Abstract
Chemical synthesis of 3-deoxy-d-manno-2-octulosonic acid (Kdo)-containing glycans, such as bacterial lipopolysaccharides (LPSs) and capsular polysaccharides (CPSs), is in high demand for the development of vaccines against pathogenic bacteria. We have recently achieved the complete α-stereoselective glycosidation of Kdo using a macrobicyclic donor tethered at the C1 and C5 positions. In this study, to expand the scope of Kdo glycosidation, we sought to protect the 4-OH group, thereby shortening the reaction time and ensuring the conversion of the glycosyl acceptor via its selective removal. The protection of the 4-OH group influenced the reactivity of the Kdo donor, and the triisopropylsilyl (TIPS) group acted as a selectively removable booster. The 4-O-TIPS donor allowed the synthesis of the α(2,4)-linked dimeric Kdo sequence, which is widely found in bacterial LPSs.
1. Introduction
3-Deoxy-d-manno-2-octulosonic acid (Kdo) constitutes bacterial glycans, such as lipopolysaccharides (LPSs) and capsular polysaccharides (CPSs), which are closely associated with the survival and virulence of pathogenic bacteria [1,2,3]. To understand and harness the functions of Kdo-containing LPSs and CPSs in bacterial infectious diseases, their synthesis is required. The chemical synthesis of structurally defined Kdo-containing glycans and their functionalized probes is highly profitable owing to their poor availability from natural sources. In particular, there has been growing interest in the field of vaccine development. However, the chemical synthesis of Kdo-containing glycans is challenging [4,5]; during the glycosylation of Kdo (referred to as Kdosidation), the stereocontrol is difficult, owing to the absence of a hydroxyl group at the position adjacent to the anomeric center. Previous studies on Kdosidation devised α-selective Kdo donors, such as those protected with bulky groups at the position diagonal to the anomeric position [6,7,8,9,10,11], appended with an auxiliary group at the C3 [12,13,14] and C5 [15] positions, and 2,3-ene derivatives as the precursor of the C3-appended intermediate [16,17,18,19,20]. Recently, DMF-assisted α-selective Kdosidation using a per-acetyl Kdo donor has been demonstrated [21].
Full α-selective Kdosidation was achieved using a macrobicyclic Kdo donor with α-configuration, which completely blocked the glycosidation at the β-face by the tether moiety between the C1 and C5 positions (Figure 1) [22]. In principle, this method does not produce any β-isomers and can achieve high yields using various acceptors, thereby facilitating the synthesis of α-Kdo-containing glycans. However, glycosidation using donor 1 required more time compared to glycosidation using a non-bicyclic Kdo donor [22]. In this study, we addressed this issue to expand the applicability of our method and revealed a profound effect of the protecting group at the C4 position.
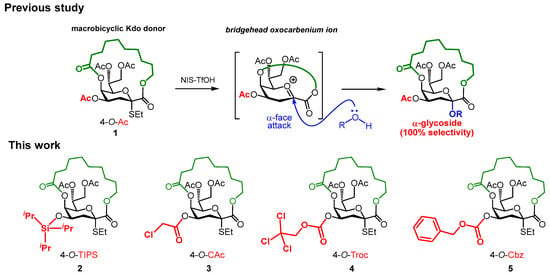
Figure 1.
Macrobicyclic Kdo donors developed in previous and present studies.
To tune the reactivity of the macrobicyclic Kdo donor, we used the effect of the hydroxyl protecting group. Based on the enormous results of the reactivities of the glycosyl donors [23,24], we presumed that the protection of the C4 and C7 positions would influence the stability of the oxocarbenium ion of Kdo. Considering the higher efficiency of the protection of the C4 hydroxyl group than that at the C7, we examined the effect of protecting groups at the C4 position. The original 4-O-acetyl (Ac) Kdo donor 1 was used as the benchmark, and the protection at the hydroxyl was diversified with selectively removable triisopropylsilyl (TIPS), chloroacetyl (CAc), 2,2,2-trichloroethoxycarbonyl (Troc), and benzyloxycarbonyl (Cbz) groups, which were expected to exert different substituent effects, affording the macrobicylic Kdo donors 2–5. Additionally, the installation of an orthogonal protecting group at this position would be useful for the synthesis of various Kdo-embedded glycans.
2. Results
2.1. Synthesis of Macrobicyclic Kdo Donors Diversified in the 4-OH Protection
To efficiently synthesize the macrobicyclic donors 2–5, the TIPS-protected donor 2 was used as a common intermediate for donors 3, 4, and 5 (Scheme 1a). To establish the α-ethylthioglycoside of Kdo, we exploited the reported intramolecular α-selective glycosidation using a dithioketal derivative to produce 6 [17]. Following the procedures reported by our group [22], compound 6 was converted into the 4,5-diol derivative 7. The TIPS group was then regioselectively introduced at 4-OH, affording 8 in an excellent yield. Next, for macrolactonization, 5-OH was esterified with the tethering unit 9 [22] by Shiina esterification to afford compound 10 in 92% yield. Treatment with 80% aqueous acetic acid followed to selectively remove the TBS protection of the tether moiety in the presence of the TIPS group, yielding 11. Compound 11 was then converted into the carboxylic acid derivative 12 via selective de-methylation using Ph3SiSH [25]. Finally, compound 12 underwent macrolactonization via the Mitsunobu reaction [22,26] to yield the TIPS-protected Kdo donor 2 in 93% yield.
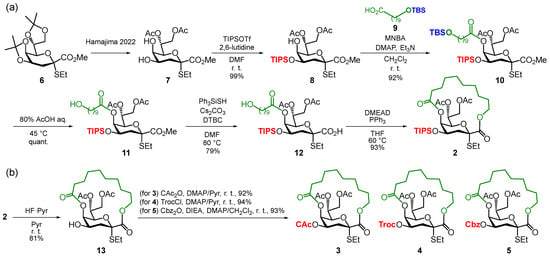
Scheme 1.
Synthesis of 4-O-modified macrobicyclic Kdo donors. (a) Synthesis of 4-O-TIPS donor 2 [22]; (b) Synthesis of 4-O-CAc, -Troc, and -Cbz donors 3, 4, and 5. TBS: tert-butyldimethylsilyl; MNBA: 2-methyl-6-nitrobenzoic anhydride; DTBC: 2,6,-di-tert-butylcresol; DMEAD: di-2-methoxyethyl azodicarboxylate.
The TIPS group of 2 was selectively removed using a fluoride anion to retrieve a free hydroxyl group at the C4 position (Scheme 1b) and synthesize the other donors. When compound 2 was treated with TBAF and acetic acid in THF, the migration of the ester groups at the O5 and O7 positions occurred randomly to give a complex mixture, whereas the use of HF·pyridine provided the 4-OH derivative 13 in high yield by decreasing the migrated byproduct. Next, the 4-OH, CAc, Troc, and Cbz groups were introduced to afford donors 3, 4, and 5, respectively, in excellent yields.
2.2. Examination of α-Glycosidation Using the Macrobicyclic Kdo Donors
The glycosylation of the 6-OH of the glucosyl acceptor 14 was examined to compare the reactivities of the macrobicyclic Kdo donors (Table 1). The protection of the group at the C4 position affected the reactivity of the Kdo donors. Among the tested donors, the TIPS group boosted the reactivity of the donor most significantly. In all cases, equimolar amounts of donor and acceptor were reacted in the presence of N-iodosuccinimide (NIS), trifluoromethanesulfonic acid (TfOH), and 3 Å molecular sieves in CH2Cl2 at −80 °C. The reaction was quenched upon complete consumption of the donor or halt of the reaction, confirmed by TLC analysis. The previously reported result of the glycosidation of 4-O-Ac Kdo donor [22] is shown in Entry 1 as the benchmark for this study. Among the tested groups, only the TIPS group was able to shorten the reaction time (by a factor of ~0.62), achieving a 76% yield of the product 16, comparable to that of Entry 1. However, the introduction of acyl protecting groups slowed down the reaction (Entries 3–5). The result of Entry 3 showed the significant effect of the electron-withdrawing CAc group on the reactivity of the macrobicyclic Kdo donor, in which the reaction time became much longer (96 h) by a factor of ~2.7. In this case, apart from the 2,3-ene derivative, the reaction produced complex side products of the donor, including a 4-OH derivative. Glycoside 17 was obtained in 17% yield, and the unreacted donor was recovered in 35% yield, indicating the significantly reduced reactivity of donor 3. Likewise, the introduction of carbonate groups, such as Troc and Cbz, retarded the reaction to some extent (Entries 4 and 5). A comparison of Troc and Cbz revealed sensitivity to the electron-withdrawing group. Although the reactions provided similar yields as those of the Kdo glycosides (20% for 18 and 28% for 19) and 2,3-ene derivatives as major side products (75% for 23 and ~70% for 24), 4-O-Troc donor 4 took much longer (68 h) than the 4-O-Cbz donor 5 (42 h). Substitution at the C4 position may affect the electron density of the sulfur atom of the leaving group and the stability of the oxocarbenium ion intermediate, through an unknown mechanism. Therefore, donors with electron-withdrawing groups required a longer period and readily underwent 1,2-elimination rather than glycosidation to produce 2,3-ene side products.

Table 1.
α−glycosylation parameters using macrobicyclic Kdo donors.
