
Crystal Structure, Topology, DFT and Hirshfeld Surface Analysis of a Novel Charge Transfer Complex (L3) of Anthraquinone and 4-{[(anthracen-9-yl)meth-yl] amino}-benzoic Acid (L2) Exhibiting Photocatalytic Properties: An Experimental and Theoretical Approach

 , ,
, ,  , , and
, , and
Abstract
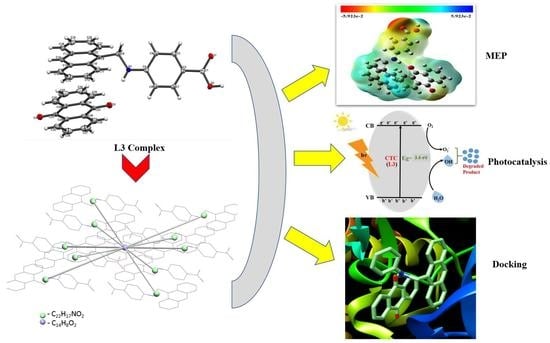
:
1. Introduction
2. Material and Methods
2.1. Reagents and Materials
2.2. Methods and Instrumentation
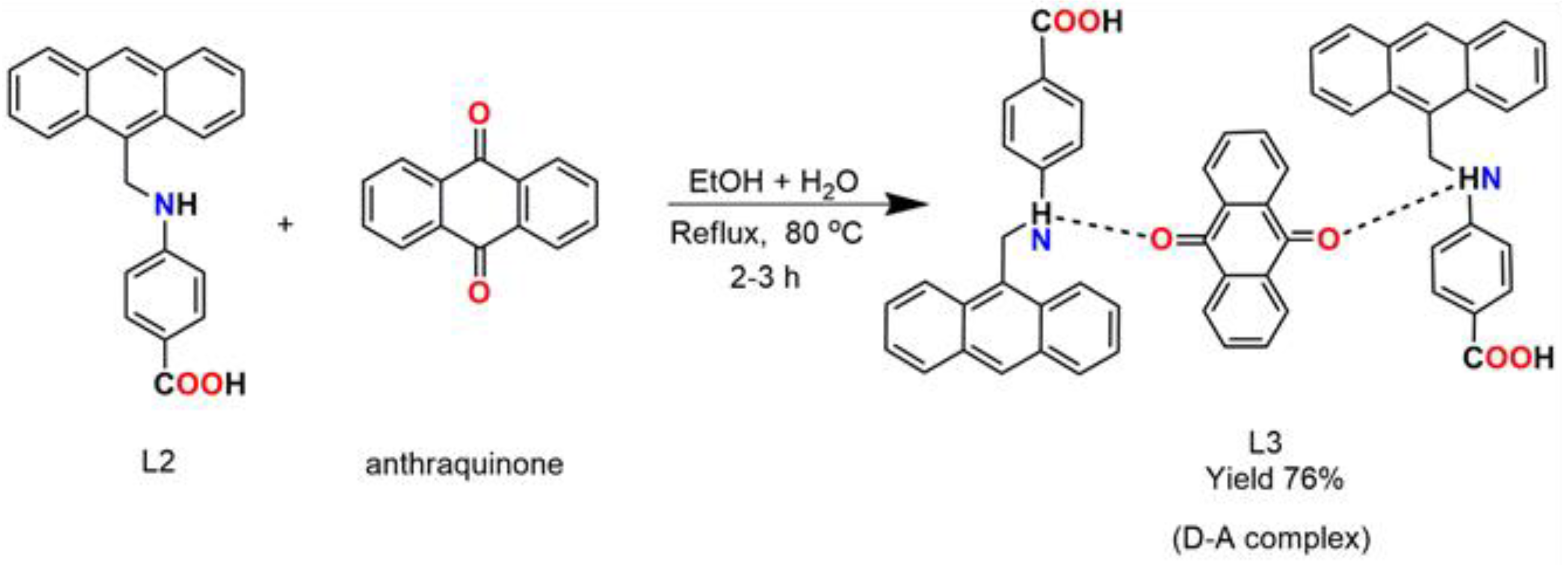
2.3. Synthesis of CT Complex (L3)
