Abstract
Five focused compound libraries (forty-nine compounds), based on prior studies in our laboratory were synthesized and screened for antibiotic and anti-fungal activity against S. aureus, E. coli, K. pneumoniae, P. aeruginosa, A. baumannii, C. albicans and C. neoformans. Low levels of activity, at the initial screening concentration of 32 μg/mL, were noted with analogues of (Z)-2-(3,4-dichlorophenyl)-3-phenylacrylonitriles which made up the first two focused libraries produced. The most promising analogues possessing additional substituents on the terminal aromatic ring of the synthesised acrylonitriles. Modifications of the terminal aromatic moiety were explored through epoxide installation flowed by flow chemistry mediated ring opening aminolysis with discreet sets of amines to the corresponding amino alcohols. Three new focused libraries were developed from substituted anilines, cyclic amines, and phenyl linked heterocyclic amines. The aniline-based compounds were inactive against the bacterial and fungal lines screened. The introduction of a cyclic, such as piperidine, piperazine, or morpholine, showed >50% inhibition when evaluated at 32 μg/mL compound concentration against methicillin-resistant Staphylococcus aureus. Examination of the terminal aromatic substituent via oxirane aminolysis allowed for the synthesis of three new focused libraries of afforded amino alcohols. Aromatic substituted piperidine or piperazine switched library activity from antibacterial to anti-fungal activity with ((Z)-2-(3,4-dichlorophenyl)-3-(4-(2-hydroxy-3-(4-methylpiperazin-1-yl)propoxy)phenyl)acrylonitrile), ((Z)-2-(3,4-dichlorophenyl)-3-(4-(2-hydroxy-3-(4-(4-hydroxyphenyl)piperazin-1-yl)propoxy)-phenyl)acrylonitrile) and ((Z)-3-(4-(3-(4-cyclohexylpiperazin-1-yl)-2-hydroxypropoxy)-phenyl)-2-(3,4-dichlorophenyl)-acrylonitrile) showing >95% inhibition of Cryptococcus neoformans var. grubii H99 growth at 32 μg/mL. While (Z)-3-(4-(3-(cyclohexylamino)-2-hydroxypropoxy)phenyl)-2-(3,4-dichlorophenyl)-acrylonitrile, (S,Z)-2-(3,4-dichlorophenyl)-3-(4-(2-hydroxy-3-(piperidin-1-yl)propoxy)phenyl)acrylonitrile, (R,Z)-2-(3,4-dichlorophenyl)-3-(4-(2-hydroxy-3-(piperidin-1-yl)propoxy)phenyl)acrylonitrile, (Z)-2-(3,4-dichlorophenyl)-3-(4-(2-hydroxy-3-(D-11-piperidin-1-yl)propoxy)phenyl)-acrylonitrile, and (Z)-3-(4-(3-(4-cyclohexylpiperazin-1-yl)-2-hydroxypropoxy)-phenyl)-2-(3,4-dichlorophenyl)-acrylonitrile 32 μg/mL against Staphylococcus aureus.
1. Introduction
The recent report detailing bacterial isolates resistant to polymyxin removes the last effective class of antibiotics against multi-drug resistant (MDR) bacteria [1]. With this loss of the last line of defenses against bacteria, the health community believes that this places us on the brink of a “post-antibiotic era” [2]. In the USA alone, >2 million people are subject to serious antibiotic resistant bacterial infections each year [3]. Across the USA and Europe, antibiotic resistant and fungal infections are responsible for annual deaths of >20,000 and 25,000, respectively [3,4]. Most concerning is the lack of efficacious compounds which treat Gram-negative bacteria [5,6,7]. There are only a handful of potential drugs which target Gram-negative bacteria in clinical trials offering significant benefits over current clinically used antibiotics [5,8,9].
Currently most of the antibiotic development is focused on improvements to existing antibiotics. This lack of structural novelty progressing to the clinic positions generational compound development as a critical strategy for the near future, but it is not known if this approach is sustainable or can addresses the growing problem of antibiotic resistance. Governments are starting to react through initiatives such as the Combating Antibiotic Resistant Bacteria Biopharmaceutical Accelerator (CARB-X) initiative, but a significant number of pharmaceutical companies are no longer pursuing antibiotic development [10,11,12]. Despite a recent resurgence in antibiotic drug discovery, to date there has been limited progress in developing novel mode of action antibiotics [5,8,9,13,14].
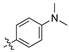
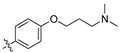
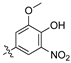
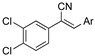
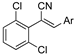
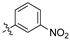
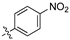
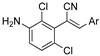
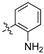
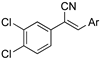
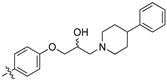
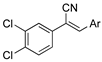
As part of a series of studies we have developed a number of classes of biologically active molecules [15,16,17,18,19,20]. With our in-house compound library, we sought to examine one of our compound series, that were originally designed to target the aryl hydrocarbon receptor (AhR) (Figure 1) [15,21,22,23], for activity against methicillin-resistant Staphylococcus aureus (MRSA), E. coli, Klebsiella pneumoniae, Acinetobacter baumannii and Pseudomonas aeruginosa; and the yeasts Candida albicans and Cryptococcus neoformans. We rationalized that the ease of synthetic access and further modification to amino alcohols, a class of compounds that spans the amphenicol and key macrolide antibiotics, positioned these compounds as logical development leads [24,25,26].
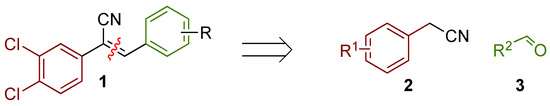
Figure 1.
An illustrative example of an AhR active ligand (1), with the proposed retrosynthesis, and potential analogue development from substituted phenyacetonitriles (2) and aldehydes (3).
2. Results
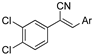
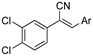
The acrylonitrile analogues 1a–1m (Library A) were generated as previously described from the Knoevenagel condensation of 3,4-dichlorophenylacetonitrile (2a) and substituted benzaldehydes (3a–m) (Supporting Information for detail) [15,21]. Screening of Library A, against the S. aureus, E. coli, K. pneumoniae, P. aeruginosa, A. baumannii bacterial and C. albicans, and C. neoformans fungal lines at an initial compound test concentration of 32 μg/mL gave the data presented in Table 1.

Table 1.
Percentage inhibition of bacteria and yeast at 32 μg/mL on treatment with Library A compounds 1a–1m.
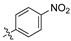
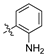
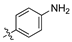
Analysis of the Library A data from (Table 1) revealed no broad-spectrum antibiotic activity 32 μg/mL with only low levels of bacteria and fungi death noted. Of note the 4-nitroanilino 1e showed 28% inhibition of P. aeruginosa; 4-anilino 1g, 37% inhibition of A. baumannii; 4-propyldimethyl amino ether 1j, 28% inhibition of A. baumannii and 3-nitrovanillin 1m, 64% inhibition of Candida albicans growth. While poor, all of the most active species retain a 4-substituent on the aromatic ring originating from the aldehydes (2).
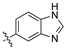
The second library, Library B, saw the use of phenylacetonitriles (2b–d; Scheme 1) and selected aldehydes spanning pyrrole carboxaldehydes (3n–q), 1H-benzo[d]imidazole carboxaldehyde (3r); and nitro-benzaldehydes (3s–w) and amino-benzaldehydes (3x, 3y) (Supporting Information) [21]. These compounds were subjected to bacterial and fungal screening as before and this data is presented in Table 2 [27].
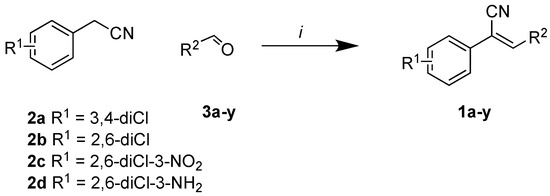
Scheme 1.
Reagents and Conditions. (i) PhCH2NMe3(OH), H2O, 50 °C, 5 h.
Table 2.
Percentage inhibition of bacteria and yeast growth at 32 μg/mL on treatment with Library B compounds 1n–1y.
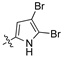
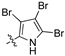
Examination of the data presented in Table 2 revealed a modest increase in activity of Library B analogues at the initial screening concentration (32 μg/mL) with 2-bromopyrrole 1o showing 37% inhibition of C. albicans growth; 2,3-dibromopyrrole 1p 32% inhibition of S. aureus growth; 1H-benzo[d]imidazole (1r), 27% inhibition of P. aeruginosa growth. With the 2,6-dichlorophenylacrylonitrile (2b) based 3′-nitrophenyl (1u), 26% inhibition of C. neoformans growth; the 2,6-dichloro-3-nitrophenylacetonitrile (2c) based 2′-nitrophenyl (1s) 29% inhibition of C. neoformans growth. The corresponding diamino analogue to (1v), compound (1y), demonstrated the best activity of any in Library B, exhibiting 48% inhibition against A. baumannii growth. These data support retention of a nitrogen substituent, with the enhanced activity possibly due to electronic effects. The activity increases were limited, and with this in mind we revisited Library A where the most active analogues suggested a preference for a 4-substituted aromatic moiety. As amino alcohol containing macrolides are known to have antibiotic properties, we rationalized that introducing such a moiety might confer enhanced activity to our lead compounds described above [24,25,26]. Access to the desired amino alcohol analogues was via introduction, and subsequent ring opening of an oxirane moiety with an amine.
