.87 (CH3); LRMS (EI) m/z 140 (M+-H2O, 18%), 116 (32), 111 (16), 101 (29), 98 (70), 85 (100), 83 (63), 57 (91), 55 (85); HRMS (EI-TOF) Calcd for C9H18O2 [M+]: 158.1307, found: 158.1295.
1-Hydroxydecan-5-one (3d). Following the general procedure, compound 3d was obtained from amide 2a as a colorless oil (1.011 g, 5.88 mmol, 98%): C10H20O2; Rf 0.48 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 3.63 (t, J = 6.1 Hz, 2H), 2.61–2.22 (m, 4H), 1.83 (s, 1H), 1.71–1.51 (m, 6H), 1.34–1.21 (m, 4H), 0.89 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 211.7 (C), 62.3 (CH2), 42.8 (CH2), 42.3 (CH2), 32.15 (CH2), 31.4 (CH2), 23.6 (CH2), 22.5 (CH2), 19.7 (CH2), 13.9 (CH3); LRMS (EI) m/z 154 (M+-H2O, 15%), 111 (24), 99 (100), 98 (90), 83 (89), 71 (85), 55 (94); HRMS (EI-TOF) Calcd for C10H20O2 [M+]: 172.1463, found: 172.1446.
9-Hydroxynon-1-en-5-one (3e). Following the general procedure, compound 3e was obtained from amide 2a as a colorless oil (655.2 mg, 4.20 mmol, 70%): C9H16O2; Rf 0.41 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 5.82–5.77 (m, 1H), 5.12–4.81 (m, 2H), 3.61 (t, J = 6.2 Hz, 2H), 2.67–2.39 (m, 4H), 2.39–2.23 (m, 2H), 2.04 (s, 1H), 1.77–1.58 (m, 2H), 1.62–1.45 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 210.6 (C), 137.2 (CH), 115.3 (CH2), 62.3 (CH2), 42.5 (CH2), 41.9 (CH2), 32.2 (CH2), 27.9 (CH2), 19.8 (CH2); LRMS (EI) m/z 138 (M+-H2O, 46%), 123 (18), 101 (23), 96 (27), 83 (52), 67 (18), 55 (100); HRMS (EI-TOF) Calcd for C9H14O [M+-H2O]: 138.1045, found: 138.1041.
5-Hydroxy-1-phenylpentan-1-one (3f). Following the general procedure, compound 3f was obtained from amide 2a as a colorless oil (587.4 mg, 3.30 mmol, 55%): C11H14O2; Rf 0.44 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 8.06–7.91 (m, 2H), 7.67–7.50 (m, 1H), 7.52–7.41 (m, 2H), 3.79–3.63 (m, 2H), 3.05 (t, J = 7.1 Hz, 2H), 1.92–1.79 (m, 2H), 1.74 (s, 1H), 1.71–1.61 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 200.5 (C), 137.2 (C), 133.1 (CH), 128.7 (CH), 128.2 (CH), 62.55 (CH2), 38.25 (CH2), 32.4 (CH2), 20.40 (CH2); LRMS (EI) m/z 178 (M+, 1%), 119 (20), 101 (45), 91 (40), 77 (100), 65 (25), 55 (65); HRMS (EI-TOF) Calcd for C11H14O2 [M+]: 178.0994, found: 178.0992.
7-Hydroxy-1-phenylheptan-3-one (3h). Following the general procedure, compound 3h was obtained from amide 2a as a colorless oil (848.4 mg, 4.20 mmol, 70%): C13H18O2; Rf 0.45 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 7.39–7.13 (m, 5H), 3.61 (t, J = 6.3 Hz, 2H), 2.92 (t, J = 7.6 Hz, 2H), 2.45 (t, J = 7.1 Hz, 2H), 1.71–1.61 (m, 3H), 1.61–1.49 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 210.3 (C), 141.1 (C), 128.5 (CH), 128.3 (CH), 126.1 (CH), 62.3 (CH2), 44.25 (CH2), 42.55 (CH2), 32.1 (CH2), 29.8 (CH2), 19.7 (CH2); LRMS (EI) m/z 206 (M+, 3%), 188 (54), 133 (37), 105 (75), 104 (28), 91 (100), 83 (19), 55 (22); HRMS (EI-TOF) Calcd for C13H18O2 [M+]: 206.1307, found: 206.1307.
8-Hydroxy-1-phenyloctan-4-one (3i). Following the general procedure, compound 3i was obtained from amide 2a as a colorless oil (1.095 g, 4.98 mmol, 83%): C14H20O2; Rf 0.50 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 7.41–7.08 (m, 5H), 3.62 (t, J = 6.2 Hz, 2H), 2.69–2.56 (m, 2H), 2.49–2.36 (m, 3H), 2.02–1.84 (m, 4H), 1.79–1.45 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 211.2 (C), 141.6 (C), 128.5 (CH), 128.5 (CH), 128.4 (CH), 126.0 (CH), 62.2 (CH2), 42.35 (CH2), 41.9 (CH2), 35.1 (CH2), 32.1 (CH2), 25.2 (CH2), 19.7 (CH2); LRMS (EI) m/z 202 (M+-H2O, 22%), 116 (20), 104 (44), 98 (100), 91 (38), 83 (12), 55 (13); HRMS (EI-TOF) Calcd for C14H20O2 [M+]: 220.1463, found: 220.1463.
1-Hydroxyoctan-4-one (12). Following the general procedure, compound 12 was obtained from amide 2b as a colorless oil (795.0 mg, 5.52 mmol, 92%): C8H16O2; Rf 0.45 (hexane/EtOAc 1:1); 1H NMR (400 MHz, CDCl3) δ 3.64 (t, J = 6.1 Hz, 2H), 2.56 (t, J = 6.9 Hz, 2H), 2.43 (t, J = 7.5 Hz, 2H), 1.89–1.80 (m, 3H), 1.66–1.46 (m, 2H), 1.35–1.27 (m, 2H), 0.94–0.85 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 212.2 (C), 62.15 (CH2), 42.7 (CH2), 39.5 (CH2), 26.6 (CH2), 26.0 (CH2), 22.4 (CH2), 13.89 (CH3); LRMS (EI) m/z 144 (M+, 2%), 126 (13), 102 (45), 97 (29), 87 (99), 85 (85), 69 (46), 58 (93), 57 (100); HRMS (EI-TOF) Calcd for C8H16O2 [M+]: 144.1150, found: 144.1153.