2.4. Photodegradation of Dye
3. Results and Discussion
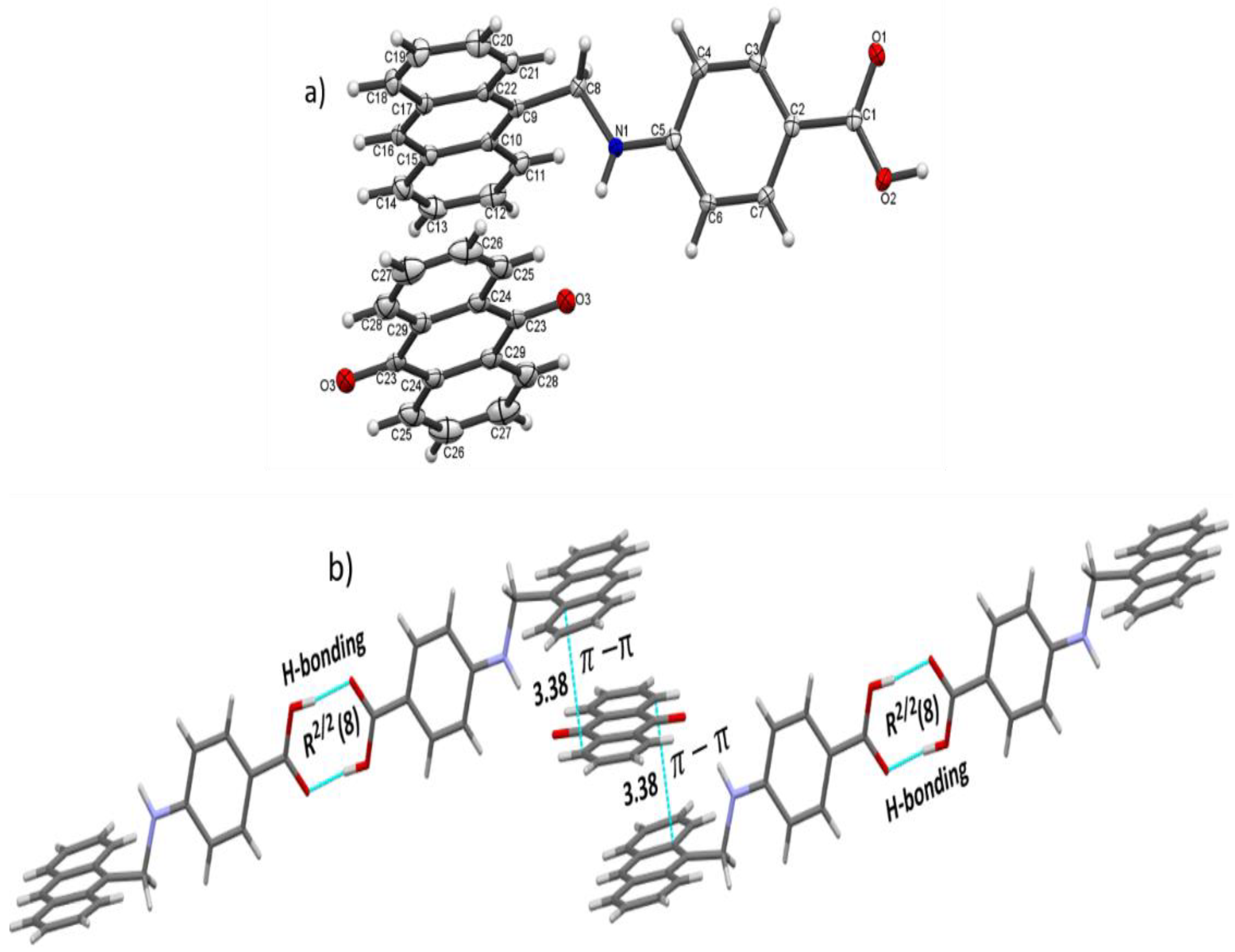
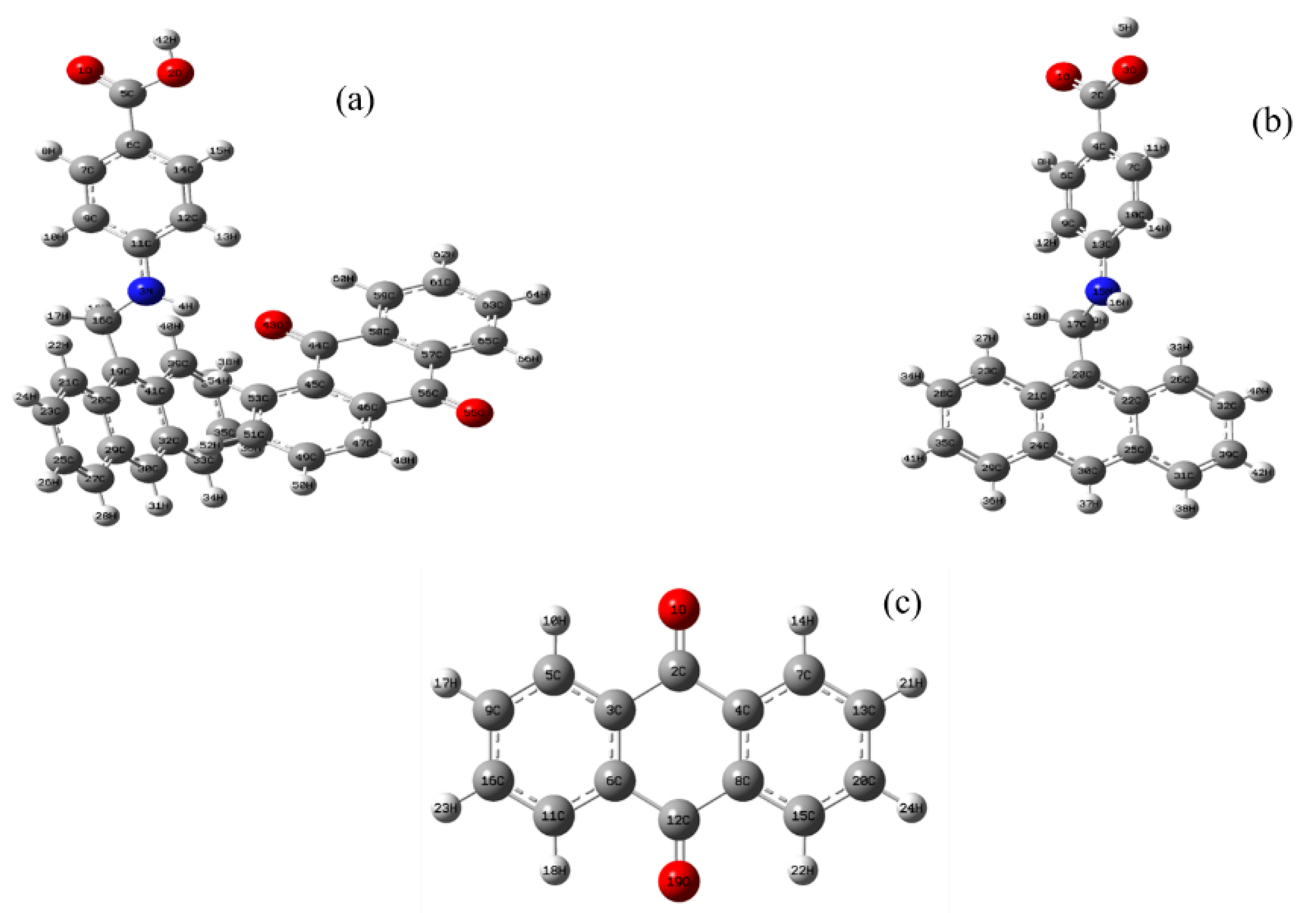