In a typical experiment, epichlorohydrin (4) was coupled to the 4-hydroxybenzaldehyde (3z), yielding epoxyether (5) (Supporting Information) [28]. Subsequent Knoevenagel condensation with 2a under batch conditions gave the key intermediate (6). The desired libraries of amino alcohols were accessed through an amine (7a–w; see Table 3, Table 4 and Table 5 for details) mediated ring opening of 6. This protocol afforded a diverse array of amino alcohols which categorized as three libraries in total (8a–w) [28]. Library C (6, 8a–8d), from simple anilines; Library D (8e–8l) from cyclic aliphatic amines; and Library E (8m–8w) from aromatic piperidines and piperazines (see Table 3, Table 4 and Table 5 for details and Scheme 2).
Table 3.
Percentage inhibition of bacteria and yeast at 32 μg/mL on treatment with Library C compounds 6, 8a–8d.
Table 4.
Percentage inhibition of bacteria and yeast at 32 μg/mL on treatment with Library D compounds 8e–8l.
Table 5.
Percentage inhibition of bacteria and yeast at 32 μg/mL on treatment with Library E compounds 8m–8x.
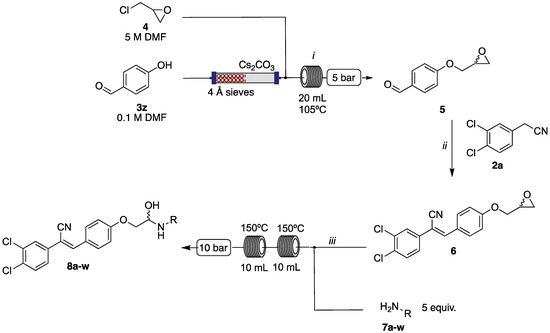
Scheme 2.
Reagents and conditions: (i) Vapourtec Easy-MedChem, 5 M 4 (anhy. DMF), 0.1 M 3z (anhy. DMF), 105 °C, 5 bar, 1.0 mL·min−1 (tr = 20 min); (ii) 1a, PhCH2NMe3(OH), H2O, 50 °C, 5h; (iii) RNH2 (7a–w) 0.05 M (EtOH), 6 0.01 M (EtOH), 150 °C, 10 bar, 1.0 mL·min−1 (tr = 20 min).
Library C retained the 3,4-dichlorophenylacetonitrile (2a), with the introduction of simple anilines via epoxide ring opening of 6 (Scheme 2). Screening of this library as before and analysis of this date revealed that no Library C analogue displayed broad spectrum activity. Analogues 6, 8a and 8d showed >20% inhibition against P. aeruginosa (Table 3).
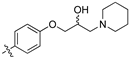
Library D (8e–8l) saw introduction of simple cyclic aliphatic and small heterocyclic rings. This minor modification had a major effect on the degree and number of active compounds (Table 4). Screening of these analogues at 32 μg/mL, and analysis of the data acquired revealed modest levels of activity with cyclohexyl 8e displaying 88% inhibition of S. aureus growth, and cyclohexyl piperidine (8f) 55% inhibition of S. aureus; piperidine (8g) 67 and 50% inhibition of S. aureus and C. albicans growth, respectively.
The introduction of the amino alcohol moiety also introduced a stereocenter with the added -OH moiety. To examine the effect of the two possible stereoisomers, optically pure (8h), (8i) and racemic (8g) where prepared. Both (8h) and (8i) returned essentially identical inhibition of S. aureus at 92 and 90% inhibition, respectively, suggesting no stereochemical preference for the -OH moiety. However, racemic (8g) displayed 20% lower S. aureus at 67% inhibition.
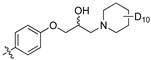
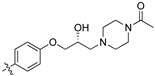
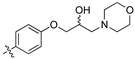
With other Library D analogues, per-deuteration of the piperidine moiety giving (8j) retained 90% inhibition of S. aureus growth. The piperazine acetamide (8k) and morpholino (8l), lost antibiotic activity but showed 53% and 35% growth inhibition of C. albicans, respectively. These latter compounds suggest that the presence of polar moieties on the heterocyclic ring are detrimental to antibiotic activity but may be favourable for fungal activity.
Having established the preferential installation of small cyclic aliphatics and heterocyclic moieties, Library E extended the substituents of these rings through the addition of a terminal aromatic moiety. The inclusion of the additional aromatic moiety saw modest activity, at the initial 32 μg/mL screening concentration, with acetophenone (8u) inhibiting P. aeruginosa growth (28%); 4-bromophenyl (8r) inhibited K. pneumoniae (28%); 4-(p-tolyl)piperazine (8p) inhibited C. neoformans (96%); and 4-methylpiperazinylphenol (8s) inhibited the C. albicans (65%) and C. neoformans (97%) fungal lines.
Having completed the initial screening of these focused libraries at 32 μg/mL, analogues showing >85% activity against at least one of the bacteria or fungi lines proceeded to full minimum inhibitory concentration (MIC) determination by broth micro-dilution dose response assay. These data are presented in Table 6.
Table 6.
Minimum inhibitory concentration (MIC) μg/mL of analogues 8e, h, i, j, q, s and x.
As can be seen from the data presented in Table 6, it is clear that the MIC values across the analogues evaluated was generally 32 μg/mL. Analogues (8e), (8h), (8i) and (8j) were all confirmed to be active with an MIC of 32 μg/mL against S. aureus, which is equipotent with the clinically used colistin, but less active than amoxicillin. These analogues did not show broad-spectrum antimicrobial activity with no activity at the highest tested concentration against the four Gram-negative strains, nor against the two yeasts tested. While this is modest, it does support the screening of biologically active scaffolds against alternative targets.
3. Discussion
Within the pool of 3,4-dichlorophenyl analogues, Library A, 4-NO2 (1e) was more active against P. aeruginosa than the corresponding 2-NO2 (1c) and 3-NO2 (1d). Modification of the 4-NO2 moiety to 4-NH2 with (1f) saw a loss of P. aeruginosa activity, but modest activity against A. baumannii. This was the sole aniline analogue to display activity. Incorporation of the N,N-dimethylamino moiety (1g) resulted in a slight potency reduction, whereas nitro-vanillin (1m) showed no antibiotic activity, but modest effects against C. albicans. Caution is required in ascribing definitive SAR outcomes in these series as they involve analysis of a single datapoint.
Modification of either the parent phenylacrylonitrile via the 2,6-dichloro or 2,6-dichloro-3-nitro or 2,6-dichloro-3-amino phenylacrylonitrile or parent aldehyde with pyrrole, imidazole and nitrophenyl substituents all displayed lower levels of activity than Library A analogues. Excepting 3′-amino (1y), with modest activity against A. baumannii. However, within this Library general trends were evident, with the di-Br (1p) the most active against S. aureus; mono-Br (1o) against C. albicans and benzimidazole (1r) against P. aeruginosa.
The development of analogues with the inclusion of the β-aminoalcohol moiety, while retaining the 3,4-dichlorophenyl moiety was readily accomplished in a series of simple flow chemistry mediated steps. However, the retention of the terminal aromatic moieties with Library C showed essentially no activity. The use of cyclic and heterocyclic amines in the oxiraine aminolysis step, gave analogues 8e–8l. This removal of the terminal aromatic moiety was rewarded with a significant increase in antibiotic activity, especially against S. aureus (8e–8j, >50% inhibition at 32 μg/mL). Individual enantiomers (8h) and (8i) were equipotent (>90%), yet the racemate (8g) showed only partial inhibition of MRSA at 32 μg/mL (67%), supporting a negative cooperability of these isomers. This stereochemical preference will be examined and reported on in a future study. Alternatively, as the deuterated racemate (8j) showed the same activity as the pure isomers (8h) and (8i), incorporation of such moieties may negate the need to isolate single pure stereoisomers. This supported further development of this series with the introduction of an aromatic moiety appended to a saturated six-membered ring (i.e., piperazine and piperidine). Incorporation of polar moieties in the terminus of the heterocyclic, viz N-Ac (8k) and morpholino (18l), were detrimental to S. aureus activity, but these analogues showed modest activity against C. albicans.