2.3. Application of the 4-O-TIPS Kdo Donor 2 to the Synthesis of α(2,4)-Linked Dimeric Kdo
The synthesis of an α(2,4)-linked dimeric Kdo was attempted, which is widely embedded in the core regions of bacterial LPSs [1], using the 4-O-TIPS Kdo donor 2 (Scheme 2). Disaccharide 16 was used as a model of Kdo glycoside and was synthesized following the aforementioned reaction conditions. First, 16 was treated with HF·pyridine in pyridine at room temperature. The reaction successfully removed TIPS without any sequential migration of acyl groups, such as those at 5-, 7-, and 8-OH, affording the disaccharide acceptor 25 in 97% yield. Subsequently, acceptor 25 was glycosylated with Kdo donor 2. As expected, upon activation with NIS-TfOH in CH2Cl2 at −80 °C, the reaction proceeded smoothly and ended in 22 h, yielding the dimeric Kdo-containing trisaccharide 26 in 70% yield.
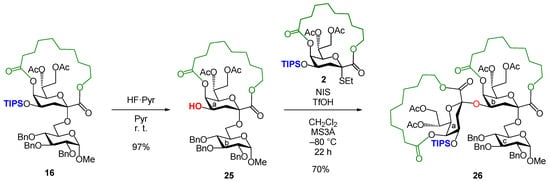
Scheme 2.
Construction of the α(2,4)−linked dimeric Kdo sequence using the 4−O−TIPS Kdo donor 2.
3. Materials and Methods
3.1. Chemicals
All chemicals were purchased from commercial suppliers and were used as received, unless otherwise noted. The molecular sieves were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan) and were pre-dried at 300 °C for 2 h in a muffle furnace followed by drying in a flask at 250 °C for 2 h under vacuum, prior to use. The dry solvents used for the reaction media (CH2Cl2, toluene, THF, MeCN, DMF, and pyridine) were purchased from Kanto Chemical Co. Inc. (Tokyo, Japan) and were used as received. Other reaction media solvents were dried over molecular sieves and used without purification.
3.2. TLC Analysis
TLC analyses were performed on Merck TLC plates (silica gel 60F254 on glass plate) (Darmstadt, Germany). Compounds were detected either by exposure to UV light (253.6 nm) or by soaking in a H2SO4 solution (10% in EtOH) or phosphomolybdic acid solution (20% in EtOH) followed by heating.
3.3. Chromatograhic Purification
Flash column chromatography separations were performed by using silica gel (80 mesh and 300 mesh; Fuji silysia Co. (Aichi, Japan)) or Biotage IsoleraTM (Uppsala, Sweden) equipped with Biotage SNAP Ultra Silica Cartridges (10 g, 25 g, 50 g, 100 g, and 340 g) (Uppsala, Sweden) or Biotage Sfär Silica HC D Cartridges (10 g, 25 g, 100 g, 200 g, and 350 g) (Uppsala, Sweden). The quantity of silica gel was typically 100 to 200 times the weight of the crude sample. Cytiva Sephadex LH-20 (Tokyo, Japan) was used for size-exclusion chromatography. Solvent systems for chromatography are specified as v/v ratios.
3.4. Structural Analysis and Acquisition of Physical Data
1H and 13C NMR spectra were recorded on an Avance III 500 spectrometer (Bruker, Billerica, MA, USA). The chemical shifts in the 1H NMR spectra are expressed in ppm (δ), relative to the Me4Si signal (0.00 ppm). The chemical shifts in the 13C NMR spectra are expressed in ppm (δ), relative to the Me4Si (0.00 ppm), CDCl3 (77.16 ppm), and adjusted CD3OD (49.00 ppm) signals. The data are presented as: chemical shift, multiplicity (s = singlet, br s = broad singlet, d = doublet, t = triplet, q = quartet, quint = quintet, dd = doublet of doublets, td = triplet of doublets, and m = multiplet and/or multiple resonances), integration, coupling constant in hertz (Hz), and position of the corresponding proton. COSY, HMBC, and HMQC methods were used to assign the NMR signals. High-resolution mass spectrometry (ESI-TOF MS) data were obtained using a micrOTOF, Bruker mass spectrometer. Optical rotations were measured with a high-sensitivity polarimeter (SEPA-500, Horiba (Kyoto, Japan)). The copies of 1H and 13C NMR spectra of the new compounds are provided in the Supplementary Material.
3.5. General Procedure for Chemical Reaction
All operations were carried out in a fume hood. All reactions were carried out under a positive pressure of argon unless otherwise noted. Evaporations and concentrations were carried out in vacuo. All heated reactions were carried out in an oil bath. All reactions below 0 °C were carried out in a low temperature reaction tank (PSL-1820, EYELA (Tokyo, Japan)).
3.6. Pretreatment for Glycosidation Reaction
A glycosyl donor (1.0–2.0 equiv.) and a glycosyl acceptor (1.0 equiv.) were mixed in a pear-shaped flask, and then residual water was azeotropically removed with dry toluene. The mixture was exposed to high vacuum for 3 h. 3Å molecular sieves (100 mg was used for 1 mL of reaction solvent) were pre-dried at 300 °C for 2 h in a muffle furnace. The pre-dried molecular sieves were added to a two-necked, round-bottomed flask and heated at 250 °C for 2 h in vacuo.
3.7. General Procedureof the Glycosidation Reaction
In a pear-shaped flask, a glycosyl donor and a glycosyl acceptor were dissolved in the reaction solvent (50 mM), and the mixture was then transferred via a cannula to a two-necked flask containing the pre-dried molecular sieves. After stirring for 1 h at −80 °C, the promoters were added to the mixture at the same temperature. The progress of the reaction was monitored using TLC analysis and/or mass spectrometry (MALDI-TOF performed with Autoflex, Bruker; matrix: CHCA). The reaction mixture was then quenched, filtered, subjected to an aqueous work-up, and concentrated. The resulting residue was purified by column chromatography. The yields of the isolated coupled products are reported.
3.8. Synthesis and Characterisation of the New Compounds
Ethyl 7,8-di-O-acetyl-3-deoxy-5-O-(10′-hydroxydecanoyl)-2-thio-4-O-triisopropylsilyl-α-d-manno-2-octulopyranosidono-1,10′-lactone (2)
A solution of 12 (1.07 g, 1.55 mmol) and DMEAD (1.45 g, 6.20 mmol) in THF (194 mL) was transferred to a solution of PPh3 (1.63 g, 6.20 mmol) in THF (705 mL) at 60 °C over a period of 2 h using a cannula. The cannula was then washed with THF (70.0 mL), and the washings were transferred to the reaction mixture. After stirring for 14 h at 60 °C and while the reaction was monitored using TLC (acetone/n-hexane = 1/4), MeOH (1.3 mL, 31.0 mmol) and AcOH (1.8 mL, 31.0 mmol) were added to the reaction mixture. The mixture was concentrated; diluted with CHCl3; washed with 2 M HCl aq., water, satd. NaHCO3 aq., and brine; dried over Na2SO4; and concentrated. The residue was purified by gel filtration column chromatography on Sephadex LH-20, using MeOH/CHCl3 (1/1) as the eluent, and column chromatography on silica gel, using EtOAc in n-hexane (10–14%) as the eluent, to yield 2 (975 mg, 93%) as a colorless syrup: [α]D +127.7° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.33 (near d, 1 H, H-5), 5.12–5.09 (m, 1 H, H-7), 4.60 (dd, 1 H, J7,8a = 2.4 Hz, Jgem = 12.3 Hz, H-8a), 4.37–4.33 (m, 3 H, H-4, H-6, CO2CH2CH2), 4.29–4.25 (m, 1 H, CO2CH2CH2), 4.15 (dd, 1 H, J7,8b = 3.6 Hz, H-8b), 2.66–2.46 (m, 2 H, SCH2), 2.38–2.26 (m, 3 H, H-3ax, OCOCH2CH2), 2.22 (dd, 1 H, J3eq,4 = 4.9 Hz, Jgem = 13.8 Hz, H-3eq), 2.08–2.04 (2 s, 6 H, 2 Ac), 1.76–1.71 (m, 2 H, CO2CH2CH2), 1.63–1.58 (m, 2 H, OCOCH2CH2), 1.49–1.26 (m, 10 H, 5 CH2), 1.20 (t, 3 H, Jvic = 7.6 Hz, SCH2CH3), 1.14–1.03 (m, 21 H, 3 iPr); 13C NMR (125 MHz, CDCl3) δ 172.6, 170.5, 169.8, 168.7, 85.3, 68.8, 67.9, 67.2, 66.5, 66.0, 62.2, 36.5, 34.2, 29.0, 28.5, 28.4, 28.0, 27.9, 26.7, 25.0, 22.5, 20.8, 18.0, 17.9, 14.0, 12.2; HRMS (ESI) m/z: [M+Na]+ Calcd for C33H58O10SSiNa 697.3412; Found 697.3412.