3.2.3. Synthesis of N-tert-Butanesulfinyl Keto Aldimines 5 and 14
General Procedure. A solution of oxalyl chloride (1.143 g, 0.772 mL, 9.0 mmol) in dry dichloromethane (20.0 mL) was stirred at −78 °C for 15 min. Then, DMSO (1.171 g, 1.065 mL, 15.0 mmol) was added dropwise. The reaction mixture was stirred for 5 min at the same temperature, and after that, a solution of the corresponding hydroxy ketone 3 or 12 (3.0 mmol) in dichloromethane (10.0 mL) was added. The resulting mixture was stirred for 15 min, and after that, Et3N (3.218 g, 4.432 mL, 31.8 mmol) was slowly added over 10 min. The reaction mixture was allowed to warm up and stirred for 2 h. Then, dichloromethane (20.0 mL) and a NH4Cl saturated aqueous solution (20.0 mL) were sequentially added. The aqueous layer was extracted with dichloromethane (3 × 25 mL), and the combined organic phases were washed with brine, dried over magnesium sulfate, and the solvent was evaporated under vacuum (15 Torr). The resulting keto aldehydes 4 were not isolated nor characterized and were used in the next step, the formation of the sulfinyl imine. A solution of the corresponding crude reaction mixture 4, (S)- or (R)-tert-butanesulfinamide 1 (0.435 g, 3.6 mmol) and titanium tetraoxide (1.605 g, 1.507 mL, 7.2 mmol) in dry THF (5.0 mL) was stirred for 12 h at 23 °C. Then the resulting mixture was hydrolyzed with brine (2.0 mL), filtered through a celite pad, and washed with ethyl acetate (3 × 10 mL). The solvent was evaporated under vacuum (15 Torr), and the residue was purified by column chromatography (silica gel, hexane/EtOAc) to yield pure products 5 and 14.
(S)-N-(5-Oxohex-1-ylidene)-tert-butanesulfinamide (5a). Following the general procedure, compound 5a was obtained from hydroxy ketone 3a as a yellow oil (462.4 mg, 2.13 mmol, 71%): C10H19NO2S; Rf 0.33 (hexane/EtOAc 3:1); [α]20D +183.1 (c 1.03, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.07 (t, J = 4.3 Hz, 1H), 2.61–2.50 (m, 4H), 2.15 (s, 3H), 2.00–1.86 (m, 2H), 1.20 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 207.9 (C), 168.8 (CH), 56.7 (C), 42.6 (CH2), 35.3 (CH2), 30.1 (CH2), 22.5 (CH3), 19.4 (CH2); LRMS (EI) m/z 160 (M+-C4H9, 4%), 112 (25), 103 (39), 70 (18), 57 (100), 55 (13), 43 (80), 41 (15); HRMS (EI-TOF) Calcd for C6H10NO2S [M+-C4H9] 160.0432, found 160.0422.
(S)-N-(5-Oxohep-1-ylidene)-tert-butanesulfinamide (5b). Following the general procedure, compound 5b was obtained from hydroxy ketone 3b as a yellow oil (416.0 mg, 1.80 mmol, 60%): C11H21NO2S; Rf 0.35 (hexane/EtOAc 3:1); [α]20D +198.2 (c 0.84, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.07 (t, J = 4.3 Hz, 1H), 2.71–2.47 (m, 4H), 2.48–2.35 (m, 2H), 2.05–1.85 (m, 2H), 1.19 (s, 9H), 1.06 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 210.8 (C), 168.9 (CH), 56.7 (C), 41.3 (CH2), 36.1 (CH2), 35.4 (CH2), 22.4 (CH3), 19.45 (CH2), 7.1 (CH3); LRMS (EI) m/z 175 (M+-C4H8, 5%), 127 (25), 111 (23), 57 (100), 56 (10), 55 (11), 43 (13), 41 (22); HRMS (EI-TOF) Calcd for C7H13NO2S [M+-C4H8] 175.0667, found 175.0667.
(S)-N-(5-Oxonon-1-ylidene)-tert-butanesulfinamide (5c). Following the general procedure, compound 5c was obtained from hydroxy ketone 3c as a yellow oil (489.7 mg, 1.89 mmol, 63%): C13H25NO2S; Rf 0.40 (hexane/EtOAc 3:1); [α]20D +131.6 (c 1.50, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.07 (t, J = 4.3 Hz, 1H), 2.59–2.47 (m, 4H), 2.40 (t, J = 7.4 Hz, 2H), 2.00–1.85 (m, 2H), 1.60–1.49 (m, 2H), 1.37–1.25 (m, 2H), 1.20 (s, 9H), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 210.5 (C), 168.9 (CH), 56.7 (C), 42.8 (CH2), 41.7 (CH2), 35.4 (CH2), 26.1 (CH2), 22.5 (CH3), 19.5 (CH2), 14.0 (CH3); LRMS (EI) m/z 203 (M+-C4H8, 13%), 155 (75), 139 (12), 113 (17), 103 (14), 98 (14), 85 (13), 70 (14), 57 (100), 56 (24), 55 (15), 43 (14), 41 (32); HRMS (EI-TOF) Calcd for C9H17NO2S [M+-C4H8] 203.0980, found 203.0987.
(S)-N-(5-Oxodec-ylidene)-tert-butanesulfinamide (5d). Following the general procedure, compound 5d was obtained from hydroxy ketone 3d as a yellow oil (491.6 mg, 1.80 mmol, 60%): C14H27NO2S; Rf 0.42 (hexane/EtOAc 3:1); [α]20D +153.6 (c 2.83, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.07 (t, J = 4.3 Hz, 1H), 2.62–2.45 (m, 4H), 2.39 (t, J = 7.4 Hz, 2H), 2.02–1.82 (m, 2H), 1.58 (s, 2H), 1.41–1.23 (m, 4H), 1.20 (s, 9H), 0.89 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 210.45 (C), 168.9 (CH), 56.7 (C), 43.0 (CH2), 41.7 (CH2), 35.4 (CH2), 31.5 (CH2), 23.7 (CH2), 22.55 (CH3), 19.4 (CH2), 14.0 (CH3); LRMS (EI) m/z 217 (M+-C4H8, 12%), 169 (75), 153 (15), 113 (12), 103 (16), 99 (15), 98 (18), 70 (19), 57 (100), 56 (33), 55 (20), 43 (34), 41 (32); HRMS (EI-TOF) Calcd for C10H19NO2S [M+-C4H8] 217.1136, found 217.1134.