Within Library E switching the ether linker position from 4- (8o) to 3- (8n) and 2- (8m) was detrimental to activity. These analogues were essentially devoid of antibacterial activity. The switch to antifungal activity was unexpected, but combined with the observed antibiotic activity strongly supports the on-going development of this class of compounds as novel antibacterial/antifungal agents. Of the analogues with antifungal activity, the piperidine (8g, 8x) is more prevalent than piperazine (8p). This is fortuitous as phenylpiperazines are known to form highly reactive iminoquinone moieties (a potential toxicophore) capable of reacting with glutathione [29,30,31,32]. Fungal active compounds show higher specificity towards the yeasts with both (8p) and (8s) inhibiting >95% C. neoformans growth at 32 μg/mL. Analogue (8s) shows promising activity against both yeasts, C. albicans and C. neoformans with 65 and 97% inhibition at 32 μg/mL, respectively. These compounds were not antibacterial.
Examination of the most active analogues across all five libraries (8e, 8h, 8i, 8j, 8s and 8x) in a broth microdilution dose response assay revealed (8e), and (8h–i) as having MIC values of 32 μg/mL (which is equivalent to an IC50 value of approximately 40 μM), which represents a significant enhancement relative to lead 1. To further explore the efficacy of these compounds, cytotoxicity screening was conducted in HEK393 cells and all evaluated analogues returned growth inhibition values >20 μM.
4. Conclusions
Five focused libraries totaling 49 compounds (plus the oxirane intermediate, 6) were rapidly assembled. With the exception of Library C, active compounds were observed across all focused libraries. The most promising antibiotic activity was observed with (8e–8i) against S. aureus. A negative cooperativity between isomers in the racemic (8e) was noted (with single enantiomers (8g) and (8i) ca 25% more active). This effect was ameliorated through the introduction of a per-deuterated piperidine moiety (8j). The introduction of aromatic substituents to the piperidine or piperazine moiety resulted in a transition from antibiotic to antifungal activity with (8p), (8s) and (8x) showing excellent activity at >95% inhibition of C. neoformans at 32 μg/mL (which is equivalent to an IC50 value of approximately 40 μM).
From this study we have identified a novel scaffold that is active against S. aureus (8e, 8h–8j), and with structural manipulation shows differential activity against C. albicans (8p, 8s and 8x).
5. Experimental
5.1. Biology
All bacteria and yeast strains were sourced from the American Type Culture Collection (ATCC) and antimicrobial assays were conducted by the Community for Open Antimicrobial Drug Discovery (CO-ADD) [33,34,35]. Relevant positive inhibitor controls for each species were included in all assays.
Compounds for inhibitory assays were prepared in DMSO and water to a final testing concentration of (i) 32 μg/mL or 10 μM for single point concentration screening, or (ii) serially diluted two-fold down the wells for 8 times with a top testing concentration of 32 μg/mL or 10 μM for minimum inhibitory concentration (MIC) dose response assays, in 384-well, non-binding surface plate (NBS, Corning 3460) for each bacterial/yeast strain to be tested, and in duplicate (n = 2), Final maximum DMSO concentration was 1%. All of the sample-preparations were carried out using liquid handling robots.
5.2. Antibacterial Assays
For both single concentration screening and dose response assays, all bacteria were cultured in Cation-adjusted Mueller Hinton broth (CAMHB) at 37 °C overnight. A sample of each culture was then diluted 40-fold in fresh broth and incubated at 37 °C for 1.5–3 h. The resultant mid-log phase cultures were diluted (CFU/mL measured by OD600), then added to each well of the compound containing plates, giving a cell density of 5 × 105 CFU/mL and a total volume of 50 μL in 384-well non-binding surface plates (NBS, Corning 3460). All of the plates were covered and incubated at 37 °C for 18 h without shaking.
Inhibition of bacterial growth was determined measuring absorbance at 600 nm (OD600), using a Tecan M1000 Pro monochromator plate reader. The percentage of growth inhibition was calculated for each well, using the negative control (media only) and positive control (bacteria without inhibitors) on the same plate as references.
The percentage of growth inhibition was calculated for each well, using the negative control (media only) and positive control (bacteria without inhibitors) on the same plate. For single point concentration assays the final recorded readout was the percentage inhibition at the concentration tested (i.e., % at 32 μg/mL or 10 μM). For the dose response MIC assay, the MIC was determined as the lowest concentration at which the growth was fully inhibited, defined by an inhibition ≥ 80% by spectrophotometric read, the equivalent 100% inhibition by eye.
5.3. Antifungal Assay
For both single concentration screening and dose response assays, fungi/yeast strains were cultured for three days on yeast extract-peptone dextrose (YPD) agar at 30 °C. A yeast suspension of 1 × 106 to 5 × 106 CFU/mL (as determined by OD530) was prepared from five colonies. The suspension was subsequently diluted and added to each well of the compound-containing plates giving a final cell density of fungi suspension of 2.5 × 103 CFU/mL and a total volume of 50 μL in 384-well non-binding surface plates (NBS, Corning 3460). All plates were covered and incubated at 35 °C for 36 h without shaking.
Growth inhibition of C. albicans was determined measuring absorbance at 630 nm (OD630), while the growth inhibition of C. neoformans was determined measuring the difference in absorbance between 600 and 570 nm (OD600–570), after the addition of resazurin (0.001% final concentration) and incubation at 35 °C for 2 h. The absorbance was measured using a Biotek Multiflo Synergy HTX plate reader.
For both yeasts, the percentage of growth inhibition was calculated for each well, using the negative control (media only) and positive control (fungi without inhibitors) on the same plate. For single point concentration assays the final recorded readout was the percentage inhibition at the concentration tested (i.e., % at 32 μg/mL or 10 μM). For the dose response MIC assay, the MIC was determined as the lowest concentration at which the growth was fully inhibited, defined by an inhibition ≥80% for C. albicans and an inhibition ≥70% for C. neoformans. Due to a higher variance in growth and inhibition, a lower threshold was applied to the data for C. neoformans.
5.4. Chemistry
General Methods
All reactions were performed using standard laboratory equipment and glassware. Solvents and reagents were purchased from Sigma Aldrich, Alfa Aesar or AK Scientific (Australia) and used as received. Organic solvents were of bulk quality. Organic solvent extracts were dried with magnesium sulfate (MgSO4), and dried under reduced pressure with either Büchi or Heidolph rotary evaporators. Melting points were recorded in open capillaries on a Stuart SMP11 Melting Point Apparatus. Where available, literature values are provided and appropriately referenced. Electrospray mass spectra were recorded using HPLC-grade 10% acetonitrile/H2O or 100% acetonitrile (with 0.1% formic acid) as carrier solvents on an Agilent Technologies 1260 Infinity UPLC system with a 6120 Quadrupole LC/MS in electrospray ionization (ESI) positive and negative modes. TLC was performed on Merck silica gel 60 F254 pre-coated aluminum plates with a thickness of 0.2 mm. Column chromatography was performed under ‘flash’ conditions on Merck silica gel 60 (230–400 mesh).
Nuclear magnetic resonance (NMR) spectroscopy was performed on a Bruker Avance III 400 MHz spectrometer, where proton NMR (1H NMR) spectra and carbon NMR (13C NMR) spectra were acquired at 400 and 100 MHz, respectively, or a Bruker Avance III 600 MHz spectrometer, where proton NMR (1H NMR) spectra and carbon NMR (13C NMR) spectra were acquired at 600 and 150 MHz, respectively. All spectra were recorded in deuterated dimethyl sulfoxide (DMSO-d6) or deuterated acetone (acetone-d6) obtained from Cambridge Isotope Laboratories Inc. Chemical shifts (δ) were measured in parts per million (ppm) and referenced against the internal reference peaks. Coupling constants (J) were measured in Hertz (Hz). NMR assignments were determined through the interpretation of one- and two-dimensional spectra. Multiplicities are denoted as singlet (s), broad singlet (bs), doublet (d), doublet of doublets (dd), doublet of doublet of doublets (ddd), triplet (t), quartet (q), triplet of doublets (td), doublet of triplets (dt) and multiplet (m). Peaks are listed in decreasing chemical shift in the following format: chemical shift (integration (1H), multiplicity (1H), coupling constant (1H). The Biotage® initiator+ was used to perform microwave reactions.
Experimental details for all previously published compounds can be found in the Supplementary Material. Spectral data for all compounds can be found in the Supplementary material.
General Procedure 1: - The aldehyde (1.05 eq., 1.05 mmol), was added to a vigorously stirred solution of water (10 mL) and heated to 50 °C. The dichlorophenylacetonitrile (1 mmol) was then slowly added forming a suspension. After 5–10 min of stirring, 40% PhCH2NMe3(OH) (7 mL) was added dropwise. After complete addition, the reaction vessel stirred at 50 °C for 5 h. After this period, the solution was filtered hot, washed with warm water and purified by either recrystallisation or column chromatography.
General Procedure 2: - (Z)-2-(3,4-dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)acrylonitrile (15, 1 eq.) was combined with the required amine (either 1.5 or 2 eq., as stated) and 20 mL ethanol. The solution was irradiated at 120 °C for 20 min. Upon chilling, the desired product was isolated via vacuum filtration.