Ethyl 7,8-di-O-acetyl-4-O-chloroacetyl-3-deoxy-5-O-(10′-hydroxydecanoyl)-2-thio-α-d-manno-2-octulopyranosidono-1,10′-lactone (3)
To a solution of 13 (60.3 mg, 116 µmol) in pyridine (1.2 mL), CAc2O (59.5 mg, 348 µmol) and DMAP (1.4 mg, 12 µmol) were added at 0 °C. After stirring for 30 min at room temperature and while the reaction was monitored by TLC (EtOAc/n-hexane = 1/2), MeOH was added at 0 °C. The mixture was co-evaporated with toluene; diluted with CHCl3; and washed with 2 M HCl aq., water, 5% NaHCO3 aq., and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel, using EtOAc in n-hexane (20%) as the eluent, to afford 3 (63.6 mg, 92%) as a white amorphous mass: [α]D +162.8° (c 0.9, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.46–5.42 (m, 1 H, H-4), 5.37–5.36 (m, 1 H, H-5), 5.22–5.19 (m, 1 H, H-7), 4.63 (dd, 1 H, J7,8a = 2.4 Hz, Jgem = 12.4 Hz, H-8a), 4.49–4.42 (m, 2 H, H-6, CO2CH2CH2), 4.24–4.20 (m, 1 H, CO2CH2CH2), 4.13 (dd, 1 H, J7,8b = 3.6 Hz, H-8b), 4.01 (d, 1 H, Jgem = 15.4 Hz, ClH2C), 3.97 (d, 1 H, ClH2C), 2.71–2.50 (m, 2 H, SCH2), 2.43–2.38 (m, 2 H, H-3ax, OCOCH2CH2), 2.34–2.27 (m, 2 H, H-3eq, OCOCH2CH2), 2.09–2.01 (2 s, 6 H, 2 Ac), 1.78–1.55 (m, 4 H, CO2CH2CH2, OCOCH2CH2), 1.46–1.25 (m, 10 H, 5 CH2), 1.21 (t, 3 H, Jvic = 7.6 Hz, SCH2CH3); 13C NMR (125 MHz, CDCl3) δ 173.3, 170.4, 169.6, 167.9, 166.3, 84.6, 69.3, 68.2, 67.7, 66.3, 64.2, 61.8, 40.6, 33.9, 32.1, 29.0, 28.5, 28.3, 28.0, 27.9, 26.8, 25.3, 22.5, 20.8, 20.7, 13.8; HRMS (ESI) m/z: [M+Na]+ Calcd for C26H39ClO11SNa 617.1794; Found 617.1790.
Ethyl 7,8-di-O-acetyl-4-O-(2,2,2-trichloroethoxycarbonyl)-3-deoxy-5-O-(10′-hydroxydecanoyl)-2-thio-α-d-manno-2-octulopyranosidono-1,10′-lactone (4)
To a solution of 13 (60.4 mg, 117 µmol) in pyridine (1.2 mL), TrocCl (47.1 µL, 351 µmol) and DMAP (1.4 mg, 12 µmol) were added at 0 °C. After stirring for 30 min at room temperature, TrocCl (47.1 µL, 351 µmol) was added, and the mixture was stirred for a total of 1.5 h as the reaction was monitored by TLC (EtOAc/n-hexane = 1/2). MeOH was added to the reaction mixture at 0 °C. The mixture was co-evaporated with toluene; diluted with CHCl3; and washed with 2 M HCl aq., water, satd. NaHCO3 aq., and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel, using EtOAc in n-hexane (20%) as the eluent, to yield 4 (76.1 mg, 94%) as a white amorphous mass: [α]D +145.5° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.52 (near d, 1 H, H-5), 5.25–5.21 (m, 1 H, H-4), 5.20–5.17 (m, 1 H, H-7), 4.84 (d, 1 H, Jgem = 11.8 Hz, CCl3CH2), 4.68 (d, 1 H, CCl3CH2), 4.64 (dd, 1 H, J7,8a = 2.4 Hz, Jgem = 12.4 Hz, H-8a), 4.47 (dd, 1 H, J5,6 = 1.1 Hz, J6,7 = 9.7 Hz, H-6), 4.45–4.21 (m, 2 H, CO2CH2CH2), 4.13 (dd, 1 H, J7,8b = 3.5 Hz, H-8b), 2.72–2.50 (m, 2 H, SCH2), 2.44 (near t, 1 H, H-3ax), 2.40–2.30 (m, 3 H, H-3eq, OCOCH2CH2), 2.09–2.02 (2 s, 6 H, 2 Ac), 1.78–1.57 (m, 4 H, CO2CH2CH2, OCOCH2CH2), 1.50–1.25 (m, 10 H, 5 CH2), 1.21 (t, 3 H, Jvic = 7.6 Hz, SCH2CH3); 13C NMR (125 MHz, CDCl3) δ 173.0, 170.4, 169.6, 167.9, 152.7, 94.1, 84.6, 72.4, 68.2, 67.7, 66.3, 63.7, 61.8, 33.8, 32.3, 29.0, 28.4, 28.3, 28.1, 28.0, 26.7, 25.2, 22.5, 20.8, 20.7, 13.8; HRMS (ESI) m/z: [M+Na]+ Calcd for C27H39Cl3O12SNa 715.1120; Found 715.1123.
Ethyl 7,8-di-O-acetyl-4-O-benzyloxycarbonyl-3-deoxy-5-O-(10′-hydroxydecanoyl)-2-thio-α-d-manno-2-octulopyranosidono-1,10′-lactone (5)
To a solution of 13 (31.0 mg, 59.8 µmol) in CH2Cl2 (1.2 mL), DIEA (30.4 µL, 179 µmol), DMAP (7.3 mg, 60 µmol), and CbzCl (25.2 µL, 179 µmol) were added at 0 °C. To complete the reaction, DIEA (30.4 µL, 179 µmol), DMAP (7.3 mg, 60 µmol), and CbzCl (25.2 µL, 179 µmol) were added after 7, 10, and 11 h. The mixture was stirred for a total of 12.5 h as the reaction was monitored by TLC (EtOAc/n-hexane = 1/2), and MeOH was added to the reaction mixture at 0 °C. The mixture was co-evaporated with toluene; diluted with CHCl3; and washed with 2 M HCl aq., water, satd. NaHCO3 aq., and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel, using EtOAc in n-hexane (30%) as the eluent, to afford 5 (36.4 mg, 93%) as a white amorphous mass: [α]D +173.8° (c 1.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.39–7.32 (m, 5 H, Ph), 5.51 (near d, 1 H, H-5), 5.22–5.15 (m, 3 H, H-4, H-7, PhCH2), 5.13 (d, 1 H, Jgem = 12.1 Hz, PhCH2), 4.64 (d, 1 H, J7,8a = 2.4 Hz, Jgem = 12.3 Hz, H-8a), 4.46 (dd, 1 H, J5,6 = 0.9 Hz, J6,7 = 9.8 Hz, H-6), 4.43–4.20 (m, 2 H, CO2CH2CH2), 4.13 (dd, 1 H, J7,8b = 3.5 Hz, H-8b), 2.70–2.49 (m, 2 H, SCH2), 2.41–2.30 (m, 4 H, H-3ax, H-3eq, OCOCH2CH2), 2.08–2.03 (2 s, 6 H, 2 Ac), 1.77–1.54 (m, 4 H, CO2CH2CH2, OCOCH2CH2), 1.51–1.23 (m, 10 H, 5 CH2), 1.20 (t, 3 H, Jvic = 7.6 Hz, SCH2CH3); 13C NMR (125 MHz, CDCl3) δ 172.9, 170.4, 169.6, 168.1, 153.9, 135.0, 128.6, 128.3, 84.7, 71.3, 70.0, 68.3, 67.7, 66.2, 64.1, 61.8, 33.9, 32.3, 29.0, 28.4, 28.0, 26.7, 25.1, 22.5, 20.8, 20.7, 13.8; HRMS (ESI) m/z: [M+Na]+ Calcd for C32H44O12SNa 675.2446; Found 675.2442.
Methyl (ethyl 7,8-di-O-acetyl-3-deoxy-2-thio-4-O-triisopropylsilyl-α-d-manno-2-octulopyranosid)onate (8)
To a solution of 7 (1.01 g, 2.66 mmol) in DMF (13.3 mL), 2,6-lutidine (930 µL, 7.98 mmol) and TIPSOTf (1.1 mL, 4.0 mmol) were added at 0 °C. After stirring for 16 h at room temperature and while the reaction was monitored by TLC (MeOH/CHCl3 = 1/10), the reaction was quenched with MeOH at 0 °C. The mixture was co-evaporated with toluene; diluted with CHCl3; and washed with 2 M HCl aq., water, satd. NaHCO3 aq., and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by column chromatography on silica gel, using EtOAc in n-hexane (20–25%) as the eluent, to afford 8 (1.41 g, 99%) as a colorless syrup: [α]D +138.4° (c 0.9, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.45–5.42 (m, 1 H, H-7), 4.75 (dd, 1 H, J7,8a = 2.4 Hz, Jgem = 12.3 Hz, H-8a), 4.30–4.26 (m, 1 H, H-4), 4.25–4.21 (m, 2 H, H-6, H-8b), 3.79 (s, 4 H, H-5, CO2CH3), 2.62–2.47 (m, 2 H, SCH2), 2.40 (s, 1 H, OH), 2.23 (dd, 1 H, J3ax,4 = 11.3 Hz, Jgem = 13.9 Hz, H-3ax), 2.14 (dd, 1 H, J3eq,4 = 5.0 Hz, H-3eq), 2.07–2.06 (2 s, 6 H, 2 Ac), 1.19 (t, 3 H, Jvic = 7.6 Hz, SCH2CH3), 1.13–1.05 (m, 21 H, 3 iPr); 13C NMR (125 MHz, CDCl3) δ 170.1, 169.8, 169.1, 85.1, 70.0, 69.8, 67.5, 66.7, 62.4, 52.7, 34.7, 22.4, 20.9, 20.8, 18.0, 14.1, 12.2; HRMS (ESI) m/z: [M+Na]+ Calcd for C24H44O9SSiNa 559.2368; Found 559.2365.