(R)-N-(5-Oxodec-ylidene)-tert-butanesulfinamide (ent-5d). Following the general procedure, compound ent-5d was obtained from hydroxy ketone 3d as a yellow oil (508.0 mg, 1.86 mmol, 62%). Physical and spectroscopic data were found to be the same as for 5d. [α]20D −183.1 (c 0.98, CH2Cl2).
(S)-N-(5-Oxonon-8-en-1-ylidene)-tert-butanesulfinamide (5e). Following the general procedure, compound 5e was obtained from hydroxy ketone 3e as a yellow oil (462.8 mg, 1.80 mmol, 60%): C13H23NO2S; Rf 0.41 (hexane/EtOAc 3:1); [α]20D +181.3 (c 0.95, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.07 (t, J = 4.3 Hz, 1H), 5.87–5.67 (m, 1H), 5.07–4.89 (m, 2H), 2.60–2.44 (m, 6H), 2.40–2.26 (m, 2H), 2.02–1.86 (m, 2H), 1.19 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 209.2 (C), 168.7 (CH), 136.95 (CH), 115.3 (CH2), 56.6 (C), 41.9 (CH2), 41.7 (CH2), 35.2 (CH2), 27.7 (CH2), 22.3 (CH3), 19.2 (CH2); LRMS (EI) m/z 201 (M+-C4H8, 21%), 153 (95), 137 (13), 111 (10), 103 (20), 99 (16), 98 (13), 96 (14), 57 (100), 56 (26), 55 (40), 41 (24); HRMS (EI-TOF) Calcd for C9H15NO2S [M+-C4H8] 201.0823, found 201.0823.
(R)-N-(5-Oxonon-8-en-1-ylidene)-tert-butanesulfinamide (ent-5e). Following the general procedure, compound ent-5e was obtained from hydroxy ketone 3e as a yellow oil (555.3 mg, 2.16 mmol, 72%). Physical and spectroscopic data were found to be the same as for 5e. [α]20D −223.2 (c 1.02, CH2Cl2).
(S)-N-(5-Oxo-5-phenylpent-1-ylidene)-tert-butanesulfinamide (5f). Following the general procedure, compound 5f was obtained from hydroxy ketone 3f as a yellow oil (419.1 mg, 1.50 mmol, 50%): C15H21NO2S; Rf 0.33 (hexane/EtOAc 3:1); [α]20D +164.7 (c 2.01, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.13 (t, J = 4.2 Hz, 1H), 8.03–7.90 (m, 2H), 7.64–7.54 (m, 1H), 7.54–7.42 (m, 2H), 3.18–3.01 (m, 2H), 2.70–2.60 (m, 2H), 2.20–2.02 (m, 2H), 1.20 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 199.4 (C), 168.9 (CH), 136.9 (C), 133.3 (CH), 128.8 (CH), 128.1 (CH), 56.7 (C), 37.6 (CH2), 35.5 (CH2), 22.5 (CH3), 19.9 (CH2); LRMS (EI) m/z 223 (M+-C4H8, 14%), 176 (12), 175 (100), 133 (12), 105 (68), 103 (23), 77 (52), 73 (10), 70 (21), 57 (82), 56 (29), 51 (11), 43 (14), 41 (24); HRMS (EI-TOF) Calcd for C11H13NO2S [M+-C4H8] 223.0667, found 223.0665.
(S)-N-(5-Oxo-7-phenylhep-1-ylidene)-tert-butanesulfinamide (5h). Following the general procedure, compound 5h was obtained from hydroxy ketone 3h as a yellow oil (507.2 mg, 1.65 mmol, 55%): C17H25NO2S; Rf 0.35 (hexane/EtOAc 3:1); [α]20D +195.5 (c 1.91, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.06 (t, J = 4.3 Hz, 1H), 7.34–7.25 (m, 2H), 7.25–7.15 (m, 3H), 2.92 (t, J = 7.6 Hz, 2H), 2.80–2.70 (m, 2H), 2.57–2.43 (m, 4H), 2.01–1.86 (m, 2H), 1.20 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 209.25 (C), 168.8 (CH), 141.0 (C), 128.6 (CH), 128.45 (CH), 128.4 (CH), 126.3 (CH2), 56.72 (C), 44.5 (CH2), 42.0 (CH2), 35.3 (CH2), 29.9 (CH2), 22.5 (CH3), 19.3 (CH2); LRMS (EI) m/z 251 (M+-C4H8, 15%), 204 (14), 203 (94), 187 (13), 105 (36), 103 (21), 99 (25), 98 (15), 91 (91), 77 (10), 70 (12), 57 (100), 56 (32), 55 (15), 43 (11), 41 (23); HRMS (EI-TOF) Calcd for C13H15NO [M+-C4H10OS] 201.1154, found 201.1152.
(S)-N-(5-Oxo-8-phenyloct-1-ylidene)-tert-butanesulfinamide (5i). Following the general procedure, compound 5i was obtained from hydroxy ketone 3i as a yellow oil (588.1 mg, 1.83 mmol, 61%): C18H27NO2S; Rf 0.39 (hexane/EtOAc 3:1); [α]20D +256.8 (c 1.93, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.07 (t, J = 4.3 Hz, 1H), 7.34–7.25 (m, 2H), 7.25–7.14 (m, 3H), 2.63 (t, J = 7.5 Hz, 2H), 2.58–2.49 (m, 2H), 2.50–2.44 (m, 2H), 2.46–2.38 (m, 2H), 1.99–1.83 (m, 4H), 1.20 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 209.9 (C), 168.7 (CH), 141.50 (C), 128.5 (CH), 128.4 (CH), 126.0 (CH), 56.61 (C), 42.0 (CH2), 41.6 (CH2), 35.25 (CH2), 35.1 (CH2), 25.2 (CH2), 22.35 (CH3), 19.26 (CH2); LRMS (EI) m/z 265 (M+-C4H8, 19%), 218 (13), 217 (85), 201 (17), 147 (11), 126 (10), 113 (93), 105 (15), 104 (58), 103 (22), 98 (14), 91 (65), 70 (12), 57 (100), 56 (25), 55 (13), 41 (22); HRMS (EI-TOF) Calcd for C14H19NO2S [M+-C4H8] 265.1136, found 265.1128.