3.1. Structure Description
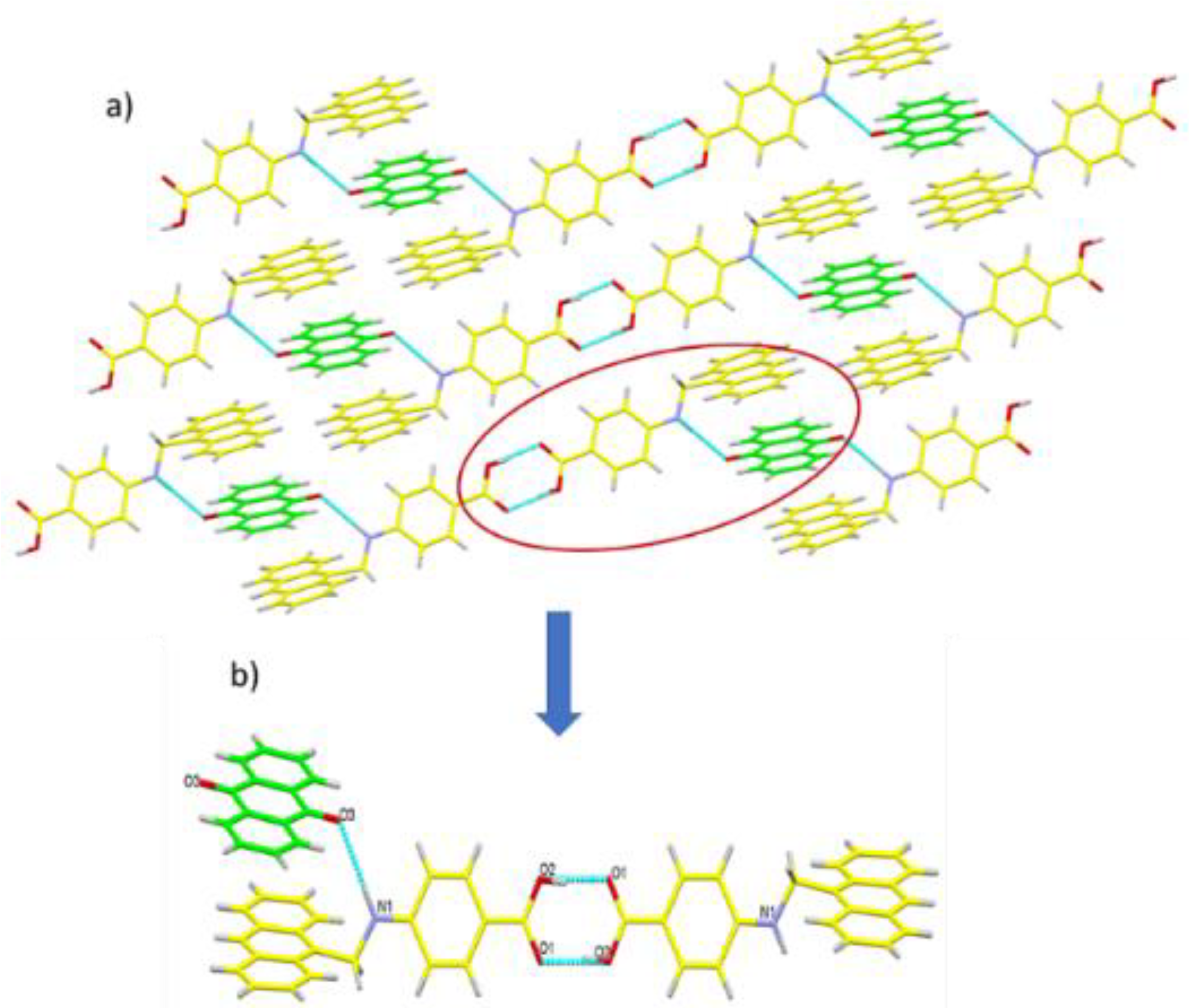
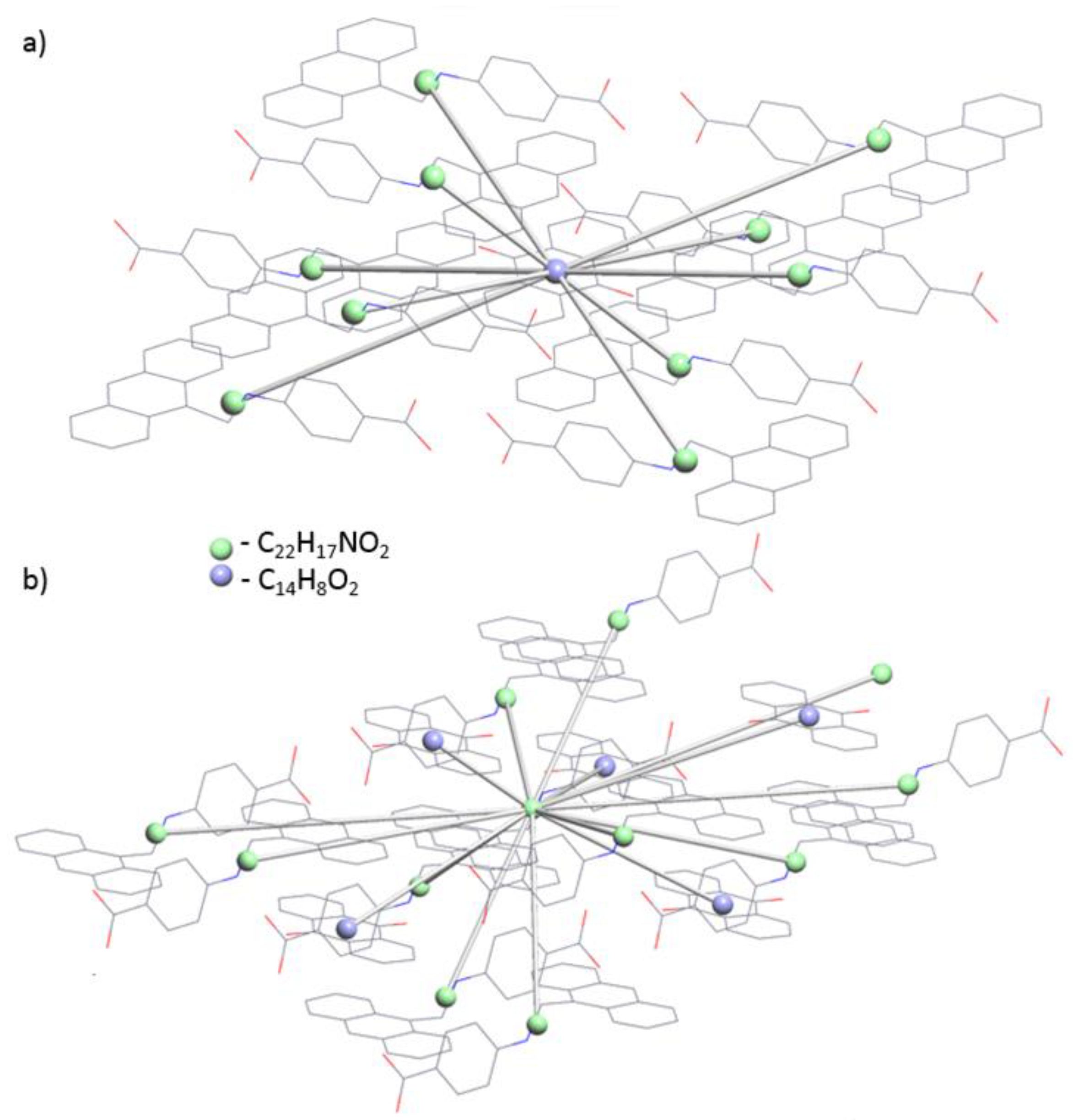