Methyl {ethyl 7,8-di-O-acetyl-5-O-[10-(tert-butyldimethylsiloxy)decanoyl]-3-deoxy-2-thio-4-O-triisopropylsilyl-α-d-manno-2-octulopyranosid}onate (10)
To a solution of 8 (80.6 mg, 150 µmol) and 9 (136 mg, 450 µmol) in CH2Cl2 (5.0 mL), Et3N (62.7 µL, 450 µmol), DMAP (55.0 mg, 450 µmol), and MNBA (136 mg, 450 µmol) were added at 0 °C. The reaction mixture was stirred for 5 h at room temperature while the reaction was monitored by TLC (EtOAc/n-hexane = 1/4). The mixture was diluted with CHCl3; washed with 2 M HCl aq., water, satd. NaHCO3 aq., and brine; dried over Na2SO4; and concentrated. The residue was purified by column chromatography on silica gel, using EtOAc/n-hexane (8–10%) as the eluent, and gel filtration column chromatography on Sephadex LH-20, using MeOH/CHCl3 (1/1) as the eluent, to yield 10 (113 mg, 92%) as a colorless syrup: [α]D +84.6° (c 0.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.34 (near d, 1 H, H-5), 5.14–5.11 (m, 1 H, H-7), 4.58 (dd, 1 H, J7,8a = 2.4 Hz, Jgem = 12.3 Hz, H-8a), 4.39–4.35 (m, 2 H, H-4, H-6), 4.14 (dd, 1 H, J7,8b = 3.7 Hz, H-8b), 3.82 (s, 3 H, CO2CH3), 3.59 (t, 2 H, Jvic = 6.7 Hz, TBSOCH2), 2.61–2.44 (m, 2 H, SCH2), 2.35–2.26 (m, 2 H, COCH2CH2), 2.24–2.16 (m, 2 H, H-3ax, H-3eq), 2.07–2.03 (2 s, 6 H, 2 Ac), 1.62–1.47 (m, 4 H, COCH2CH2, TBSOCH2CH2), 1.27 (br s, 10 H, 5 CH2), 1.19 (t, 3 H, Jvic = 7.6 Hz, SCH2CH3), 1.10–1.03 (m, 21 H, 3 iPr), 0.89 (s, 9 H, tBu), 0.04 (s, 6 H, Si(CH3)2); 13C NMR (125 MHz, CDCl3) δ 172.8, 170.5, 169.7, 169.0, 85.3, 68.9, 67.8, 66.8, 66.4, 63.3, 62.2, 52.8, 35.9, 34.1, 32.9, 29.5, 29.4, 29.3, 29.2, 26.0, 25.8, 24.7, 22.5, 20.8, 18.4, 18.0, 14.0, 12.2, -5.2; HRMS (ESI) m/z: [M+Na]+ Calcd for C40H76O11SSi2Na 843.4539; Found 843.4540.
Methyl [ethyl 7,8-di-O-acetyl-3-deoxy-5-O-(10-hydroxydecanoyl)-2-thio-4-O-triisopropylsilyl-α-d-manno-2-octulopyranosid]onate (11)
A solution of 10 (1.65 g, 2.01 mmol) in 80% AcOH aq. (20.1 mL) was stirred for 30 min at 45 °C while the reaction was monitored by TLC (EtOAc/n-hexane = 1/2). The reaction mixture was neutralized using satd. NaHCO3 aq. at 0 °C. The mixture was diluted with CHCl3; washed with satd. NaHCO3 aq. and brine; dried over Na2SO4; and concentrated. The residue was purified by column chromatography on silica gel, using EtOAc/n-hexane (40%) as the eluent, to afford 11 (1.51 g, quant.) as a colorless syrup: [α]D +92.8° (c 1.3, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.34 (near d, 1 H, H-5), 5.14–5.11 (m, 1 H, H-7), 4.59 (dd, 1 H, J7,8a = 2.5 Hz, Jgem = 12.3 Hz, H-8a), 4.39–4.35 (m, 2 H, H-4, H-6), 4.14 (dd, 1 H, J7,8b = 3.6 Hz, H-8b), 3.82 (s, 3 H, CO2CH3), 3.63 (t, 2 H, Jvic = 6.7 Hz, HOCH2), 2.61–2.44 (m, 2 H, SCH2), 2.35–2.26 (m, 2 H, COCH2CH2), 2.24–2.16 (m, 2 H, H-3ax, H-3eq), 2.07–2.03 (2 s, 6 H, 2 Ac), 1.61–1.50 (m, 4 H, COCH2CH2, HOCH2CH2), 1.39–1.26 (m, 11 H, 5 CH2), 1.19 (t, 3 H, Jvic = 7.6 Hz, SCH2CH3), 1.14–1.03 (m, 21 H, 3 iPr); 13C NMR (125 MHz, CDCl3) δ 172.8, 170.5, 169.7, 169.0, 85.3, 68.9, 67.9, 66.8, 66.4, 63.1, 62.2, 52.8, 35.9, 34.1, 32.8, 29.35, 29.34, 29.2, 29.1, 25.7, 24.7, 22.5, 20.8, 18.0, 14.0, 12.2; HRMS (ESI) m/z: [M+Na]+ Calcd for C34H62O11SSiNa 729.3674; Found 729.3673.
Ethyl 7,8-di-O-acetyl-3-deoxy-5-O-(10-hydroxydecanoyl)-2-thio-4-triisopropylsilyl-α-d-manno-2-octulopyranosylonic acid (12)
To a solution of 11 (1.53 g, 2.17 mmol) in DMF (27.1 mL), 2,6-di-tert-butyl-p-cresol (95.6 mg, 434 µmol), Ph3SiSH (1.90 g, 6.51 mmol), and Cs2CO3 (1.91 g, 5.86 mmol) were added at room temperature. After stirring for 4.5 h at 80 °C and while the reaction was monitored by TLC (MeOH/CHCl3 = 1/5), the solution was co-evaporated with toluene. The resulting mixture was diluted with EtOAc; washed with 2 M HCl aq., water, and brine; dried over Na2SO4; and concentrated. The residue was purified by column chromatography on silica gel, using MeOH in CHCl3 (2.5–10%) as the eluent, to afford 12 (1.19 g, 79%) as a white powder: [α]D +64.8° (c 1.1, MeOH); 1H NMR (500 MHz, CD3OD) δ 5.33 (near d, 1 H, H-5), 5.07–5.05 (m, 1 H, H-7), 4.58 (dd, 1 H, J7,8a = 2.1 Hz, Jgem = 12.3 Hz, H-8a), 4.43–4.39 (m, 2 H, H-4, H-6), 4.21 (dd, 1 H, J7,8b = 3.8 Hz, H-8b), 3.54 (t, 2 H, Jvic = 6.7 Hz, HOCH2), 2.65–2.50 (m, 2 H, SCH2), 2.30–2.26 (m, 2 H, COCH2CH2, H-3eq), 2.04–1.99 (2 s, 6 H, 2 Ac), 1.59–1.50 (m, 4 H, COCH2CH2, HOCH2CH2), 1.31–1.29 (m, 10 H, 5 CH2), 1.21 (t, 3 H, Jvic = 7.5 Hz, SCH2CH3), 1.08 (br s, 21 H, 3 iPr); 13C NMR (125 MHz, CD3OD) δ 174.3, 172.3, 171.5, 70.3, 69.5, 68.8, 68.3, 63.3, 63.0, 38.1, 35.1, 33.7, 30.5, 30.4, 30.2, 26.9, 25.9, 23.8, 20.9, 20.7, 18.51, 18.49, 14.4, 13.5; HRMS (ESI) m/z: [M−H]− Calcd for C33H59O11SSi 691.3553; Found 691.3551.
Ethyl 7,8-di-O-acetyl-3-deoxy-5-O-(10′-hydroxydecanoyl)-2-thio-α-d-manno-2-octulopyranosidono-1,10′-lactone (13)
To a solution of 2 (32.9 mg, 48.8 µmol) in pyridine (1.0 mL), HF pyridine (100 µL) was added at 0 °C. After stirring for 19 h at room temperature, HF pyridine (100 µL) was added, and the mixture was stirred for a total of 42 h while the reaction was monitored by TLC (EtOAc/n-hexane = 1/2). Satd. NaHCO3 aq. was added to the reaction mixture at 0 °C. The mixture was diluted with CHCl3; washed with satd. NaHCO3 aq. and brine; dried over Na2SO4; and concentrated. The residue was purified by column chromatography on silica gel, using EtOAc in n-hexane (30%) as the eluent, to yield 13 (20.5 mg, 81%) as a white amorphous mass: [α]D +144.5° (c 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.24–5.21 (m, 2 H, H-5, H-7), 4.63 (dd, 1 H, J7,8a = 2.4 Hz, Jgem = 12.3 Hz, H-8a), 4.45–4.38 (m, 2 H, CO2CH2CH2, H-6), 4.35–4.30 (m, 1 H, H-4), 4.24–4.20 (m, 1 H, CO2CH2CH2), 4.16 (dd, 1 H, J7,8b = 3.9 Hz, H-8b), 2.67–2.48 (m, 2 H, SCH2), 2.47–2.32 (m, 2 H, OCOCH2CH2), 2.29 (dd, 1 H, J3eq,4 = 4.6 Hz, Jgem = 13.9 Hz, H-3eq), 2.24 (d, 1 H, J4,OH = 3.4 Hz, OH), 2.16 (dd, 1 H, J3ax,4 = 12.1 Hz, H-3ax), 2.08–2.04 (2 s, 6 H, 2 Ac), 1.77–1.57 (m, 4 H, CO2CH2CH2, OCOCH2CH2), 1.51–1.25 (m, 10 H, 5 CH2), 1.20 (t, 3 H, Jvic = 7.6 Hz, SCH2CH3); 13C NMR (125 MHz, CDCl3) δ 174.9, 170.5, 169.6, 168.4, 85.1, 68.3, 67.8, 66.3, 66.1, 62.1, 35.1, 34.1, 29.0, 28.5, 28.3, 28.1, 28.0, 26.8, 25.3, 22.6, 20.8, 20.7, 13.9; HRMS (ESI) m/z: [M+Na]+ Calcd for C24H38O10SNa 541.2078; Found 541.2077.