(S)-N-(4-Oxooct-1-ylidene)-tert-butanesulfinamide (14). Following the general procedure, compound 14 was obtained from hydroxy ketone 12 as a yellow oil (500.4 mg, 2.04 mmol, 68%): C12H23NO2S; Rf 0.33 (hexane/EtOAc 3:1); [α]20D +124.3 (c 1.10, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.11 (t, J = 2.6 Hz, 1H), 2.96–2.63 (m, 4H), 2.52–2.38 (m, 2H), 1.61–1.47 (m, 2H), 1.37–1.25 (m, 2H), 1.15 (s, 9H), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 208.8 (C), 167.8 (CH), 56.7 (C), 42.7 (CH2), 37.0 (CH2), 29.9 (CH2), 25.9 (CH2), 22.3 (CH3), 13.8 (CH2); LRMS (EI) m/z 189 (M+-C4H8, 11%), 141 (70), 127 (15), 99 (21), 98 (14), 83 (16) 57 (100), 56 (31), 55 (14), 41 (32); HRMS (EI-TOF) Calcd for C8H14NO2S [M+-C4H9] 188.0762, found 188.0753.
3.2.4. Synthesis of N-tert-Butanesulfinyl Amino Diketones 6 and 15
General Procedure. To a solution of 3-oxobutanoic acid (40.9 mg, 0.044 mL, 0.4 mmol) in THF (2.0 mL) at 0 °C was added a 1.0 M solution of LiOEt in THF (0.50 mL, 0.5 mmol). The reaction mixture was allowed to reach 10 °C, and the corresponding N-tert-butanesulfinyl keto aldimine 5 or 14 (0.2 mmol) was added, and stirring was continued for 2 h. If the starting imine 5 or 14 was not consumed after 2 h (TLC), a 1.0 M solution of LiOEt in THF (0.25 mL, 0.25 mmol) was added, and the resulting mixture was stirred for 12 h at 23 °C. After that, the reaction was hydrolyzed with water (10 mL), extracted with ethyl acetate (3 × 10 mL), dried over magnesium sulfate, and the solvent was evaporated (15 Torr). The residue was purified by column chromatography (silica gel, hexane/EtOAc) to yield pure products 6 and 15.
(SS,S)-4-(tert-Butanesulfinylamino)decane-2,8-dione (6b). Following the general procedure, compound 6b was obtained from keto aldimine 5b as a yellow oil (32.4 mg, 0.112 mmol, 56%): C14H27NO3S; Rf 0.31 (hexane/EtOAc 1:3); [α]20D +48.0 (c 0.38, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 3.98 (d, J = 9.0 Hz, 1H), 3.63–3.35 (m, 1H), 2.95–2.71 (m, 2H), 2.44–2.34 (m, 4H), 2.14 (s, 3H), 1.77–1.39 (m, 4H), 1.19 (s, 9H), 1.04 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 211.2 (C), 208.0 (C), 55.9 (C), 53.4 (CH), 48.9 (CH2), 41.6 (CH2), 35.95 (CH2), 35.0 (CH2), 31.0 (CH3), 22.6 (CH3), 20.25 (CH2), 7.8 (CH3); LRMS (EI) m/z 233 (M+-C4H8, 2%), 215 (19), 175 (11), 167 (19), 127 (100), 111 (11), 109 (10), 57 (74), 56 (14), 43 (35), 41 (16); HRMS (EI-TOF) Calcd for C14H27NO3S [M+] 289.1712, found 289.1699.
(SS,S)-4-(tert-Butanesulfinylamino)dodecane-2,8-dione (6c). Following the general procedure, compound 6c was obtained from keto aldimine 5c as a yellow oil (41.2 mg, 0.130 mmol, 65%): C16H31NO2S; Rf 0.36 (hexane/EtOAc 1:3); [α]20D +47.3 (c 1.39, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 4.02 (d, J = 9.0 Hz, 1H), 3.52–3.48 (m, 1H), 3.05–2.66 (m, 2H), 2.47–2.33 (m, 4H), 2.15 (s, 3H), 1.64–1.44 (m, 5H), 1.38–1.22 (m, 3H), 1.21 (s, 9H), 0.90 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 211.1 (C), 208.3 (C), 56.1 (C), 53.6 (CH), 49.0 (CH2), 42.8 (CH2), 42.1 (CH2), 35.1 (CH2), 31.1 (CH3), 26.1 (CH2), 22.8 (CH3), 22.5 (CH2), 20.3 (CH2), 14.0 (CH3); LRMS (EI) m/z 261 (M+-C4H8, 2%), 243 (39), 221 (10), 203 (13), 195 (16), 156 (20), 155 (100), 153 (10), 147 (12), 139 (17), 113 (16), 97 (13), 85 (35), 73 (15), 70 (10), 57 (60), 56 (14), 43 (29), 41 (19); HRMS (EI-TOF) Calcd for C12H23NO3S [M+-C4H8] 261.1399, found 261.1384.
(SS,S)-4-(tert-Butanesulfinylamino)tridecane-2,8-dione (6d). Following the general procedure, compound 6d was obtained from keto aldimine 5d as a yellow oil (41.8 mg, 0.126 mmol, 63%): C17H33NO3S; Rf 0.39 (hexane/EtOAc 1:3); [α]20D +47.1 (c 1.01, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 4.01 (d, J = 9.0 Hz, 1H), 3.53–3.49 (m, 1H), 2.97–2.73 (m, 2H), 2.46–2.32 (m, 4H), 2.15 (s, 3H), 1.67–1.41 (m, 5H), 1.44–1.21 (m, 5H), 1.21 (s, 9H), 0.89 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 210.95 (C), 208.1 (C), 55.9 (C), 53.4 (CH), 48.9 (CH2), 42.9 (CH2), 42.0 (CH2), 35.0 (CH2), 31.4 (CH2), 31.0 (CH3), 23.5 (CH2), 22.65 (CH3), 22.4 (CH2), 20.2 (CH2), 13.89 (CH3); LRMS (EI) m/z 275 (M+-C4H8, 1%), 257 (22), 170 (12), 169 (100), 153 (17), 99 (26), 71 (13), 57 (33), 56 (14), 43 (34), 41 (12); HRMS (EI-TOF) Calcd for C17H33NO3S [M+] 311.2181, found 331.2182.