General Procedure 3: - Using a Vapourtec RS-400 equipped with fraction collection kit and auto-sampler, a 2.0 mL sample loop was charged with a 0.4 M solution of epoxide (15) in toluene. An additional 2 mL sample loop was charged with a 2.8 M amine solution in ethanol (as stated). The solutions were flowed together and the resulting stream was then passed through two PFA coil reactors in series at 150 °C, 10 bar back pressure and 0.5 mL·min−1 (residence time 40 min). The resulting reaction mixture was collected, concentrated in vacuo and purified as described.
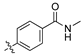
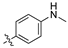
(Z)-4-(2-cyano-2-(3,4-dichlorophenyl)vinyl)-N-methylbenzamide (1a).
4-Formylbenzoic acid (0.300 g, 2 mmol) and oxalyl chloride (1.3 eq., 0.330 g, 2.6 mmol (1.3 mL 2.0 M DCM solution)) were combined with 15 mL DCM. DMF (1.5 mL) was added dropwise, the flask was stoppered and the reaction left to stir at ambient temperature until TLC analysis indicated reaction was complete (1 h). The solvent was removed in vacuo and the resultant brown oil was carried directly through to the next step.
4-Formylbenzoyl chloride (0.336 g, 2 mmol), K2CO3 (2 eq., 0.553 g, 4 mmol) and methylamine (1.2 eq., 210 µL 40 wt.% aqueous solution, 2.4 mmol) were combined with 5 mL diethyl ether and stirred vigorously at RT overnight. The reaction was quenched with 1 M HCl (10 mL), diluted with diethyl ether (30 mL), washed with sat. NaHCO3 (3 × 20 mL), brine (20 mL), dried over MgSO4 and the solvent removed in vacuo.
The desired product ((Z)-4-(2-cyano-2-(3,4-dichlorophenyl)vinyl)-N-methylbenzamide, 1a) was subsequently prepared according to general procedure 1, from the freshly prepared 4-formyl-N-methylbenzamide (1.05 eq., 95 mg, 0.56 mmol) and 3,4-dichlorophenyl acetonitrile (103 mg, 0.55 mmol). The crude solid was taken up in 40 mL 2:1 diethyl ether:EtOAc, washed with water (3 × 20 mL), 1 M NaOH (3 × 10 mL) and brine (2 × 20 mL). The organic extract was dried over MgSO4 and the solvent removed in vacuo to afford the desired product as a tan solid (148 mg, 81%), m.p.: 222–225 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.58 (q, J = 4.2 Hz, NH), 8.25 (s, 1H), 8.08 (d, J = 2.2 Hz, 1H), 8.00 (d, J = 8.5 Hz, 2H), 7.97 (d, J = 8.5 Hz, 2H), 7.82 (d, J = 8.5 Hz, 1H), 7.75 (dd, J = 8.5, 2.3 Hz, 1H), 2.81 (d, J = 4.5 Hz, 3H); 13C NMR (151 MHz, DMSO-d6) δ 165.8, 143.9, 136.3, 135.7, 134.3, 132.1, 132.0, 131.3, 129.2 (2C), 127.6 (2C), 127.5, 126.3, 117.2, 109.3, 26.3; IR υmax/cm−1: 3314 (NH), 2931 (C=C), 2216 (CN), 1634 (C=O), 816 (C-Cl); LRMS (ESI+) m/z: 331 (C13H13Cl2N2O2) [M+H]; HRMS: Exact mass calculated for C13H13Cl2N2O2 [M+H], 331.0339.
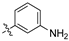
(Z)-3-(2-aminophenyl)-2-(3,4-dichlorophenyl)acrylonitrile (1f).
(Z)-2-(3,4-Dichlorophenyl)-3-(2-nitrophenyl)acrylonitrile (2c, 1.239 g, 3.88 mmol) and iron powder (9 equiv, 1.517 g, 27.16 mmol) were combined with a mixture of glacial acetic acid (4 mL), ethanol (4 mL), and water (2 mL) and exposed to ultrasonic irradiation at 30 °C for 7 h. The reaction mixture was diluted with ethanol (30 mL) and filtered through a pad of Celite, before concentration under reduced pressure to afford the desired product as an off-white solid (650 mg, 58%), m.p.: 157–159 °C. 1H NMR (400 MHz, acetone-d6): δ 7.92 (s, 1H), 7.77 (d, J = 1.2 Hz, 1H), 7.73 (d, J = 5.2 Hz, 1H), 7.70 (d, J = 5.6 Hz, 1H), 7.60 (d, J = 5.6 Hz, 1H), 7.56 (m, 2H), 7.26 (td, J = 9.2, 4.8, 0.4 Hz, 1H); 13C NMR (101 MHz, acetone-d6): δ 173.0, 156.5, 148.3, 139.4, 138.5, 133.2, 132.3, 132.0, 132.0, 130.7, 130.1, 128.7, 124.6, 123.7, 123.2; IR υmax/cm−1: 3073 (NH2), 2970 (NH2), 2219 (CN), 821 (C-Cl); LRMS (ESI−) m/z: 288 (C15H9Cl2N2) [M-H]; HRMS: Exact mass calculated for C15H11Cl2N2 [M+H], 289.0924.
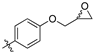
(Z)-2-(3,4-Dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)-acrylonitrile (6) [21].
4-Hydroxybenzaldehyde (3z) (305 mg, 2.5 mmol) in 25 mL acetonitrile was passed through a series of Omnifit columns, the first containing molecular sieves, the second contained cesium carbonate and sand (50%:50% w/w) at a flow rate of 0.5 mL/min. The reactant stream was then met by a secondary stream containing epichlorohydrin (17 mL, with 20 mL DMF and 63 mL acetonitrile, flow rate 0.5 mL/min), and together the reactant streams were passed through a Vapourtec static mixer reactor (20mL) heated at 105 °C and held at 5 bar back pressure. At the completion of the reaction, the product solution was evaporated under reduced pressure, diluted with ethyl acetate (50 mL), washed with water (3 × 50mL), and then brine (1 × 50 mL), dried over MgSO4 and the solvent removed under reduced pressure to afford 4-(oxiran-2-ylmethoxy)benzaldehyde (5) as a light brown oil (364 mg, 82%).
The desired product ((Z)-2-(3,4-dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)-acrylonitrile (6)) was then prepared according to general procedure 1 from the freshly prepared (4-(oxiran-2-ylmethoxy)benzaldehyde (5, 1.05 eq., 12.177 g, 68 mmol) and 3,4-dichlorophenylacetonitrile (1a, 12.109 g, 65 mmol) The crude solid was recrystallized from ethanol to afford the desired product (6) as a yellow solid (15.805 g, 70%), m.p.: 121–124 °C. 1H NMR (400 MHz, acetone-d6) δ 8.02 (d, J = 8.8 Hz, 2H), 7.97 (s, 1H), 7.94 (d, J = 1.4 Hz, 1H), 7.73–7.68 (m, 2H), 7.15 (d, J = 8.8 Hz, 2H), 4.47 (dd, J = 11.4, 2.6 Hz, 1H), 4.00 (dd, J = 11.4, 6.4 Hz, 1H), 3.36 (td, J = 6.6, 2.6 Hz, 1H), 2.87 (dd, J = 4.8, 4.4 Hz, 1H), 2.75 (dd, J = 5.2, 2.4 Hz, 1H); 13C NMR (101 MHz, acetone-d6) δ 162.0, 144.6, 136.4, 133.5, 132.8, 132.5 (2C), 132.0, 128.2, 127.5, 126.5, 118.5, 116.0 (2C), 106.6, 70.4, 50.4, 44.4; IR υmax/cm−1: 3063 (C=C), 2210 (CN), 1255 (arom. C-O), 811 (C-Cl); LRMS: (ESI−) m/z: 368 (C18H12Cl2NNaO2) [M+Na-H].
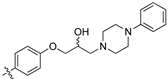
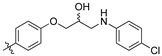
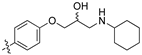
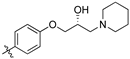
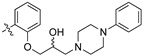
(S,Z)-2-(3,4-Dichlorophenyl)-3-(4-(2-hydroxy-3-(piperidin-1-yl)propoxy)phenyl)acrylonitrile (8h).
4-Hydroxybenzaldehyde (3z, 1.071 g, 8.77 mmol), cesium carbonate (1.5 eq., 4.287 g, 13.2 mmol) and [S]-(+)-glycidyl-3-nitrobenzene sulfonate (1.1 eq., 2.500 g, 9.64 mmol) were combined with 30 mL DMF. Heated to 80 °C for 4 h. Reaction was diluted with water (50 mL) and EtOAc (30 mL), washed with water (5 × 30 mL) and brine (30 mL). The aqueous layer was extracted with EtOAc (3 × 30 mL). Organic extracts combined, dried over MgSO4 and concentrated under reduced pressure to afford the desired product as a pale brown oil. Yield not calculated and material carried directly through.