Methyl 6-O-[7,8-di-O-acetyl-3-deoxy-5-O-(10′-hydroxydecanoyl)-4-O-triisopropylsilyl-α-d-manno-2-octulopyranosylono-1,10′-lactone]-2,3,4-tri-O-benzyl-α-d-glucopyranoside (16) and 7,8-di-O-acetyl-2,6-anhydro-3-deoxy-5-O-(10′-hydroxydecanoyl)-4-O-triisopropylsilyl-d-manno-2-octenosono-1,10′-lactone (21)
In this step, 3 Å molecular sieves (110 mg) and NIS (19.3 mg, 85.7 µmol) were added to a solution of 2 (38.5 mg, 57.1 µmol) and 14 (26.5 mg, 57.1 µmol) in CH2Cl2 (1.1 mL) at room temperature. After stirring for 1 h at −80 °C, TfOH (1.0 µL, 11 µmol) was added to the mixture at −80 °C. The reaction mixture was stirred for 22 h at −80 °C while the reaction was monitored by TLC (acetone/toluene = 1/14, developed twice). The reaction was quenched with Et3N, and the mixture was filtered through a pad of Celite. The pad was then washed with CHCl3. The combined filtrate and washings were washed with satd. Na2S2O3 aq. and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by gel filtration column chromatography on Sephadex LH-20, using MeOH/CHCl3 (1/1) as the eluent, and column chromatography on silica gel, using EtOAc in n-hexane (12%) as the eluent, to afford 16 (46.9 mg, 76%) as a colorless syrup and 21 (6.9 mg, 20%) as a colorless syrup. Compound 16: [α]D +36.0° (c 4.9, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.36–7.25 (m, 15 H, 3 Ph), 5.33 (near d, 1 H, H-5a), 5.07–5.04 (m, 1 H, H-7a), 4.99 (d, 1 H, Jgem = 11.0 Hz, PhCH2), 4.89 (d, 1 H, Jgem = 11.0 Hz, PhCH2), 4.81 (d, 1 H, PhCH2), 4.78 (d, 1 H, Jgem = 12.2 Hz, PhCH2), 4.67 (d, 1 H, PhCH2), 4.59 (d, 1 H, J1,2 = 3.5 Hz, H-1b), 4.52 (d, 1 H, PhCH2), 4.49 (dd, 1 H, J7,8a = 2.2 Hz, Jgem = 12.2 Hz, H-8aa), 4.34–4.27 (m, 2 H, H-4a, CO2CH2CH2), 4.11–4.05 (m, 3 H, H-6a, H-8ba, CO2CH2CH2), 4.01 (t, 1 H, J2,3 = J3,4 = 9.2 Hz, H-3b), 3.83 (near t, 1 H, H-5b), 3.78 (near dd, 1 H, H-6ab), 3.46 (dd, 1 H, H-2b), 3.42 (dd, 1 H, J5,6b = 7.5 Hz, H-6bb), 3.37 (s, 3 H, OCH3), 3.25 (dd, 1 H, J4,5 = 10.0 Hz, H-4b), 2.39–2.28 (m, 2 H, OCOCH2CH2), 2.14 (near dd, 1 H, H-3eqa), 2.07–2.02 (m, 4 H, H-3axa, Ac), 1.87 (s, 3 H, Ac), 1.70–1.61 (m, 4 H, CO2CH2CH2, OCOCH2CH2), 1.54–1.29 (m, 10 H, 5 CH2), 1.02 (br s, 21 H, 3 iPr); 13C NMR (125 MHz, CDCl3) δ 160.6, 158.3, 157.6, 155.3, 126.4, 125.83, 125.76, 116.3, 116.2, 115.9, 115.73, 115.67, 115.5, 115.4, 87.0, 85.4, 69.9, 67.5, 66.1, 63.5, 62.8, 61.0, 57.6, 56.7, 56.1, 55.0, 53.9, 53.5, 51.2, 50.1, 42.5, 24.1, 22.1, 16.1, 15.8, 15.6, 15.3, 14.2, 12.6, 8.6, 8.5, 5.7; HRMS (ESI) m/z: [M+Na]+ Calcd for C59H84O16SiNa 1099.5421; Found 1099.5421. Compound 21: [α]D −40.6° (c 0.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.90 (t, 1 H, J3,4 = 1.8 Hz, H-3), 5.49–5.48 (m, 1 H, H-5), 5.12–5.09 (m, 1 H, H-7), 4.81–4.79 (m, 1 H, H-4), 4.61 (dd, 1 H, J7,8a = 2.5 Hz, Jgem = 12.4 Hz, H-8a), 4.37–4.33 (m, 1 H, CO2CH2CH2), 4.30 (d, 1 H, J6,7 = 9.7 Hz, H-6), 4.29–4.24 (m, 2 H, H-8b, CO2CH2CH2), 2.39–2.28 (m, 2 H, OCOCH2CH2), 2.09–2.07 (2 s, 6 H, 2 Ac), 1.74–1.71 (m, 2 H, CO2CH2CH2), 1.64–1.25 (m, 12 H, 6 CH2), 1.14–1.07 (m, 21 H, 3 iPr); 13C NMR (125 MHz, CDCl3) δ 172.7, 170.6, 169.5, 161.6, 143.0, 112.5, 73.3, 67.8, 66.5, 65.1, 63.5, 62.0, 34.4, 29.1, 29.0, 28.5, 28.3, 27.5, 27.3, 25.3, 20.8, 20.7, 17.9, 12.2; HRMS (ESI) m/z: [M+Na]+ Calcd for C31H52O10SiNa 635.3222; Found 635.3222.
Methyl 6-O-[7,8-di-O-acetyl-4-O-chloroacetyl-3-deoxy-5-O-(10′-hydroxydecanoyl)-α-d-manno-2-octulopyranosylono-1,10′-lactone]-2,3,4-tri-O-benzyl-α-d-glucopyranoside (17)
Next, 3 Å molecular sieves (100 mg) and NIS (17.3 mg, 77.0 µmol) were added to a solution of 3 (30.5 mg, 51.3 µmol) and 14 (23.8 mg, 51.3 µmol) in CH2Cl2 (1.0 mL) at room temperature. After stirring for 1 h at −80 °C, TfOH (0.9 µL, 0.01 mmol) was added to the mixture at −80 °C. The reaction mixture was stirred for 96 h at −80 °C while the reaction was monitored by TLC (acetone/toluene = 1/10, developed twice). The reaction was quenched with Et3N, and the mixture was filtered through a pad of Celite. The pad was then washed with CHCl3. The combined filtrate and washings were washed with satd. Na2S2O3 aq. and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by gel filtration column chromatography on Sephadex LH-20, using MeOH/CHCl3 (1/1) as the eluent, and column chromatography on silica gel, using EtOAc in n-hexane (20%) as the eluent, to afford 17 (8.9 mg, 17%) as a colorless syrup: [α]D +60.7° (c 0.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.37–7.25 (m, 15 H, 3 Ph), 5.40–5.36 (m, 1 H, H-4a), 5.33 (m, 1 H, H-5a), 5.15–5.12 (m, 1 H, H-7a), 4.99 (d, 1 H, Jgem = 10.8 Hz, PhCH2), 4.91 (d, 1 H, Jgem = 11.2 Hz, PhCH2), 4.80 (d, 1 H, PhCH2), 4.79 (d, 1 H, Jgem = 12.2 Hz, PhCH2), 4.67 (d, 1 H, PhCH2), 4.59 (d, 1 H, J1,2 = 3.6 Hz, H-1b), 4.56–4.51 (m, 2 H, PhCH2, H-8aa), 4.31–4.27 (m, 1 H, CO2CH2CH2), 4.19 (dd, 1 H, J5,6 = 1.1 Hz, J6,7 = 9.5 Hz, H-6a), 4.15–4.11 (m, 1 H, CO2CH2CH2), 4.05 (dd, 1 H, J7,8b = 4.7 Hz, Jgem = 12.4 Hz, H-8ba), 4.02–3.97 (m, 2 H, H-3b, ClH2C), 3.95 (d, 1 H, Jgem = 15.3 Hz, ClH2C), 3.87–3.82 (m, 2 H, H-5b, H-6ab), 3.48–3.43 (m, 2 H, H-2b, H-6bb), 3.42 (s, 3 H, OCH3), 3.25 (dd, 1 H, J3,4 = 9.0 Hz, J4,5 = 10.2 Hz, H-4b), 2.44–2.28 (m, 2 H, OCOCH2CH2), 2.22–2.16 (m, 2 H, H-3axa, H-3eqa), 1.98–1.57 (2 s, 6 H, 2 Ac), 1.76–1.57 (m, 4 H, OCOCH2CH2, CO2CH2CH2), 1.48–1.25 (m, 10 H, 5 CH2); 13C NMR (125 MHz, CDCl3) δ 173.1, 170.4, 169.7, 166.7, 152.7, 138.6, 138.1, 138.0, 128.5, 128.44, 128.41, 128.1, 128.03, 127.99, 127.9, 127.8, 127.7, 98.5, 97.7, 94.1, 82.2, 79.8, 78.2, 75.8, 75.0, 73.3, 71.8, 69.6, 68.4, 68.0, 66.4, 63.8, 63.5, 62.0, 55.1, 33.9, 32.2, 29.7, 28.4, 27.9, 27.8, 27.7, 26.5, 25.0, 20.8, 20.7; HRMS (ESI) m/z: [M+Na]+ Calcd for C52H65ClO17Na 1019.3802; Found 1019.3798.