(RS,R)-4-(tert-Butanesulfinylamino)tridecane-2,8-dione (ent-6d). Following the general procedure, compound ent-6d was obtained from keto aldimine ent-5d as a yellow oil (35.1 mg, 0.106 mmol, 53%). Physical and spectroscopic data were found to be the same as for 6d. [α]20D −49.3 (c 0.89, CH2Cl2).
(SS,S)-4-(tert-Butanesulfinylamino)dodec-11-ene-2,8-dione (6e). Following the general procedure, compound 6e was obtained from keto aldimine 5e as a yellow oil (34.7 mg, 0.110 mmol, 55%): C16H29NO3S; Rf 0.38 (hexane/EtOAc 1:3); [α]20D +52.9 (c 0.52, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 5.79 (dd, J = 17.0, 10.3 Hz, 1H), 5.09–4.93 (m, 2H), 4.09 (d, J = 9.1 Hz, 1H), 3.56–3.44 (m, 1H), 2.85 (qd, J = 17.9, 5.2 Hz, 2H), 2.49 (t, J = 7.4 Hz, 2H), 2.42 (t, J = 6.8 Hz, 2H), 2.37–2.26 (m, 2H), 2.15 (s, 3H), 1.81–1.41 (m, 4H), 1.21 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 209.9 (C), 208.2 (C), 137.1 (CH), 115.4 (CH2), 56.1 (C), 53.6 (CH), 49.0 (CH2), 42.2 (CH2), 41.95 (CH2), 35.1 (CH2), 31.1 (CH3), 27.85 (CH2), 22.8 (CH3), 20.3 (CH2); LRMS (EI) m/z 259 (M+-C4H8, 11%), 241 (42), 201 (11), 193 (19), 177 (54), 154 (11), 153 (86), 141 (29), 137 (15), 135 (17), 111 (14), 99 (11), 97 (17), 95 (23), 83 (100), 69 (11), 57 (86), 56 (26), 55 (84), 43 (48), 41 (30); HRMS (EI-TOF) Calcd for C12H21NO3S [M+-C4H8] 259.1242, found 259.1235.
(RS,R)-4-(tert-Butanesulfinylamino)dodec-11-ene-2,8-dione (ent-6e). Following the general procedure, compound ent-6e was obtained from keto aldimine ent-5e as a yellow oil (31.5 mg, 0.100 mmol, 50%). Physical and spectroscopic data were found to be the same as for 6e. [α]20D −54.6 (c 0.89, CH2Cl2).
(SS,S)-5-(tert-Butanesulfinylamino)-1-phenyloctane-1,7-dione (6f). Following the general procedure, compound 6f was obtained from keto aldimine 5f as a yellow oil (40.5 mg, 0.120 mmol, 60%): C18H27NO3S; Rf 0.31 (hexane/EtOAc 3:1); [α]20D +46.2 (c 1.14, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.95–7.82 (m, 2H), 7.57–7.31 (m, 3H), 4.07–3.98 (m, 1H), 3.59–3.44 (m, 1H), 2.93 (t, J = 6.9 Hz, 2H), 2.84–2.80 (m, 2H), 2.09 (s, 3H), 1.94–1.44 (m, 4H), 1.14 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 208.0 (C), 199.8 (C), 136.8 (C), 133.0 (CH), 128.6 (CH), 127.9 (CH), 55.9 (C), 53.4 (CH), 48.9 (CH2), 37.85 (CH2), 35.05 (CH2), 30.9 (CH3), 22.6 (CH3), 20.6 (CH2); LRMS (EI) m/z 281 (M+-C4H8, 5%), 199 (20), 175 (11), 105 (100), 77 (17), 57 (20), 43 (11); HRMS (EI-TOF) Calcd for C14H19NO3S [M+-C4H8] 281.1086, found 281.1076.
(SS,S)-4-(tert-Butanesulfinylamino)-10-phenyldecane-2,8-dione (6h). Following the general procedure, compound 6h was obtained from keto aldimine 5h as a yellow oil (44.6 mg, 0.122 mmol, 61%): C20H31NO3S; Rf 0.34 (hexane/EtOAc 1:3); [α]20D +39.7 (c 0.58, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.36–7.06 (m, 5H), 4.03 (d, J = 9.1 Hz, 1H), 3.48 (d, J = 4.8 Hz, 1H), 2.93–2.87 (m, 2H), 2.91–2.74 (m, 2H), 2.75–2.66 (m, 2H), 2.40 (t, J = 6.8 Hz, 2H), 2.15 (s, 3H), 1.79–1.59 (m, 2H), 1.62–1.48 (m, 2H), 1.20 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 209.7 (C), 208.1 (C), 141.0 (C), 128.5 (CH), 128.3 (CH), 126.1 (CH), 56.0 (C), 53.45 (CH), 48.9 (CH2), 44.3 (CH2), 42.3 (CH2), 34.9 (CH2), 31.0 (CH3), 29.7 (CH2), 22.7 (CH3), 20.15 (CH2); LRMS (EI) m/z 292 (M+-C4H9O, 6%), 291 (30), 243 (19), 204 (16), 203 (100), 187 (14), 133 (19), 105 (44), 103 (10), 99 (15), 97 (13), 91 (74), 57 (51), 56 (19), 43 (35), 41 (16); HRMS (EI-TOF) Calcd for C16H22NO3S [M+-C4H8] 308.1320, found 308.1324.