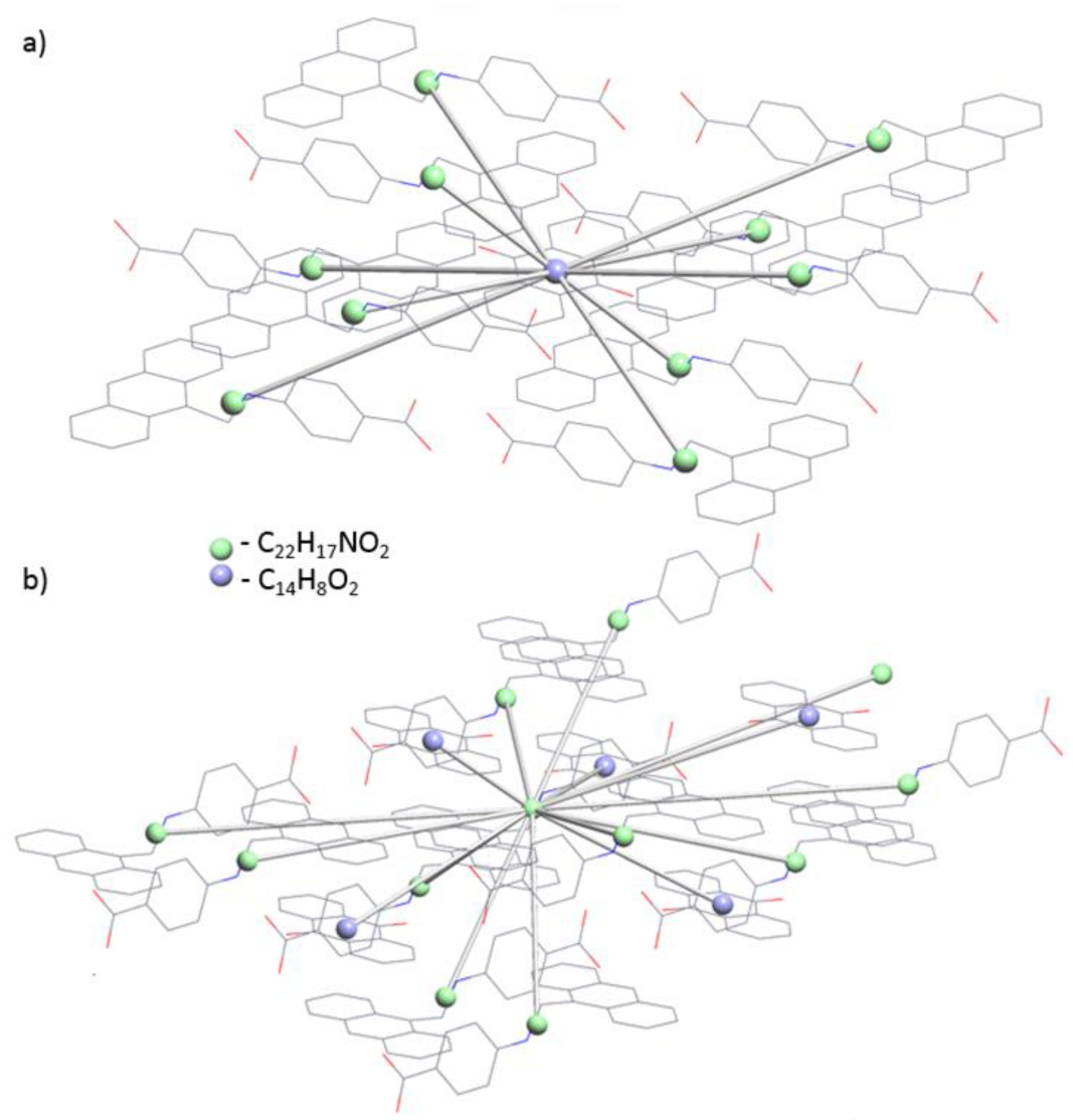
3.2. Topological Analysis
3.3. Optimized Geometrical Parameters
3.4. PXRD Studies and TGA Analysis