Methyl 6-O-[7,8-di-O-acetyl-3-deoxy-5-O-(10′-hydroxydecanoyl)-4-O-(2,2,2-trichloroethoxycarbonyl)-α-d-manno-2-octulopyranosylono-1,10′-lactone]-2,3,4-tri-O-benzyl-α-d-glucopyranoside (18) and 7,8-di-O-acetyl-2,6-anhydro-3-deoxy-5-O-(10′-hydroxydecanoyl)-4-O-(2,2,2-trichloroethoxycarbonyl)-d-manno-2-octenosono-1,10′-lactone (23)
Next, 3 Å molecular sieves (100 mg) and NIS (17.1 mg, 75.9 µmol) were added to a solution of 4 (35.0 mg, 50.6 µmol) and 14 (23.5 mg, 50.6 µmol) in CH2Cl2 (1.0 mL) at room temperature. After stirring for 1 h at −80 °C, TfOH (0.9 µL, 0.01 mmol) was added to the mixture at −80 °C. The reaction mixture was stirred for 68 h at −80 °C while the reaction was monitored by TLC (acetone/toluene = 1/10, developed twice). The reaction was quenched with Et3N, and the mixture was filtered through a pad of Celite. The pad was then washed with CHCl3. The combined filtrate and washings were washed with satd. Na2S2O3 aq. and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by gel filtration column chromatography on Sephadex LH-20, using MeOH/CHCl3 (1/1) as the eluent, and column chromatography on silica gel, using EtOAc in n-hexane (20%) as the eluent, to give 18 (11.1 mg, 20%) as a colorless syrup and 23 (24.0 mg, 75%) as a white amorphous mass. Compound 18: [α]D +59.1° (c 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.37–7.26 (m, 15 H, 3 Ph), 5.49 (br s, 1 H, H-5a), 5.18–5.14 (m, 1 H, H-4a), 5.13–5.10 (m, 1 H, H-7a), 4.99 (d, 1 H, Jgem = 10.8 Hz, PhCH2), 4.92 (d, 1 H, Jgem = 11.1 Hz, PhCH2), 4.82–4.78 (m, 3 H, PhCH2, CCl3CH2), 4.69–4.65 (m, 2 H, PhCH2, CCl3CH2), 4.59 (d, 1 H, J1,2 = 3.6 Hz, H-1b), 4.56–4.51 (m, 2 H, PhCH2, H-8aa), 4.32–4.28 (m, 1 H, CO2CH2CH2), 4.17 (dd, 1 H, J5,6 = 1.0 Hz, J6,7 = 9.5 Hz, H-6a), 4.14–4.10 (m, 1 H, CO2CH2CH2), 4.05 (dd, 1 H, J7,8b = 4.6 Hz, Jgem = 12.4 Hz, H-8ba), 4.00 (t, 1 H, J2,3 = J3,4 = 9.3 Hz, H-3b), 3.88–3.82 (m, 2 H, H-5b, H-6ab), 3.49–3.43 (m, 2 H, H-2b, H-6bb), 3.41 (s, 1 H, OCH3), 3.26 (near t, 1 H, H-4b), 2.42–2.25 (m, 3 H, OCOCH2CH2, H-3eqa), 2.22 (near t, 1 H, H-3axa), 1.99–1.89 (2 s, 6 H, 2 Ac), 1.75–1.58 (m, 4 H, OCOCH2CH2, CO2CH2CH2), 1.49–1.25 (m, 10 H, 5 CH2); 13C NMR (125 MHz, CDCl3) δ 173.1, 170.4, 169.7, 166.7, 152.7, 138.6, 138.1, 138.0, 128.5, 128.44, 128.41, 128.1, 128.04, 127.99, 127.9, 127.8, 127.7, 98.5, 97.7, 94.1, 82.2, 79.8, 78.2, 75.8, 75.0, 73.3, 71.8, 69.6, 68.4, 68.0, 66.4, 63.8, 63.5, 62.0, 55.1, 33.9, 32.2, 29.7, 28.4, 27.9, 27.8, 27.7, 26.5, 25.0, 20.8, 20.7; HRMS (ESI) m/z: [M+Na]+ Calcd for C53H65Cl3O18Na 1117.3129; Found 1117.3125. Compound 23: [α]D −4.4° (c 2.5, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.95 (t, 1 H, J3,4 = 2.0 Hz, H-3), 5.64–5.62 (m, 1 H, H-5), 5.61–5.59 (m, 1 H, H-4), 5.25–5.22 (m, 1 H, H-7), 4.84 (d, 1 H, Jgem = 11.8 Hz, CCl3CH2), 4.70 (d, 1 H, CCl3CH2), 4.66 (dd, 1 H, J7,8a = 2.5 Hz, Jgem = 12.4 Hz, H-8a), 4.47–4.43 (m, 1 H, CO2CH2CH2), 4.39 (d, 1 H, J6,7 = 9.6 Hz, H-6), 4.26 (dd, 1 H, J7,8b = 3.9 Hz, H-8b), 4.25–4.20 (m, 1 H, CO2CH2CH2), 2.40–2.30 (m, 2 H, OCOCH2CH2), 2.10–2.06 (2 s, 6 H, 2 Ac), 1.79–1.72 (m, 2 H, CO2CH2CH2), 1.65–1.56 (m, 2 H, OCOCH2CH2), 1.46–1.26 (m, 10 H, 5 CH2); 13C NMR (125 MHz, CDCl3) δ 173.1, 170.5, 169.4, 160.7, 153.0, 145.3, 105.9, 94.0, 72.9, 69.9, 67.4, 66.9, 61.7, 60.1, 34.0, 29.1, 28.5, 28.2, 27.6, 27.2, 25.5, 20.8, 20.7; HRMS (ESI) m/z: [M+Na]+ Calcd for C25H33Cl3O12Na 653.0930; Found 653.0935.
Methyl 6-O-[7,8-di-O-acetyl-4-O-benzyloxycarbonyl-3-deoxy-5-O-(10′-hydroxydecanoyl)-α-d-manno-2-octulopyranosylono-1,10′-lactone]-2,3,4-tri-O-benzyl-α-d-glucopyranoside (19) and 7,8-di-O-acetyl-2,6-anhydro-4-O-benzyloxycarbonyl-3-deoxy-5-O-(10′-hydroxydecanoyl)-d-manno-2-octenosono-1,10′-lactone (24)
Next, 3 Å molecular sieves (100 mg) and NIS (16.7 mg, 74.3 µmol) were added to a solution of 5 (32.3 mg, 49.5 µmol) and 14 (23.0 mg, 49.5 µmol) in CH2Cl2 (1.0 mL) at room temperature. After stirring for 1 h at −80 °C, TfOH (0.9 µL, 0.01 mmol) was added to the mixture at −80 °C. The reaction mixture was stirred for 42 h at −80 °C while the reaction was monitored by TLC (acetone/toluene = 1/10, developed twice). The reaction was quenched with Et3N, and the mixture was filtered through a pad of Celite. The pad was washed with CHCl3. The combined filtrate and washings were washed with satd. Na2S2O3 aq. and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by gel filtration column chromatography on Sephadex LH-20, using MeOH/CHCl3 (1/1) as the eluent, and column chromatography on silica gel, using EtOAc in n-hexane (22%) and EtOAc in n-hexane (35%) as the eluent, to afford 19 (14.8 mg, 28%) and 24 (approximately 20.4 mg, approximately 70%) as colorless syrups. Compound 19: [α]D +61.0° (c 1.5, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.38–7.25 (m, 20 H, 4 Ph), 5.14 (near d, 1 H, H-5a), 5.19–5.09 (m, 4 H, H-4a, H-7a, PhCH2), 4.98 (d, 1 H, Jgem = 10.8 Hz, PhCH2), 4.91 (d, 1 H, Jgem = 11.1 Hz, PhCH2), 4.81 (d, 1 H, PhCH2), 4.79 (d, 1 H, Jgem = 12.1 Hz, PhCH2), 4.67 (d, 1 H, PhCH2), 4.59 (d, 1 H, J1,2 = 3.6 Hz, H-1b), 4.54 (d, 1 H, PhCH2), 4.53 (dd, 1 H, J7,8a = 2.3 Hz, Jgem = 12.3 Hz, H-8aa), 4.30–4.26 (m, 1 H, CO2CH2CH2), 4.17 (dd, 1 H, J5,6 = 1.1 Hz, J6,7 = 9.5 Hz, H-6a), 4.13–4.09 (m, 1 H, CO2CH2CH2), 4.05 (dd, 1 H, J7,8b = 4.5 Hz, H-8ba), 4.00 (t, 1 H, J2,3 = J3,4 = 9.2 Hz, H-3b), 3.86–3.82 (m, 2 H, H-5b, H-6ab), 3.49–3.43 (m, 2 H, H-2b, H-6bb), 3.41 (s, 3 H, OCH3), 3.26 (dd, 1 H, J4,5 = 10.2 Hz, H-4b), 2.41–2.29 (m, 2 H, OCOCH2CH2), 2.22 (dd, 1 H, J3eq,4 = 5.1 Hz, Jgem = 12.7 Hz, H-3eqa), 2.16 (t, 1 H, H-3axa), 2.00–1.88 (2 s, 6 H, 2 Ac), 1.75–1.61 (m, 4 H, CO2CH2CH2, OCOCH2CH2), 1.48–1.20 (m, 10 H, 5 CH2); 13C NMR (125 MHz, CDCl3) δ 173.1, 170.4, 169.7, 166.8, 153.9, 138.6, 138.1, 138.0, 135.0, 128.6, 128.53, 128.50, 128.43, 128.39, 128.3, 128.1, 128.03, 127.98, 127.9, 127.74, 127.68, 98.6, 97.7, 82.2, 79.8, 78.3, 75.8, 75.0, 73.3, 70.6, 69.9, 69.6, 68.5, 68.0, 66.3, 64.2, 63.5, 62.1, 55.2, 34.0, 32.3, 28.4, 27.9, 27.8, 27.6, 26.5, 25.0, 20.8, 20.7; HRMS (ESI) m/z: [M+Na]+ Calcd for C58H70O18Na 1077.4454; Found 1077.4453. Compound 24: [α]D −7.0° (c 1.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.40–7.33 (m, 5 H, Ph), 5.92 (t, 1 H, J3,4 = 1.9 Hz, H-3), 5.62 (near d, 1 H, H-5), 5.57–5.55 (m, 1 H, H-4), 5.23–5.21 (m, 2 H, H-7, PhCH2), 5.16 (d, 1 H, Jgem = 12.1 Hz, PhCH2), 4.65 (dd, 1 H, J7,8a = 2.4 Hz, Jgem = 12.4 Hz, H-8a), 4.44–4.40 (m, 1 H, CO2CH2CH2), 4.38 (d, 1 H, J6,7 = 9.6 Hz, H-6), 4.27–4.20 (m, 2 H, H-8b, CO2CH2CH2), 2.40–2.29 (m, 2 H, OCOCH2CH2), 2.09–2.06 (2 s, 6 H, 2 Ac), 1.77–1.69 (m, 2 H, CO2CH2CH2), 1.62–1.51 (m, 2 H, OCOCH2CH2), 1.46–1.25 (m, 10 H, 5 CH2); 13C NMR (125 MHz, CDCl3) δ 173.0, 170.5, 169.4, 160.9, 154.2, 144.9, 134.9, 128.6, 128.3, 106.7, 73.0, 70.2, 69.0, 67.5, 66.8, 61.8, 60.4, 34.1, 29.1, 28.4, 28.3, 27.6, 27.2, 25.5, 20.8, 20.7; HRMS (ESI) m/z: [M+Na]+ Calcd for C30H38O12Na 613.2255; Found 613.2256.