(S)-4-(oxiran-2-ylmethoxy)benzaldehyde (1.1 eq., 1.603 g, 8.77 mmol) was combined with 15 mL water and heated to 50 °C for 10 min while stirring. 3,4-Dichlorophenylacetonitrile (2a, 1.483 g, 7.97 mmol) was then added and the reaction stirred for a further 10 min. 10 mL Benzyltrimethylammonium hydroxide (40 wt % aq. solution) was added dropwise and the reaction left to stir at 50 °C for 6 h. Filtered under vacuum, washed with water (2 × 10 mL) and recrystallized from ethanol to afford the desired product as a yellow solid (1.7198 g, 62% over 2 steps).
The desired product (8h) was then prepared according to general procedure 3 from (S,Z)-2-(3,4-dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)acrylonitrile (346 mg, 1 mmol) and piperidine (1.5 eq., 148 μL, 1.5 mmol) to afford the desired compound as a yellow solid (305 mg, 74%), m.p.: 100–102 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.09 (s, 1H), 8.00 (d, J = 1.9 Hz, 1H), 7.96 (d, J = 8.7 Hz, 2H), 7.76 (d, J = 8.5 Hz, 1H), 7.68 (dd, J = 8.5, 1.9 Hz, 1H), 7.13 (d, J = 8.8 Hz, 2H), 4.86 (s, 1H), 4.10–4.07 (m, 1H), 4.00–3.95 (m, 2H), 2.41–2.31 (m, 5H), 1.50–1.47 (m, 4H), 1.37 (d, J = 4.8 Hz, 2H); 13C NMR (151 MHz, DMSO-d6) δ 161.2, 144.3, 134.9, 132.0, 131.5 (2C), 131.2, 131.1, 127.0, 125.76, 125.74, 117.9, 115.1 (2C), 104.3, 71.5, 66.4, 61.7, 54.8 (2C), 25.7 (2C), 24.0; IR υmax/cm−1: 3300 cm−1 (OH), 2933 cm−1 (N-CH2), 2216 cm−1 (CN), 1182 cm−1 (C-O), 813 cm−1 (C-Cl); LRMS: (ESI+) m/z: 431 (C23H25Cl2N2O2) [M+H]; HRMS: Exact mass calculated for C23H25Cl2N2O2 [M+H], 431.1288.
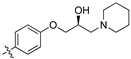
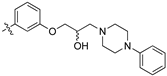
(R,Z)-2-(3,4-Dichlorophenyl)-3-(4-(2-hydroxy-3-(piperidin-1-yl)propoxy)phenyl)acrylonitrile (8i).
4-Hydroxybenzaldehyde (3z, 0.978 g, 8.00 mmol), cesium carbonate (1.5 eq., 3.910 g, 12.0 mmol) and [R]-(−)-glycidyl-3-nitrobenzene sulfonate (1.1 eq., 2.282 g, 8.80 mmol) were combined with 30 mL DMF. Heated to 80 °C for 4 h. Reaction was diluted with water (50 mL) and EtOAc (30 mL), washed with water (5 × 30 mL) and brine (30 mL). Aqueous back-extracted with EtOAc (3 × 30 mL). Organic extracts combined, dried over MgSO4 and concentrated under reduced pressure to afford the desired product as a pale brown oil (1.279 g, 90%).
(R)-4-(oxiran-2-ylmethoxy)benzaldehyde (1.05 eq., 1.511 g, 6.46 mmol) was combined with 15 mL water and heated to 50 °C for 10 min while stirring. 3,4-Dichlorophenylacetonitrile (2a, 1.144 g, 6.15 mmol) was then added, and the reaction stirred for a further 10 min. 10 mL benzyltrimethylammonium hydroxide (40 wt% aq. solution) was then added dropwise and the reaction left to stir at 50 °C for 6 h. Filtered under vacuum, washed with water (2 × 10 mL) and recrystallized from ethanol to afford the desired product as a yellow solid (1.5043 g, 71%).
The desired product (8i) was then prepared according to general procedure 3 from (R,Z)-2-(3,4-dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)acrylonitrile (346 mg, 1 mmol) and piperidine (1.5 eq., 148 µL, 1.5 mmol) to afford the desired compound as a yellow solid (264 mg, 64%), m.p.: 101–103 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.10 (s, 1H), 8.01 (d, J = 2.1 Hz, 1H), 7.96 (d, J = 8.8 Hz, 2H), 7.77 (d, J = 8.5 Hz, 1H), 7.69 (dd, J = 8.5, 2.1 Hz, 1H), 7.13 (d, J = 8.8 Hz, 2H), 4.86 (d, J = 3.3 Hz, 1H), 4.10–4.05 (m, 1H), 4.00–3.95 (m, 2H), 2.41–2.31 (m, 5H), 1.50–1.47 (m, 4H), 1.37 (d, J = 4.7 Hz, 2H); 13C NMR (151 MHz, DMSO-d6) δ 161.2, 144.3, 135.0, 132.0, 131.5 (2C), 131.2, 131.1, 127.0, 125.77, 125.75, 117.9, 115.1 (2C), 104.3, 71.5, 66.4, 61.7, 54.8 (2C), 25.7 (2C), 24.0; IR υmax/cm−1: 3366 cm−1 (OH), 2932 cm−1 (N-CH2), 2211 cm−1 (CN), 1182 cm−1 (C-O), 813 cm−1 (C-Cl); LRMS: (ESI+) m/z: 431 (C23H25Cl2N2O2) [M+H]; HRMS: Exact mass calculated for C23H25Cl2N2O2 [M+H], 431.1288.
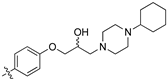
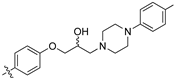
(Z)-2-(3,4-Dichlorophenyl)-3-(4-(2-hydroxy-3-(4-methylpiperazin-1-yl)propoxy)phenyl)acrylonitrile (8p) [15].
Prepared according to general procedure 3 from (Z)-2-(3,4-dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)acrylonitrile (6, 346 mg, 1 mmol) and 1-methyl-4-phenylpiperazine (1.5 eq., 264 mg, 1.5 mmol) to afford the desired compound as a bright yellow solid (448 mg, 89%), m.p.: 147–150 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.10 (s, 1H), 8.00 (d, J = 1.9 Hz, 1H), 7.96 (d, J = 8.7 Hz, 2H), 7.76 (d, J = 8.5 Hz, 1H), 7.68 (dd, J = 8.5, 1.9 Hz, 1H), 7.14 (d, J = 8.7 Hz, 2H), 7.01 (d, J = 8.2 Hz, 2H), 6.81 (d, J = 8.4 Hz, 2H), 4.97 (d, J = 4.2 Hz, 1H), 4.12 (d, J = 6.7 Hz, 1H), 4.00 (d, J = 8.1 Hz, 2H), 3.06–3.05 (m, 4H), 2.64–2.56 (m, 4H), 2.43 (dd, J = 12.7, 5.6 Hz, 1H), 2.19 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 161.2, 149.0, 144.3, 135.0, 132.0, 131.5 (2C), 131.2, 131.1, 129.4 (2C), 127.5, 127.0, 125.81, 125.78, 117.9, 115.6 (2C), 115.2 (C2), 104.4, 71.4, 66.5, 60.9, 53.5 (2C), 48.8 (2C), 20.0; IR υmax/cm−1: 3333 (OH), 2816 (N-CH2), 2208 (CN), 1182 (C-O), 817 (C-Cl); LRMS (ESI+) m/z: 522 (C29H30Cl2N3O2) [M+H]; HRMS: Exact mass calculated for C29H30Cl2N3O2 [M+H], 522.1710. Found 522.1713.
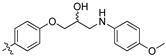
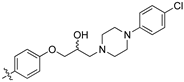
(Z)-3-(4-(3-(4-(4-Chlorophenyl)piperazin-1-yl)-2-hydroxypropoxy)phenyl)-2-(3,4-dichlorophenyl)-acrylonitrile (8q) [15].
Prepared according to general procedure 3 from (Z)-2-(3,4-dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)acrylonitrile (6, 200 mg, 0.58 mmol) and 1-(4-chlorophenyl)piperazine (2 eq., 227 mg, 1.16 mmol) to afford the desired compound as a cream colored solid (250 mg, 82%), m.p.: 159–162 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.10 (s, 1H), 8.01 (d, J = 2.0 Hz, 1H), 7.96 (d, J = 8.8 Hz, 2H), 7.77 (d, J = 8.5 Hz, 1H), 7.69 (dd, J = 8.5, 2.1 Hz, 1H), 7.21 (d, J = 8.9 Hz, 2H), 7.14 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 9.0 Hz, 2H), 4.99 (d, J = 2.6 Hz, 1H), 4.12–4.11 (m, 1H), 4.05–4.00 (m, 2H), 3.12 (t, J = 4.3 Hz, 4H), 2.65–2.57 (m, 4H), 2.44 (dd, J = 13.1, 4.4 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.2, 149.9, 144.3, 135.0, 132.1, 131.6 (2C), 131.24, 131.15, 128.6 (2C), 127.0, 125.83, 125.81, 122.3, 117.9, 116.8 (2C), 115.2 (2C), 104.4, 71.4, 66.5, 60.9, 53.3 (2C), 48.1 (2C); IR υmax/cm−1: 3355 (br, OH), 2830 (N-CH2), 2216 (CN), 1260 (C-O), 815 (C-Cl); LRMS: (ESI+) m/z: 542 (C28H27Cl3N3O2) [M+H); HRMS: Exact mass calculated for (C28H27Cl3N3O2 [M+H], 542.1163. Found 542.1147.