(SS,S)-4-(tert-Butanesulfinylamino)-11-phenylundecane-2,8-dione (6i). Following the general procedure, compound 6i was obtained from ketoaldimine 5i as a yellow oil (38.7 mg, 0.102 mmol, 51%): C21H33NO3S; Rf 0.35 (hexane/EtOAc 1:3); [α]20D +46.8 (c 1.49, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.37–7.06 (m, 5H), 3.99 (d, J = 9.1 Hz, 1H), 3.55–3.40 (m, 1H), 2.97–2.74 (m, 2H), 2.61 (t, J = 7.6 Hz, 2H), 2.46–2.33 (m, 4H), 2.14 (s, 3H), 1.98–1.82 (m, 2H), 1.75–1.38 (m, 4H), 1.20 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 210.4 (C), 208.1 (C), 141.5 (C), 128.5 (CH), 128.4 (CH), 126.0 (CH), 55.9 (C), 53.4 (CH), 48.9 (CH2), 42.1 (CH2), 42.0 (CH2), 35.1 (CH2), 35.0 (CH2), 31.0 (CH3), 25.2 (CH2), 22.7 (CH3), 20.2 (CH2); LRMS (EI) m/z 323 (M+-C4H8, 1%), 305 (51), 218 (18), 217 (100), 201 (26), 153 (27), 147 (60), 113 (78), 105 (20), 104 (67), 91 (31), 91 (89), 70 (15), 57 (92), 56 (27), 43 (66), 41 (33); HRMS (EI-TOF) Calcd for C17H24NO3S [M+-C4H8] 322.1520, found 322.1524.
(SS,S)-4-(tert-Butanesulfinylamino)undecane-2,7-dione (15). Following the general procedure, compound 15 was obtained from keto aldimine 14 as a yellow oil (40.6 mg, 0.134 mmol, 67%): C15H29NO2S; Rf 0.29 (hexane/EtOAc 1:3); [α]20D +39.3 (c 0.80, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 4.05 (d, J = 9.6 Hz, 1H), 3.61–3.38 (m, 1H), 2.88 (qd, J = 17.9, 5.0 Hz, 2H), 2.63–2.49 (m, 2H), 2.44–2.34 (m, 2H), 2.16 (s, 3H), 1.89–1.77 (m, 2H), 1.64–1.47 (m, 2H), 1.33–1.26 (m, 2H), 1.22 (s, 9H), 0.90 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 210.7 (C), 208.2 (C), 56.1 (C), 53.6 (CH), 49.4 (CH2), 42.8 (CH2), 39.45 (CH2), 31.1 (CH3), 29.4 (CH2), 26.1 (CH2), 22.8 (CH3), 22.4 (CH2), 13.9 (CH3); LRMS (EI) m/z 247 (M+-C4H8, 8%), 204 (12), 198 (25), 155 (26), 140 (20), 113 (28), 111 (17), 105 (100), 57 (80), 56 (18), 43 (19); HRMS (EI-TOF) Calcd for C11H19NO3S [M+-C4H10] 245.1086, found 245.1086.
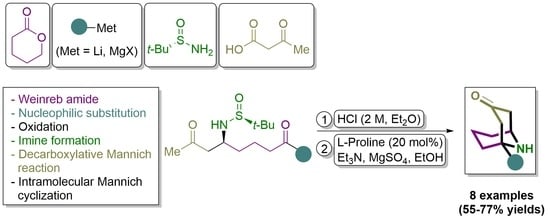
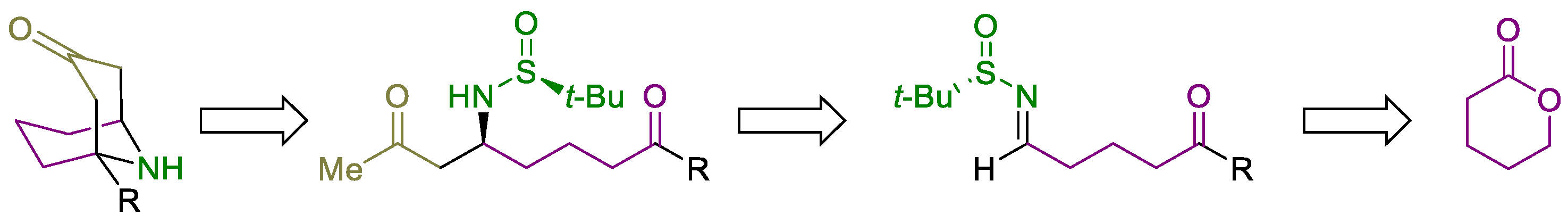
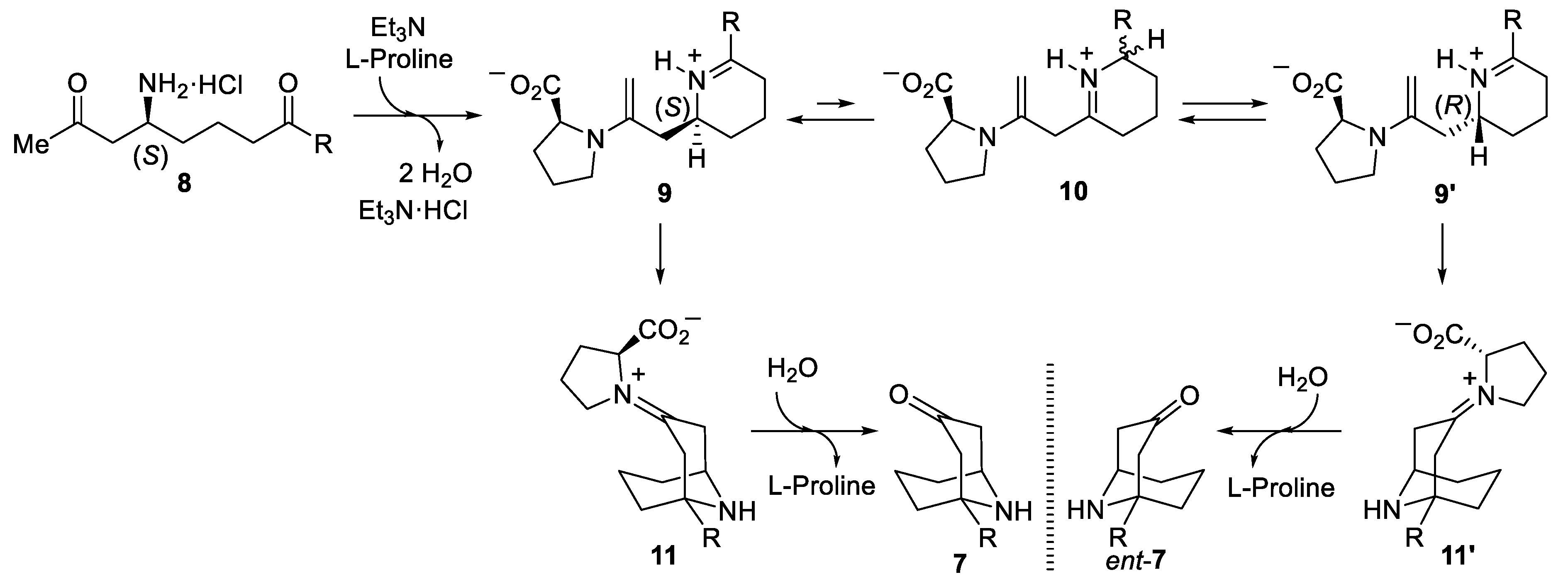
3.2.5. Synthesis of 1-Substituted 9-Azabicyclo[3.3.1]nonan-3-ones 7
General Procedure. To a solution of the corresponding amino diketone derivative 6 (0.2 mmol) in MeOH (2.0 mL), a 2.0 M solution of HCl was added into Et2O (1.0 mL, 2.0 mmol) at 0 °C. After stirring for 30 min at this temperature, the disappearance of the starting reagent and the subsequent formation of the ammonium chloride was monitored by TLC. After that, the solvents were removed under vacuum (15 Torr), and EtOH, 2.0 mL), triethylamine (20.3 g, 0.028 mL, 0.2 mmol), L-proline (4.6 mg, 0.04 mmol) and anhydrous magnesium sulfate (24.0 mg, 0.2 mmol) were successively added to the flask containing the dry ammonium salt, and the resulting reaction mixture was stirred for 12 h at 23 °C. After that, the reaction was hydrolyzed with a sodium bicarbonate saturated aqueous solution (10 mL), extracted with dichloromethane (3 × 10 mL), dried over magnesium sulfate, and the solvents were evaporated (15 Torr). The residue was purified by column chromatography (silica gel, hexane/EtOAc) to yield pure products 7.