Methyl 6-O-[7,8-di-O-acetyl-3-deoxy-5-O-(10′-hydroxydecanoyl)-α-d-manno-2-octulopyranosylono-1,10′-lactone]-2,3,4-tri-O-benzyl-α-d-glucopyranoside (25)
To a solution of 16 (44.1 mg, 41.0 µmol) in pyridine (0.8 mL), HF pyridine (82.0 µL) was added at 0 °C. After stirring for 24 h at room temperature, HF pyridine (82.0 µL) was added, and the mixture was stirred for a total of 42 h while the reaction was monitored by TLC (EtOAc/n-hexane = 2/3). Satd. NaHCO3 aq. was added to the reaction mixture at 0 °C. The mixture was diluted with CHCl3, washed with satd. NaHCO3 aq. and brine, dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel, using EtOAc in n-hexane (40%) and acetone in toluene (9%) as eluents, to afford 25 (36.6 mg, 97%) as a colorless syrup: [α]D +43.4° (c 1.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.37–7.26 (m, 15 H, 3 Ph), 5.20 (br s, 1 H, H-5a), 5.16–5.13 (m, 1 H, H-7a), 4.99 (d, 1 H, Jgem = 10.8 Hz, PhCH2), 4.91 (d, 1 H, Jgem = 11.1 Hz, PhCH2), 4.80 (d, 1 H, PhCH2), 4.79 (d, 1 H, Jgem = 12.2 Hz, PhCH2), 4.67 (d, 1 H, PhCH2), 4.57 (d, 1 H, J1,2 = 3.6 Hz, H-1b), 4.55–4.52 (m, 2 H, H-8aa, PhCH2), 4.31–4.27 (m, 1 H, CO2CH2CH2), 4.24–4.22 (m, 1 H, H-4a), 4.12–4.07 (m, 3 H, H-6a, H-8ba, CO2CH2CH2), 4.00 (t, 1 H, J2,3 = J3,4 = 9.2 Hz, H-3b), 3.83–3.80 (m, 2 H, H-5b, H-6ab), 3.49–3.41 (m, 2 H, H-2b, H-6bb), 3.38 (s, 3 H, OCH3), 3.29 (near t, 1 H, H-4b), 2.47–2.32 (m, 2 H, OCOCH2CH2), 2.19 (dd, 1 H, J3eq,4 = 4.7 Hz, Jgem = 12.8 Hz, H-3eqa), 2.12 (d, 1 H, J4,OH = 3.2 Hz, OHb), 2.01 (s, 3 H, Ac), 1.94–1.89 (m, 4 H, H-3axa, Ac), 1.71–1.63 (m, 4 H, CO2CH2CH2, OCOCH2CH2), 1.47–1.25 (m, 10 H, 5 CH2); 13C NMR (125 MHz, CDCl3) δ 174.9, 170.5, 169.7, 167.2, 138.6, 138.1, 138.0, 128.5, 128.42, 128.39, 128.1, 128.0, 127.9, 127.8, 127.75, 127.67, 98.9, 97.7, 82.2, 79.8, 78.1, 75.8, 74.9, 73.3, 69.6, 68.5, 68.3, 67.7, 66.2, 65.4, 62.3, 55.0, 34.9, 34.2, 28.4, 27.9, 27.8, 27.6, 26.5, 25.1, 20.8, 20.7; HRMS (ESI) m/z: [M+Na]+ Calcd for C50H64O16Na 943.4087; Found 943.4082.
Methyl 6-O-{7,8-di-O-acetyl-4-O-[7,8-di-O-acetyl-3-deoxy-5-O-(10′-hydroxydecanoyl)-4-O-triisopropylsilyl-α-d-manno-2-octulopyranosylono-1,10′-lactone]-3-deoxy-5-O-(10′’-hydroxydecanoyl)-α-d-manno-2-octulopyranosylono-1,10′’-lactone}-2,3,4-tri-O-benzyl-α-d-glucopyranoside (26)
3 Å Molecular sieves (80 mg) and NIS (28.6 mg, 127 µmol) were added to a solution of 2 (56.9 mg, 84.4 µmol) and 25 (38.8 mg, 42.2 µmol) in CH2Cl2 (0.8 mL) at room temperature. After stirring for 1 h at −80 °C, TfOH in MeCN (7.4 µL, 8.44 µmol, 10-fold dilution) was added to the mixture at −80 °C. The reaction mixture was stirred for 22 h at −80 °C while the reaction was monitored by TLC (acetone/toluene = 1/14, developed twice). The reaction was quenched with Et3N, and the mixture was filtered through a pad of Celite. The pad was washed with CHCl3, and the combined filtrate and washings were washed with satd. Na2S2O3 aq. and brine, dried over Na2SO4, and concentrated. The resulting residue was purified by column chromatography on silica gel, using EtOAc in n-hexane (25%) as the eluent, to afford 26 (45.4 mg, 70%) as a colorless syrup: [α]D +77.8° (c 2.7, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.38–7.24 (m, 15 H, 3 Ph), 5.32 (br s, 1 H, H-5a), 5.30 (br s, 1 H, H-5b), 5.05–4.97 (m, 3 H, H-7a, H-7b, PhCH2), 4.94 (d, 1 H, Jgem = 11.2 Hz, PhCH2), 4.85 (dd, 1 H, J7,8a = 2.5 Hz, Jgem = 12.4 Hz, H-8ab), 4.80–4.73 (m, 3 H, H-4b, PhCH2), 4.67 (d, 1 H, Jgem = 12.1 Hz, PhCH2), 4.60 (d, 1 H, J1,2 = 3.5 Hz, H-1c), 4.53 (d, 1 H, PhCH2), 4.47 (dd, 1 H, J7,8a = 2.3 Hz, Jgem = 12.3 Hz, H-8aa), 4.38–4.30 (m, 2 H, CO2CH2CH2), 4.19–4.15 (m, 2 H, H-4a, H-6b), 4.12–3.98 (m, 6 H, CO2CH2CH2, H-6a, H-8ba, H8bb, H-3c), 3.78–3.72 (m, 2 H, H-5c, H-6ac), 3.47–3.43 (m, 2 H, H-2c, H-6bc), 3.41 (s, 3 H, OCH3), 3.22 (near t, 1 H, H-4c), 2.38–2.22 (m, 4 H, OCOCH2CH2), 2.19–2.13 (m, 2 H, H-3axa, H-3eqb), 2.10 (s, 3 H, Ac), 2.02–1.97 (m, 8 H, H-3eqa, H-3axb, 2 Ac), 1.82 (s, 3 H, Ac), 1.71–1.57 (m, 8 H, CO2CH2CH2, OCOCH2CH2), 1.47–1.25 (m, 20 H, 10 CH2), 1.03 (br s, 21 H, 3 iPr); 13C NMR (125 MHz, CDCl3) δ 172.6, 172.5, 171.0, 170.6, 169.9, 169.8, 167.6, 167.0, 138.7, 138.2, 128.43, 128.37, 128.3, 128.1, 127.94, 127.85, 127.8, 127.6, 98.9, 98.0, 97.5, 82.2, 80.0, 78.1, 75.7, 74.8, 73.2, 69.8, 69.5, 68.7, 68.5, 68.3, 67.2, 66.3, 66.2, 66.1, 65.5, 63.3, 62.3, 61.0, 55.1, 36.0, 34.3, 34.3, 34.2, 28.5, 28.4, 28.1, 27.9, 27.8, 27.7, 27.6, 27.4, 26.8, 26.3, 24.9, 20.81, 20.77, 20.76, 20.6, 18.0, 17.9, 12.2; HRMS (ESI) m/z: [M+Na]+ Calcd for C81H116O26SiNa 1555.7416; Found 1555.7416.