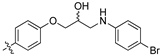
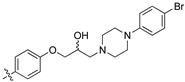
(Z)-3-(4-(3-(4-(4-Bromophenyl)piperazin-1-yl)-2-hydroxypropoxy)phenyl)-2-(3,4-dichlorophenyl)acrylonitrile (8r) [15].
Prepared according to general procedure 3 from (Z)-2-(3,4-dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)acrylonitrile (6, 200 mg, 0.58 mmol) and 1-(4-bromophenyl)piperazine (2 eq., 279 mg, 1.16 mmol) to afford the desired compound as a cream coloured solid (326 mg, 96%), m.p.: 162–165 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.10 (s, 1H), 8.00 (s, 1H), 7.96 (d, J = 8.6 Hz, 2H), 7.76 (d, J = 8.6 Hz, 1H), 7.68 (d, J = 6.8 Hz, 1H), 7.33 (d, J = 8.8 Hz, 2H), 7.14 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 4.99 (s, 1H), 4.11 (d, J = 6.5 Hz, 1H), 4.01–3.99 (m, 2H), 3.12 (s, 4H), 2.63–2.56 (m, 4H), 2.46–2.41 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.2, 150.2, 144.3, 134.9, 132.0, 131.5 (2C), 131.4 (2C), 131.2, 131.1, 127.0, 125.80, 125.77, 117.9, 117.2 (2C), 115.1 (2C), 109.9, 104.4, 71.4, 66.5, 60.9, 53.3 (2C), 47.9 (2C); IR υmax/cm−1: 3300 (OH), 2830 cm (N-CH2), 2216 (CN), 1259 cm (C-O), 814 cm (C-Cl); LRMS: (ESI+) m/z: 588 (C28H27BrCl2N3O2) [M+H]; HRMS: Exact mass calculated for (C28H27BrCl2N3O2) [M+H], 588.0638. Found 588.0617.
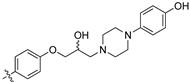
(Z)-2-(3,4-Dichlorophenyl)-3-(4-(2-hydroxy-3-(4-(4-hydroxyphenyl)piperazin-1-yl)propoxy)-phenyl)acrylonitrile (8s).
Prepared according to general procedure 3 from (Z)-2-(3,4-dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)acrylonitrile (6, 346 mg, 1.0 mmol) and 1-(4-hydroxyphenyl)piperazine (1.5 eq., 267 mg, 1.5 mmol). The resultant residue was recrystallised from methanol to afford the desired compound as a brown solid (330 mg, 63%), m.p.: 162–164 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.81 (bs, OH), 8.09 (s, 1H), 8.00 (d, J = 2.3 Hz, 1H), 7.96 (d, J = 8.9 Hz, 2H), 7.76 (d, J = 8.5 Hz, 1H), 7.68 (dd, J = 8.5, 2.3 Hz, 1H), 7.14 (d, J = 8.9 Hz, 2H), 6.77–6.76 (m, 2H), 6.65–6.63 (m, 2H), 4.97 (s, 1H), 4.12–4.10 (m, 1H), 4.02–3.98 (m, 2H), 2.95 (t, J = 4.7 Hz, 4H), 2.60 (ddd, J = 18.1, 10.4, 5.0 Hz, 4H), 2.42 (dd, J = 12.6, 6.1 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 161.2, 150.8, 144.3, 144.2, 134.9, 132.0, 132.5 (2C), 131.2, 131.1, 127.0, 125.79, 125.77, 117.9, 117.7 (2C), 115.4 (2C), 115.1 (2C), 104.4, 71.4, 66.5, 60.9, 53.7 (2C), 50.1 (2C); IR υmax/cm−1: 3366 (OH), 2212 (CN), 1370 (OH), 1182 (C-O), 815 (C-Cl); LRMS (ESI+) m/z: 524 (C29H28Cl2N3O3) [M+H]; HRMS: Exact mass calculated for (C28H28Cl2N3O3) [M+H], 524.1403.
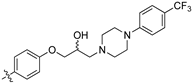
(Z)-2-(3,4-Dichlorophenyl)-3-(4-(2-hydroxy-3-(4-(4-(trifluoromethyl)phenyl)piperazin-1-yl)propoxy)-phenyl)acrylonitrile (8t) [15].
Prepared according to general proceure 3 from (Z)-2-(3,4-dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)acrylonitrile (6, 346 mg, 1 mmol) and 1-(4-trifluoromethylphenyl)piperazine (2 eq., 461 mg, 2 mmol) to afford the desired compound as a pale yellow solid (486 mg, 84%), m.p.: 159–161 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.09 (s, 1H), 8.00 (d, J = 1.8 Hz, 1H), 7.96 (d, J = 8.7 Hz, 2H), 7.75 (d, J = 8.5 Hz, 1H), 7.68 (dd, J = 8.5, 1.8 Hz, 1H), 7.48 (d, J = 8.6 Hz, 2H), 7.14 (d, J = 8.7 Hz, 2H), 7.04 (d, J = 8.6 Hz, 2H), 5.00 (d, J = 3.3 Hz, 1H), 4.12–4.11 (m, 1H), 4.02–3.99 (m, 2H), 3.27 (s, 4H), 2.64–2.55 (m, 4H), 2.47–2.42 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.2, 153.3, 144.3, 134.9, 132.1, 131.5, 131.2, 131.1, 127.0, 126.1 (q, J = 3.4 Hz, 2C), 125.83, 125.78, 125.0 (q, J = 272.7 Hz), 117.9, 117.8 (q, J = 32.2 Hz), 115.2 (2C), 114.1 (2C), 104.4, 71.4, 66.6, 60.9, 53.2 (2C), 47.1 (2C); 19F NMR (376 MHz, DMSO-d6, CF3CO2H) δ -56.1; IR υmax/cm−1: 3300 (OH), 2208 (CN), 1224 (C-F), 1179 (C-F), 1103 (C-O), 819 (C-Cl); LRMS (ESI+) m/z: 576 (C29H27Cl2F3N3O2) [M+H]; HRMS: Exact mass calculated for (C29H27Cl2F3N3O2) [M+H], 576.4442. Found 576.4311.
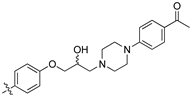
(Z)-2-(3,4-Dichlorophenyl)-3-(4-(3-(4-(4-acetamidophenyl)piperazin-1-yl)-2-hydroxypropoxy)phenyl)-acrylonitrile (8u) [15].
Prepared according to general procedure 3 from (Z)-2-(3,4-dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)acrylonitrile (6, 193 mg, 0.56 mmol) and 4′-piperazineacetophenone (2 eq., 227 mg, 1.11 mmol) to afford the desired compound as an orange solid (240 mg, 78%), m.p.: 149–153 °C. 1H NMR (400 MHz, acetone-d6) δ 8.02 (dd, J = 8.8, 1.6 Hz, 2H), 7.98 (s, 1H), 7.94 (d, J = 1.4 Hz, 1H), 7.86 (dd, J = 8.8, 1.6 Hz, 1H), 7.75–7.69 (m, 2H), 7.15 (dd, J = 8.8, 1.6 Hz, 2H), 6.99 (dd, J = 8.8, 1.6 Hz, 2H) 4.24 (dd, J = 8.8, 3.6 Hz, 1H), 4.20–4.16 (m, 1H), 4.13 (dd, J = 8.9, 5.2 Hz, 1H), 3.41 (t, J = 5.1 Hz, 4H), 2.78–2.58 (m, 6H), 2.45 (s, 3H); 13C NMR (101 MHz, acetone-d6) δ 195.8, 162.5, 155.2, 144.7, 136.5, 133.5, 132.7, 132.5 (2C), 132.0, 130.9 (2C), 128.4, 128.2, 127.2, 126.5, 118.5, 116.0 (2C), 114.2 (2C), 106.3, 72.1, 67.6, 61.7, 54.3 (2C), 48.1 (2C), 26.1; IR υmax/cm−1: 3100 (br, OH), 2981 cm-1 (CH3), 2890 (N-CH2), 2210 (CN) 1660 (C=O), 1272 (C-O), 818 (C-Cl); LRMS: (ESI+) m/z: 550 (C30H30Cl2N3O3) [M+H]; HRMS: Exact mass calculated for C30H30Cl2N3O3 [M+H], 550.1659. Found 550.1671.
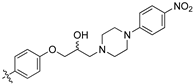
(Z)-3-(4-(3-(4-(4-Nitrophenyl)piperazin-1-yl)-2-hydroxypropoxy)phenyl)-2-(3,4-dichlorophenyl)acrylonitrile (8v) [15].