(1R,5S)-1-Ethyl-9-azabicyclo[3.3.1]nonan-3-one (7b). Following the general procedure, compound 7b was obtained from amino diketone derivative 6b as a yellow oil (25.1 mg, 0.150 mmol, 75%): C10H17NO; 2.5:97.5 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 70 °C and 70–200 °C (4 °C/min), p = 101 kPa): tminor = 26.75 min, tmajor = 26.89 min]; Rf 0.55 (CH2Cl2/EtOH 10:1); [α]20D −5.6 (c 1.23, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 3.78 (s, 1H), 2.70 (dd, J = 16.3, 6.8 Hz, 1H), 2.43 (d, J = 16.9 Hz, 2H), 2.30 (d, J = 16.5 Hz, 1H), 1.94–1.77 (m, 1H), 1.77–1.35 (m, 6H), 1.22–1.08 (m, 2H), 0.94 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 210.2 (C), 55.6 (C), 50.6 (CH2), 49.9 (CH), 46.2 (CH2), 36.7 (CH2), 35.7 (CH2), 31.2 (CH2), 17.70 (CH2), 7.15 (CH3); LRMS (EI) m/z 167 (M+, 43%), 125 (31), 124 (100), 110 (53), 96 (33), 82 (20); HRMS (EI-TOF) Calcd for C10H17NO [M+] 167.1310, found 167.1311.
(1R,5S)-1-Butyl-9-azabicyclo[3.3.1]nonan-3-one (7c). Following the general procedure, compound 7c was obtained from amino diketone derivative 6c as a yellow oil (27.4 mg, 0.140 mmol, 70%): C12H21NO; 93:7 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 70 °C and 70–200 °C (4 °C/min), p = 101 kPa): tmajor = 35.84 min, tminor = 36.64 min]; Rf 0.56 (CH2Cl2/EtOH 10:1); [α]20D −8.0 (c 0.51, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 3.87–3.69 (m, 2H), 2.72 (dd, J = 16.4, 6.8 Hz, 1H), 2.50–2.30 (m, 3H), 1.73–1.47 (m, 6H), 1.38–1.21 (m, 6H), 0.99–0.87 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 209.8 (C), 55.4 (C), 50.7 (CH2), 49.5 (CH), 45.7 (CH2), 43.6 (CH2), 35.8 (CH2), 30.75 (CH2), 24.7 (CH2), 23.1 (CH2), 17.45 (CH2), 14.0 (CH3); LRMS (EI) m/z 195 (M+, 21%), 166 (22), 153 (100), 152 (77), 138 (33), 110 (58), 96 (96); HRMS (EI-TOF) Calcd for C12H21NO [M+] 195.1623, found 195.1633.
(1
R,5
S)-1-Pentyl-9-azabicyclo[3.3.1]nonan-3-one, (−)-Adaline (
7d). Following the general procedure, compound
7d was obtained from amino diketone derivative
6d as a yellow oil (32.2 mg, 0.154 mmol, 77%): C
13H
23NO; 86:14 er [GC (CP-Chirasil-Dex CB column, T
inlet = 275 °C, T
detector = 250 °C, T
column = 70 °C and 70–200 °C (4 °C/min),
p = 101 kPa): t
major = 38.21 min, t
minor = 38.90 min];
Rf 0.57 (CH
2Cl
2/EtOH 10:1); [α]
20D −8.6 (
c 1.49, CH
2Cl
2) [lit. [
33] −11.4 (
c 0.74, CHCl
3)];
1H NMR (400 MHz, CDCl
3) δ 3.74 (s, 1H), 2.65 (dd,
J = 16.2, 6.7 Hz, 1H), 2.49–2.34 (m, 2H), 2.29 (d,
J = 16.1 Hz, 1H), 1.82–1.13 (m, 15H), 0.95–0.84 (m, 3H);
13C NMR (100 MHz, CDCl
3) δ 210.3 (C), 55.3 (C), 51.0 (CH
2), 49.6 (CH), 46.0 (CH
2), 44.1 (CH
2), 36.1 (CH
2), 32.4 (CH
2), 31.0 (CH
2), 22.6 (CH
2), 22.3 (CH
2), 17.65 (CH
2), 14.1 (CH
3); LRMS (EI)
m/
z 209 (M
+, 13%), 180 (17), 166 (99), 153 (87), 138 (21), 124 (16), 110 (53), 96 (100); HRMS (EI-TOF) Calcd for C
13H
23NO [M
+] 209.1780, found 209.1777.