4. Conclusions
The attachment of an electron-donating TIPS group at the 4-OH position boosted the reactivity of the macrobicylic Kdo donor in comparison with the prototype 4-O-Ac Kdo donor 1. Furthermore, the chemoselectivity of the removal of the TIPS group allowed the conversion of the Kdo donor into a 4-OH Kdo acceptor after the glycosidation reaction. The 4-O-TIPS Kdo donor 2 expands the scope of macrobicyclic Kdo donors in the chemical synthesis of α-Kdo-containing glycans.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28010102/s1, copies of 1H and 13C NMR spectra of the new compounds.
Author Contributions
Conceptualization, S.H. and H.A.; investigation, S.H.; resources, T.I.; data curation, S.H.; writing—original draft preparation, S.H. and H.A.; writing—review and editing, N.K., H.-N.T., A.I., H.I. and T.I.; supervision, H.A.; project administration, H.A.; funding acquisition, S.H., N.K. and H.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by KAKENHI Grant Nos. JP18H0392 (H.A.), JP22K054353 (T.I.), JP20K15412 (N.K.), and JP21J14376 (S.H.); JSPS Core-to-Core Program Grant No. JPJSCCA20200007 (H.A.); JST CREST Grant No. JRMJCR18H2 (H.A.); JST FOREST Grant No. JPMJFR2004 (N.K.); the Mizutani Foundation for Glycoscience Grant No. 170067 (H.A.), SUNBOR Grant from the Suntory Foundation for Life Sciences (N.K.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Di Lorenzo, F.; Duda, K.A.; Lanzetta, D.R.; Silipo, A.; De Castro, C.; Molinaro, A. A journey from structure to function of bacterial lipopolysaccharides. Chem. Rev. 2022, 122, 15767. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, C.; Wear, S.S.; Sande, C. Assembly of bacterial capsular polysaccharides and exopolysaccharides. Annu. Rev. Microbiol. 2020, 74, 521–543. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, L.; Gabrielli, L.; Bini, D.; Russo, L.; Shaikh, N. Kdo: A critical monosaccharide for bacteria viability. Nat. Prod. Rep. 2010, 27, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, T.K.; Mong, K.K.T. Glycosylation chemistry of 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo) donors. Isr. J. Chem. 2015, 55, 285–296. [Google Scholar] [CrossRef]
- Kosma, P. Progress in Kdo-glycoside chemistry. Tetrahedron Lett. 2016, 57, 2133–2142. [Google Scholar] [CrossRef]
- Imoto, M.; Kusunose, N.; Matsuura, Y.; Kusumoto, S.; Shiba, T. Preparation of novel pyranosyl fluorides of 3-deoxy-D-manno-2-octulosonic acid (KDO) feasible for synthesis of KDO α-glycosides. Tetrahedron Lett. 1987, 28, 6277–6280. [Google Scholar] [CrossRef]
- Yoshizaki, H.; Fukuda, N.; Sato, K.; Oikawa, M.; Fukase, K.; Suda, Y.; Kusu-moto, S. First total synthesis of the Re-type lipopoly-saccharide. Angew. Chem. Int. Ed. 2001, 40, 1475–1480. [Google Scholar] [CrossRef]
- Ichiyanagi, T.; Fukunaga, M.; Tagashira, R.; Hayashi, S.; Nanjo, M.; Yamasaki, R. A new Kdo derivative for the synthesis of an inner-core disaccharide of lipopolysaccharides and lipooligosaccharides. Tetrahedron 2011, 67, 5964–5971. [Google Scholar] [CrossRef]
- Shimoyama, A.; Saeki, A.; Tanimura, N.; Tsutsui, H.; Miyake, K.; Suda, Y.; Fujimoto, Y.; Fukase, K. Chemical synthesis of Helicobacter pylori lipopolysaccharide partial structures and their selective proinflammatory responses. Chem. Eur. J. 2011, 17, 14464–14474. [Google Scholar] [CrossRef]
- Shimoyama, A.; Fujimoto, Y.; Fukase, K. Stereoselective glycosylation of 3-deoxy-d-manno-2-octulosonic acid with batch and microfluidic methods. Synlett 2011, 2359–2362. [Google Scholar]
- Huang, J.-S.; Huang, W.; Meng, X.; Wang, X.; Gao, P.-C.; Yang, J.-S. Stereoselective synthesis of α-3-deoxy-d-manno-oct-2-ulosonic acid (α-Kdo) glycosides using 5,7-O-di-tert-butylsilylene-protected Kdo ethyl thioglycoside donors. Angew. Chem. Int. Ed. 2015, 54, 10894–10898. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Akamatsu, S.; Achiwa, K. A new synthesis of α-glycosidically-linked disaccharides using 2 α-chloro-3 β-phenylthio KDO derivatives. Chem. Pharm. Bull. 1990, 38, 279–281. [Google Scholar] [CrossRef]
- Pokorny, B.; Kosma, P. Synthesis of Chlamydia lipopolysaccharide haptens through the use of α-specific 3-iodo-Kdo fluoride glycosyl donors. Chem. Eur. J. 2015, 21, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Pokorny, B.; Kosma, P. First and stereoselective synthesis of an α-(2→5)-linked disaccharide of 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo). Org. Lett. 2015, 17, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhou, Y.-Y.; Pan, X.-L.; Zhou, X.-Y.; Lei, J.-C.; Liu, D.-M.; Chu, Y.; Yang, J.-S. Stereodirecting effect of C5-carboxylate substituents on the glycosylation stereochemistry of 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo) thioglycoside donors: Stereoselective synthesis of α- and β-Kdo glycosides. J. Am. Chem. Soc. 2018, 140, 3574–3582. [Google Scholar] [CrossRef]
- Ikeda, K.; Akamatsu, S.; Achiwa, K. A new and stereospecific approach to Kdo-containing disaccharides using phenylselenyl triflate. Carbohydr. Res. 1989, 189, C1–C4. [Google Scholar] [CrossRef]
- van der Klein, P.A.M.; Boons, G.J.P.H.; Veeneman, G.H.; van der Marel, G.A.; van Boom, J.H. Iodonium ion promoted cyclization: A convenient approach to glycosyl donors of 3-deoxy-d-manno-2-octulosonic acid (KDO). Synlett 1990, 311–313. [Google Scholar] [CrossRef]
- Tanaka, H.; Takahashi, D.; Takahashi, T. Stereoselective synthesis of oligo-α(2,8)-3-deoxy-d-manno-2-octulosonic acid derivatives. Angew. Chem. Int. Ed. 2006, 45, 770–773. [Google Scholar] [CrossRef]
- Mannerstedt, K.; Ekelöf, K.; Oscarson, S. Evaluation of thioglycosides of Kdo as glycosyl donors. Carbohydr. Res. 2007, 342, 631–637. [Google Scholar] [CrossRef]
- Yoshida, F.; Yoshinaka, H.; Tanaka, H.; Hanashima, S.; Yamaguchi, Y.; Ishihara, M.; Saburomaru, M.; Kato, Y.; Saito, R.; Ando, H.; et al. Synthesis of the core oligosaccharides of lipooligosaccharides from Campylobacter jejuni: A putative cause of Guillain-Barré syndrome. Chem. Eur. J. 2019, 25, 796–805. [Google Scholar] [CrossRef]
- Lou, Q.; Hua, Q.; Zhang, L.; Yang, Y. Dimethylformamide-modulated Kdo glycosidation for stereoselective synthesis of α-Kdo glycosides. Org. Lett. 2020, 22, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Hamajima, S.; Komura, N.; Tanaka, H.-N.; Imamura, A.; Ishida, H.; Noguchi, H.; Ichiyanagi, T.; Ando, H. Full stereocontrol in α-glycosidation of 3-deoxy-d-manno-2-octulosonic acid (Kdo) using macrobicyclic glycosyl donors. Org. Lett. 2022, 24, 8672–8676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ollmann, I.R.; Ye, X.-S.; Wischnat, R.; Baasov, T.; Wong, C.-H. Programmable one-pot oligosaccharide synthesis. J. Am. Chem. Soc. 1999, 121, 734–753. [Google Scholar] [CrossRef]
- Chang, C.-W.; Wu, C.-H.; Lin, M.-H.; Liao, P.-H.; Chang, C.-C.; Chuang, H.-H.; Lin, S.-C.; Lam, S.; Verma, V.P.; Hsu, C.-P.; et al. Establishment of guideline for the control of glycosylation reactions and intermediates by quantitative assessment of reactivity. Angew. Chem. Int. Ed. 2019, 58, 16775–16779. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, A.; Ito, Y. Preparation of sialyl donors carrying functionalized ester substituents: Effect on the selectivity of glycosylation. Synlett 2003, 1339–1343. [Google Scholar] [CrossRef]
- Kurihara, T.; Nakajima, Y.; Mitsunobu, O. Synthesis of lactones and cycloalkanes. Cyclization of ω-hydroxy acids and ethyl α-cyano-ω-hydroxycarboxylates. Tetrahedron Lett. 1976, 17, 2455–2458. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).