Prepared according to general procedure 3 from (Z)-2-(3,4-dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)acrylonitrile (6, 346 mg, 1 mmol) and 1-(4-nitrophenyl)piperazine (1.5 eq., 308 mg, 1.5 mmol) to afford the desired compound as a bright yellow solid (437 mg, 79%), m.p.: 90–92 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.10 (s, 1H), 8.04 (d, J = 9.2 Hz, 2H), 8.00 (s, 1H), 7.96 (d, J = 8.5 Hz, 2H), 7.76 (d, J = 8.3 Hz, 1H), 7.68 (d, J = 8.3 Hz, 1H), 7.14 (d, J = 8.5 Hz, 2H), 7.02 (d, J = 9.2 Hz, 2H), 5.02 (d, J = 4.1 Hz, 1H), 4.12 (d, J = 6.8 Hz, 1H), 4.01 (dd, J = 16.0, 6.8 Hz, 2H), 3.45 (s, 4H), 2.63–2.54 (m, 5H), 2.45 (dd, J = 12.6, 6.0 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.2, 154.7, 144.3, 136.8, 134.9, 132.0, 131.5 (2C), 131.2, 131.1, 127.0, 125.8, 125.7 (2C), 117.9, 115.1 (2C), 112.6 (2C), 104.4, 71.3, 66.6, 60.7, 53.0 (2C), 46.4 (2C); IR υmax/cm−1: 3337 (OH), 2835 (N-CH2), 2212 (CN), 1589 (NO2), 1332 (NO2), 1244 (C-O), 827 (C-Cl); LRMS: (ESI+) m/z: 553 (C28H27Cl2N4O4) [M+H]; HRMS: Exact mass calculated for (C28H27Cl2N4O4) [M+H], 553.1404. Found 553.1408.
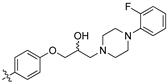
(Z)-2-(3,4-Dichlorophenyl)-3-(4-(3-(4-(2-fluorophenyl)piperazin-1-yl)-2-hydroxypropoxy)phenyl)-acrylonitrile (8w)[15].
Prepared according to general procedure 3 from (Z)-2-(3,4-dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)acrylonitrile (6, 200 mg, 0.58 mmol) and 1-(2-fluorophenyl)piperazine (2 eq., 208 mg, 1.16 mmol) to afford the desired compound as a pale yellow solid (225 mg, 74%), m.p.: 119–121 °C. 1H NMR (400 MHz, acetone-d6) δ 8.02 (d, J = 8.8 Hz, 2H), 7.98 (s, 1H), 7.94 (d, J = 1.6 Hz, 1H), 7.73–7.67 (m, 2H), 7.15 (d, J = 8.8, Hz, 2H), 7.11–7.01 (m, 3H), 6.98–6.93 (m, 1H) 4.23 (dd, J = 9.1, 3.4 Hz, 1H), 4.21–4.16 (m, 1H), 4.12 (dd, J = 9.1, 5.3 Hz, 1H), 3.12 (t, J = 4.8 Hz, 4H), 2.82–2.59 (m, 6H); 13C NMR (101 MHz, acetone-d6) δ 162.6, 156.5 (d, J = 245.4 Hz), 144.7, 141.23 (d, J = 8.5 Hz), 136.5, 133.5, 132.7, 132.5 (2C), 132.0, 128.2, 127.2, 126.5, 125.5 (d, J = 3.0 Hz), 123.1 (d, J = 8.1 Hz), 120.0 (d, J = 3.0 Hz), 118.5, 116.7 (d, J = 21.2 Hz), 116.0 (2C), 106.3, 72.2, 67.5, 61.8, 54.6 (2C), 51.36 (d, J = 3.3 Hz, 2C); 19F NMR (376 MHz, acetone-d6, CF3CO2H) δ -124.4; IR υmax/cm−1: 3200 (br, OH), 2825 (N-CH2), 2214 (CN), 1262 (C-O), 818 (C-Cl); LRMS: (ESI+) m/z: 526 (C28H27Cl2FN3O2) [M+H]; HRMS: Exact mass calculated for C28H27Cl2FN3O2 [M+H], 526.1459. Found 526.1465.
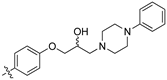
(Z)-3-(4-(3-(4-Cyclohexylpiperazin-1-yl)-2-hydroxypropoxy)-phenyl)-2-(3,4-dichlorophenyl)-acrylonitrile (8x) [15].
Prepared according to general procedure 3 from (Z)-2-(3,4-dichlorophenyl)-3-(4-(oxiran-2-ylmethoxy)phenyl)acrylonitrile (6, 346 mg, 1 mmol) and 4-phenylpiperidine (2 eq., 340 mg, 2 mmol) to afford the desired compound as a pale yellow solid (448 mg, 87%), m.p.: 170–172 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.10 (s, 1H), 8.01 (d, J = 2.2 Hz, 1H), 7.97 (d, J = 8.8 Hz, 2H), 7.76 (d, J = 8.5 Hz, 1H), 7.68 (dd, J = 8.5, 2.2 Hz, 1H), 7.30–7.22 (m, 4H), 7.18–7.14 (m, 3H), 4.91 (d, J = 4.1 Hz, 1H), 4.13–4.11 (m, 1H), 4.00–3.98 (m, 2H), 3.00 (dd, J = 26.9, 10.9 Hz, 2H), 2.49–2.35 (m, 3H), 2.12 (dt, J = 11.3, 8.4 Hz, 2H), 1.70–1.60 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 161.2, 146.3, 144.3, 135.0, 132.0 (2 overlapping signals), 131.5 (2C), 131.2, 131.1, 128.3 (2C), 127.0, 126.7 (2C), 126.0, 125.8, 117.9, 115.2 (2C), 104.3, 71.5, 66.6, 61.3, 54.6 (d, J = 20.8 Hz, 2C), 41.8, 33.2 (d, J = 3.0 Hz, 2C); IR υmax/cm−1: 3422 (OH), 2808 (N-CH2), 2207 (CN), 1263 (C-O), 810 (C-Cl); LRMS: (ESI+) m/z: 507 (C29H29Cl2N2O2) [M+H]; HRMS: Exact mass calculated for C29H29Cl2N2O2 [M+H], 507.1601. Found 507.1620.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27072050/s1, synthesis detail for compounds generated; chiral HPLC chromatograms for 8g, 8h and 8i and 1H and 13C NMR spectra for all compounds [36,37,38,39].
Author Contributions
Conceptualization, A.M., P.J.L. and M.A.C.; methodology, P.J.C., J.R.B., A.G.E., J.Z., M.A.T.B. and M.A.C.; formal analysis, A.M., P.J.L. and J.R.B.; investigation, P.J.C., J.R.B., A.G.E. and J.Z.; resources, A.M., P.J.L., M.A.T.B. and M.A.C.; writing—original draft preparation, A.M. and J.R.B.; writing—review and editing, A.M., P.J.C., M.A.T.B., A.G.E., J.Z., M.A.C. and P.J.L.; supervision, A.M. and P.J.L.; project administration, A.M. funding acquisition, A.M. and M.A.C. All authors have read and agreed to the published version of the manuscript.
Funding
J.R.B. is the recipient of an Australian Government RTP scholarship. Funding from the Australian Cancer Research Foundation and the Ramaciotti Foundation is gratefully acknowledged. Antimicrobial screening was performed by CO-ADD (The Community for Antimicrobial Drug Discovery), funded by the Wellcome Trust (UK) and The University of Queensland. MATB, AGE and JZ were supported by Wellcome Trust Strategic Grant 104797/Z/14/Z. Bacteria and yeast strains were acquired from the American Type Culture Collection (ATCC).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All pertinent data has been supplied in the Supporting Information that accompanies this article.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
References
- Liu, Y.-Y.; Wang, Y.; Walsh, T.R.; Yi, L.-X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Shore, C.K.; Coukell, A. Roadmap for antibiotic discovery. Nat. Microbiol. 2016, 1, 16083. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2019; US Department of Health and Human Services, CDC: Atlanta, GA, USA, 2019. [CrossRef] [Green Version]
- Report E Joint Technical. The Bacterial Challenge: Time to React 2009. Available online: http://ecdc.europa.eu/en/publications/Publications/0909_TER_The_Bacterial_Challenge_Time_to_React.pdf (accessed on 12 December 2021).
- Silver, L.L. Challenges of Antibacterial Discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef] [Green Version]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Jampilek, J. Design and Discovery of New Antibacterial Agents: Advances, Perspectives, Challenges. Curr. Med. Chem. 2019, 25, 4972–5006. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Cooper, M.A. Antibiotics in the clinical pipeline in 2011. J. Antibiot. 2011, 64, 413–425. [Google Scholar] [CrossRef]
- Silver, L.L. Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 2007, 6, 41–55. [Google Scholar] [CrossRef]
- Plackett, B. No money for new drugs. Nature 2020, 586, S50–S52. [Google Scholar] [CrossRef]
- McKenna, M. The antibiotic gamble. Nature 2020, 584, 338–341. [Google Scholar] [CrossRef]
- CarB-X. Available online: http://www.carb-x.org (accessed on 12 December 2021).