(1S,5R)-1-Pentyl-9-azabicyclo[3.3.1]nonan-3-one, (+)-Adaline (ent-7d). Following the general procedure, compound ent-7b was obtained from amino diketone derivative ent-6d as a yellow oil (31.8 mg, 0.152 mmol, 76%). Physical and spectroscopic data were found to be the same as for 7d. 23:77 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 70 °C and 70–200 °C (4 °C/min), p = 101 kPa): tminor = 38.50 min, tmajor = 38.87 min]; [α]20D +8.0 (c 2.70, CH2Cl2).
(1S,5S)-1-(But-3-en-1-yl)-9-azabicyclo[3.3.1]nonan-3-one (7e). Following the general procedure, compound 7e was obtained from amino diketone derivative 6e as a yellow oil (21.3 mg, 0.110 mmol, 55%): C12H19NO; 88:12 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 70 °C and 70–200 °C (4 °C/min), p = 101 kPa): tmajor = 36.31 min, tminor = 37.02 min]; Rf 0.55 (CH2Cl2/EtOH 10:1); [α]20D −8.5 (c 1.25, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 5.90–5.70 (m, 1H), 5.15–4.89 (m, 2H), 4.71–4.67 (m, 2H), 3.80 (t, J = 5.3 Hz, 1H), 2.84–2.60 (m, 1H), 2.55–2.31 (m, 3H), 2.20–2.06 (m, 2H), 1.78–1.46 (m, 7H); 13C NMR (100 MHz, CDCl3) δ 209.4 (C), 138.0 (CH), 115.3 (CH2), 55.5 (C), 50.7 (CH2), 49.4 (CH), 45.6 (CH2), 42.75 (CH2), 35.8 (CH2), 30.7 (CH2), 27.1 (CH2), 17.5 (CH2); LRMS (EI) m/z 193 (M+, 11%), 150 (100), 136 (93), 122 (35), 110 (44), 96 (51), 82 (38), 67 (22), 55 (39); HRMS (EI-TOF) Calcd for C12H19NO [M+] 193.1467, found 193.1471.
(1R,5R)-1-(But-3-en-1-yl)-9-azabicyclo[3.3.1]nonan-3-one (ent-7e). Following the general procedure, compound ent-7e was obtained from amino diketone derivative ent-6e as a yellow oil (20.1 mg, 0.104 mmol, 52%). Physical and spectroscopic data were found to be the same as for 7e. 19.5:80.5 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 70 °C and 70–200 °C (4 °C/min), p = 101 kPa): tminor = 36.61 min, tmajor = 37.06 min]; [α]20D + 7.9 (c 1.57, CH2Cl2).
(1S,5S)-1-(2-Phenylethyl)-9-azabicyclo[3.3.1]nonan-3-one (7h). Following the general procedure, compound 7h was obtained from amino diketone derivative 6h as a yellow oil (30.2 mg, 0.124 mmol, 62%): C16H21NO; 92.5:7.5 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 70 °C and 70–200 °C (4 °C/min), p = 101 kPa): tmajor = 53.44 min, tminor = 53.70 min]; Rf 0.58 (CH2Cl2/EtOH 10:1); [α]20D −5.5 (c 1.70, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.43–7.03 (m, 5H), 3.73 (s, 1H), 2.78–2.09 (m, 7H), 1.86–1.43 (m, 8H); 13C NMR (100 MHz, CDCl3) δ 210.5 (C), 141.9 (C), 128.7 (CH), 128.4 (CH), 126.15 (CH), 55.2 (C), 51.4 (CH2), 49.75 (CH), 46.4 (CH2), 46.3 (CH2), 36.4 (CH2), 31.35 (CH2), 29.3 (CH2), 17.81 (CH2); LRMS (EI) m/z 243 (M+, 73%), 200 (47), 186 (100), 184 (42), 91 (61); HRMS (EI-TOF) Calcd for C16H21NO [M+] 243.1623, found 243.1621.
(1R,5S)-1-(3-Phenylpropyl)-9-azabicyclo[3.3.1]nonan-3-one (7i). Following the general procedure, compound 7i was obtained from amino diketone derivative 6i as a yellow oil (34.5 mg, 0.134 mmol, 67%): C17H23NO; 91.3:8.7 er [GC (CP-Chirasil-Dex CB column, Tinlet = 275 °C, Tdetector = 250 °C, Tcolumn = 100 °C and 100–200 °C (4 °C/min), p = 101 kPa): tmajor = 66.97 min, tminor = 67.30 min]; Rf 0.61 (CH2Cl2/EtOH 10:1); [α]20D −7.8 (c 0.69, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.42–7.02 (m, 5H), 3.82–3.59 (m, 1H), 2.68–2.12 (m, 7H), 1.76–1.35 (m, 10H); 13C NMR (100 MHz, CDCl3) δ 211.1 (C), 142.0 (C), 128.45 (CH), 125.97 (CH), 54.75 (C), 51.5 (CH2), 49.7 (CH), 46.6 (CH2), 44.1 (CH2), 36.5 (CH2), 36.3 (CH2), 31.5 (CH2), 24.7 (CH2), 17.8 (CH2); LRMS (EI) m/z 257 (M+, 5%), 214 (24), 154 (10), 153 (100), 110 (22), 96 (89), 91 (20); HRMS (EI-TOF) Calcd for C17H23NO [M+] 257.1780, found 257.1780.
Copies of
1H-NMR,
13C-NMR, DEPT spectra of compounds
5,
6,
7,
14 and
15, as well as chiral GC chromatograms of compounds
7 are available is
Supplementary Materials.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