- León-Buitimea, A.; Garza-Cárdenas, C.R.; Garza-Cervantes, J.A.; Lerma-Escalera, J.A.; Morones-Ramírez, J.R. The Demand for New Antibiotics: Antimicrobial Peptides, Nanoparticles, and Combinatorial Therapies as Future Strategies in Antibacterial Agent Design. Front. Microbiol. 2020, 11, 1669. [Google Scholar] [CrossRef]
- Fuller, A.A.; Dounay, A.B.; Schirch, D.; Rivera, D.G.; Hansford, K.A.; Elliott, A.G.; Zuegg, J.; Cooper, M.A.; Blaskovich, M.A.T.; Hitchens, J.R.; et al. Multi-Institution Research and Education Collaboration Identifies New Antimicrobial Compounds. ACS Chem. Biol. 2020, 15, 3187–3196. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.R.; Russell, C.C.; Gilbert, J.; McCluskey, A.; Sakoff, J.A. Amino alcohol acrylonitriles as broad spectrum and tumour selective cytotoxic agents. RSC Med. Chem. 2021, 12, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.C.; Stevens, A.; Young, K.A.; Baker, J.R.; McCluskey, S.N.; Khazandi, M.; Pi, H.; Ogunniyi, A.; Page, S.W.; Trott, D.J.; et al. Discovery of 4,6-bis(2-((E)-benzylidene)hydrazinyl)pyrimidin-2-Amine with Antibiotic Activity. Chemistryopen 2019, 8, 896–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, C.C.; Stevens, A.; Pi, H.; Khazandi, M.; Ogunniyi, A.D.; Young, K.A.; Baker, J.R.; McCluskey, S.N.; Page, S.W.; Trott, D.J.; et al. Gram-Positive and Gram-Negative Antibiotic Activity of Asymmetric and Monomeric Robenidine Analogues. ChemMedChem 2018, 13, 2573–2580. [Google Scholar] [CrossRef]
- Pi, H.; Nguyen, H.; Venter, H.; Boileau, A.R.; Woolford, L.; Garg, S.; Page, S.W.; Russell, C.C.; Baker, J.R.; McCluskey, A.; et al. In vitro Activity of Robenidine Analog NCL195 in Combination With Outer Membrane Permeabilizers Against Gram-Negative Bacterial Pathogens and Impact on Systemic Gram-Positive Bacterial Infection in Mice. Front. Microbiol. 2020, 11, 1556. [Google Scholar] [CrossRef]
- Baker, J.R.; Pollard, B.L.; Lin, A.J.S.; Gilbert, J.; Paula, S.; Zhu, X.; Sakoff, J.A.; McCluskey, A. Modelling and Phenotypic Screening of NAP-6 and 10-Cl-BBQ, AhR Ligands Displaying Selective Breast Cancer Cytotoxicity in Vitro. ChemMedChem 2021, 16, 1499–1512. [Google Scholar] [CrossRef]
- Sun, J.; Baker, J.R.; Russell, C.C.; Cossar, P.J.; Pham, H.N.T.; Sakoff, J.A.; Scarlett, C.J.; McCluskey, A. Novel Cytotoxic 1,2,3-Triazoles as Potential new Leads Targeting the S100A2-p53 Complex. ChemMedChem 2021, 16. [Google Scholar] [CrossRef]
- Baker, J.R.; Russell, C.C.; Gilbert, J.; Sakoff, J.A.; McCluskey, A. Amino Alcohol Acrylonitriles as Activators of the Aryl Hydrocarbon Receptor Pathway: An Unexpected MTT Phenotypic Screening Outcome. ChemMedChem 2020, 15, 490–505. [Google Scholar] [CrossRef]
- Stanton, D.T.; Baker, J.R.; McCluskey, A.; Paula, S. Development and interpretation of a QSAR model for in vitro breast cancer (MCF-7) cytotoxicity of 2-phenylacrylonitriles. J. Comput. Aid. Mol. Des. 2021, 35, 613–628. [Google Scholar] [CrossRef]
- Baker, J.R.; Sakoff, J.A.; McCluskey, A. The aryl hydrocarbon receptor (AhR) as a breast cancer drug target. Med. Res. Rev. 2020, 40, 972–1001. [Google Scholar] [CrossRef]
- Mishra, S.; Patel, S. Design, Synthesis, and Anti-bacterial Activity of Novel Deoxycholic Acid-Amino Alcohol Conjugates. Med Chem. 2020, 16, 385–391. [Google Scholar] [CrossRef]
- Myers, A.G.; Clark, R.B. Discovery of Macrolide Antibiotics Effective against Multi-Drug Resistant Gram-Negative Pathogens. ACC Chem Res. 2021, 54, 1635–1645. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Y.; Ke, M.; Liu, M.; Zhan, P.; Xiao, Y.C.; Chen, F. Unified Strategy to Amphenicol Antibiotics: Asymmetric Synthesis of (-)-Chloramphenicol, (-)-Azidamphenicol, and (+)-Thiamphenicol and Its (+)-3-Floride. J. Org. Chem. 2020, 85, 15360–15367. [Google Scholar] [CrossRef] [PubMed]
- Blaskovich, M.A.T.; Hansford, K.A.; Gong, Y.; Butler, M.S.; Muldoon, C.; Huang, J.X.; Ramu, S.; Silva, A.B.; Cheng, M.; Kavanagh, A.M.; et al. Protein-inspired antibiotics active against vancomycin- and daptomycin-resistant bacteria. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossar, P.J.; Baker, J.; Cain, N.; McCluskey, A. In situ epoxide generation by dimethyldioxirane oxidation and the use of epichlorohydrin in the flow synthesis of a library of β-amino alcohols. R. Soc. Open Sci. 2018, 5, 171190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; et al. Synthesis, Structure–Activity Relationships, and in Vivo Efficacy of the Novel Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor 5-Chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) Currently in Phase 1 and Phase 2 Clinical Trials. J. Med. Chem. 2013, 56, 5675–5690. [Google Scholar] [CrossRef]
- Liebler, D.C. Protein Damage by Reactive Electrophiles: Targets and Consequences. Chem. Res. Toxicol. 2008, 21, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.C.; Watt, A.P.; Nicoll-Griffith, D.A.; Baillie, T.A. Drug−Protein Adducts: An Industry Perspective on Minimizing the Potential for Drug Bioactivation in Drug Discovery and Development. Chem. Res. Toxicol. 2004, 17, 3–16, Erratum in Chem. Res. Toxicol. 2005, 18, 1777. [Google Scholar] [CrossRef]
- Baillie, T.A. Metabolism and Toxicity of Drugs. Two Decades of Progress in Industrial Drug Metabolism. Chem. Res. Toxicol. 2008, 21, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Hansford, K.A.; Blaskovich, M.A.; Cooper, M.A. Chemical philanthropy: A path forward for antibiotic discovery? Future Med. Chem. 2016, 8, 925–929. [Google Scholar] [CrossRef] [Green Version]
- Blaskovich, M.A.T.; Zuegg, J.; Elliott, A.; Cooper, M. Helping Chemists Discover New Antibiotics. ACS Infect. Dis. 2015, 1, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Desselle, M.; Neale, R.; Hansford, K.A.; Zuegg, J.; Elliott, A.; Cooper, M.A.; Blaskovich, M.A. Institutional profile: Community for Open Antimicrobial Drug Discovery—Crowdsourcing new antibiotics and antifungals. Future Sci. OA 2017, 3, FSO171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossar, P.J.M.C.; Gordon, C.P.; Ambrus, J.I.; Lewis, P.J.; McCluskey, A. Identification and validation of small molecule modulators of the NusB-NusE interaction. Bioorg. Med. Chem. Lett. 2017, 27, 162–167. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, A.; Daniel, J.A.; Hadzic, G.; Chau, N.; Clayton, E.L.; Mariana, A.; Whiting, A.; Gorgani, N.N.; Lloyd, J.R.; Quan, A.; et al. Building a Better Dynasore: The Dyngo Compounds Potently Inhibit Dynamin and Endocytosis. Traffic 2013, 14, 1272–1289. [Google Scholar] [CrossRef]
- Baker, J.R.; Gilbert, J.; Paula, S.; Zhu, X.; Sakoff, J.A.; McCluskey, A. Dichlorophenylacrylonitriles as AhR Ligands That Display Selective Breast Cancer Cytotoxicity in vitro. ChemMedChem 2018, 13, 1447–1458. [Google Scholar] [CrossRef]
- Tarleton, M.; Gilbert, J.; Robertson, M.J.; McCluskey, A.; Sakoff, J.A. Library synthesis and cytotoxicity of a family of 2-phenylacrylonitriles and discovery of an estrogen dependent breast cancer lead compound. MedChemComm 2011, 2, 31–37. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).