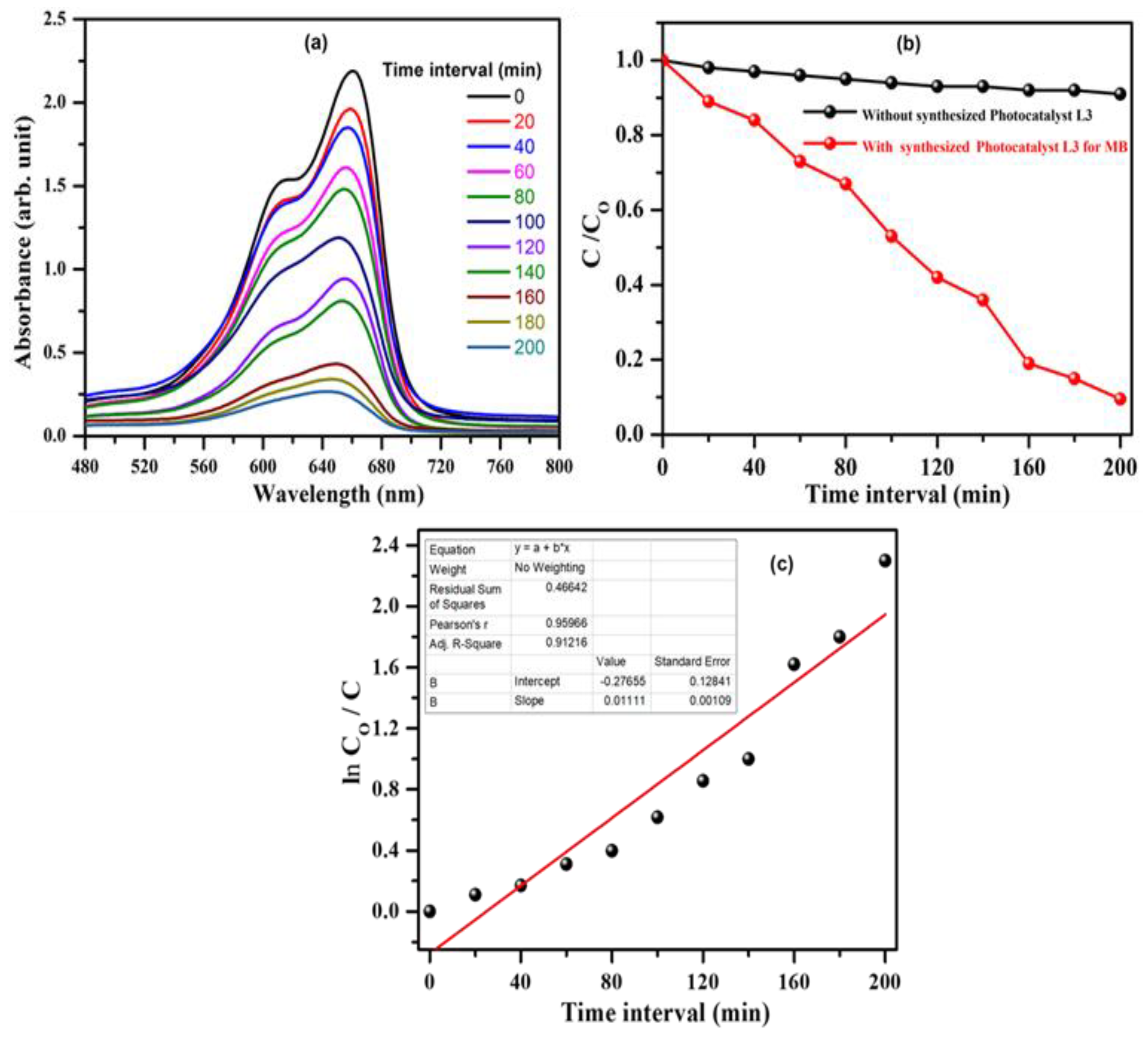
3.5. Photocatalytic Activity: Experimental Approach
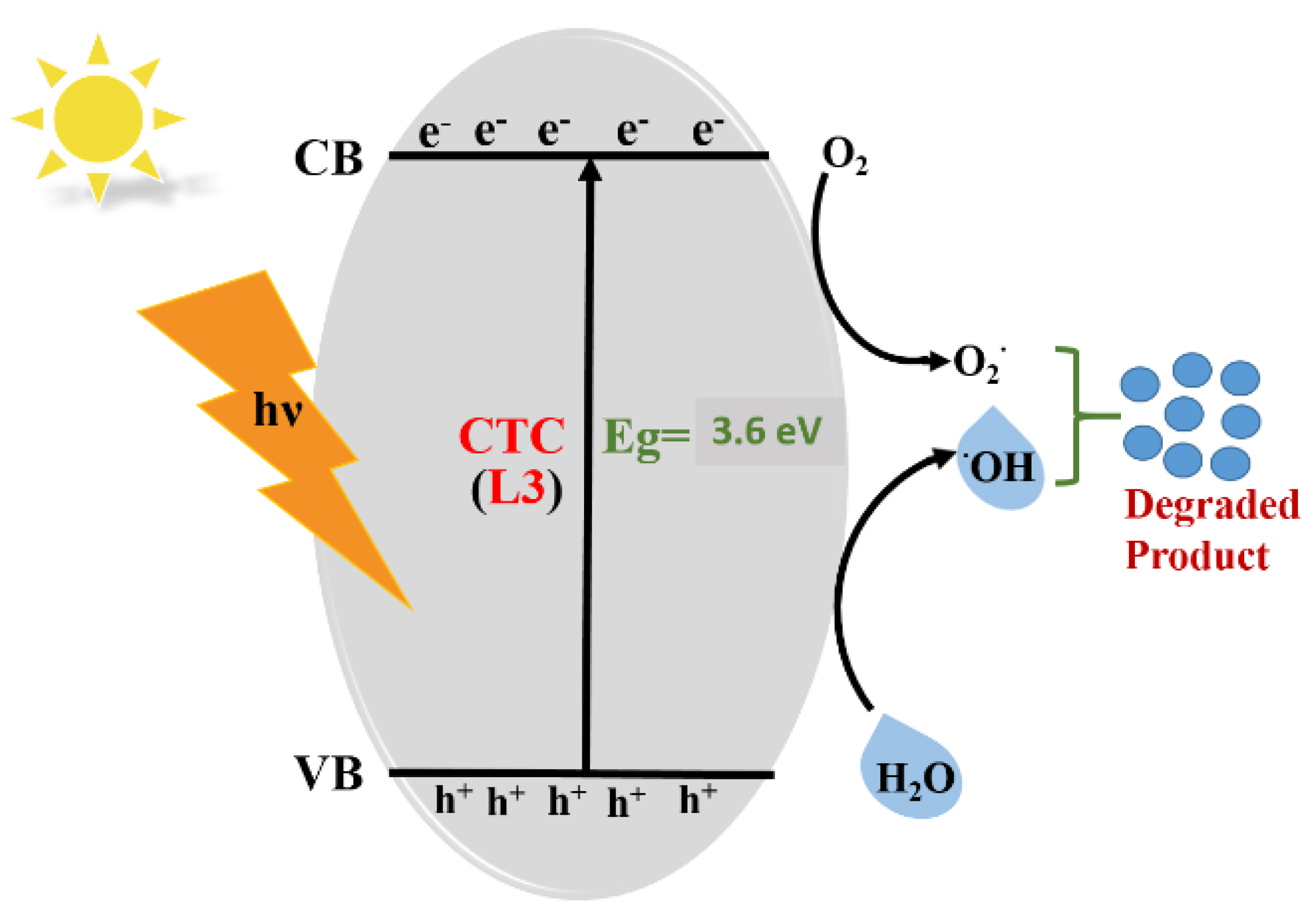
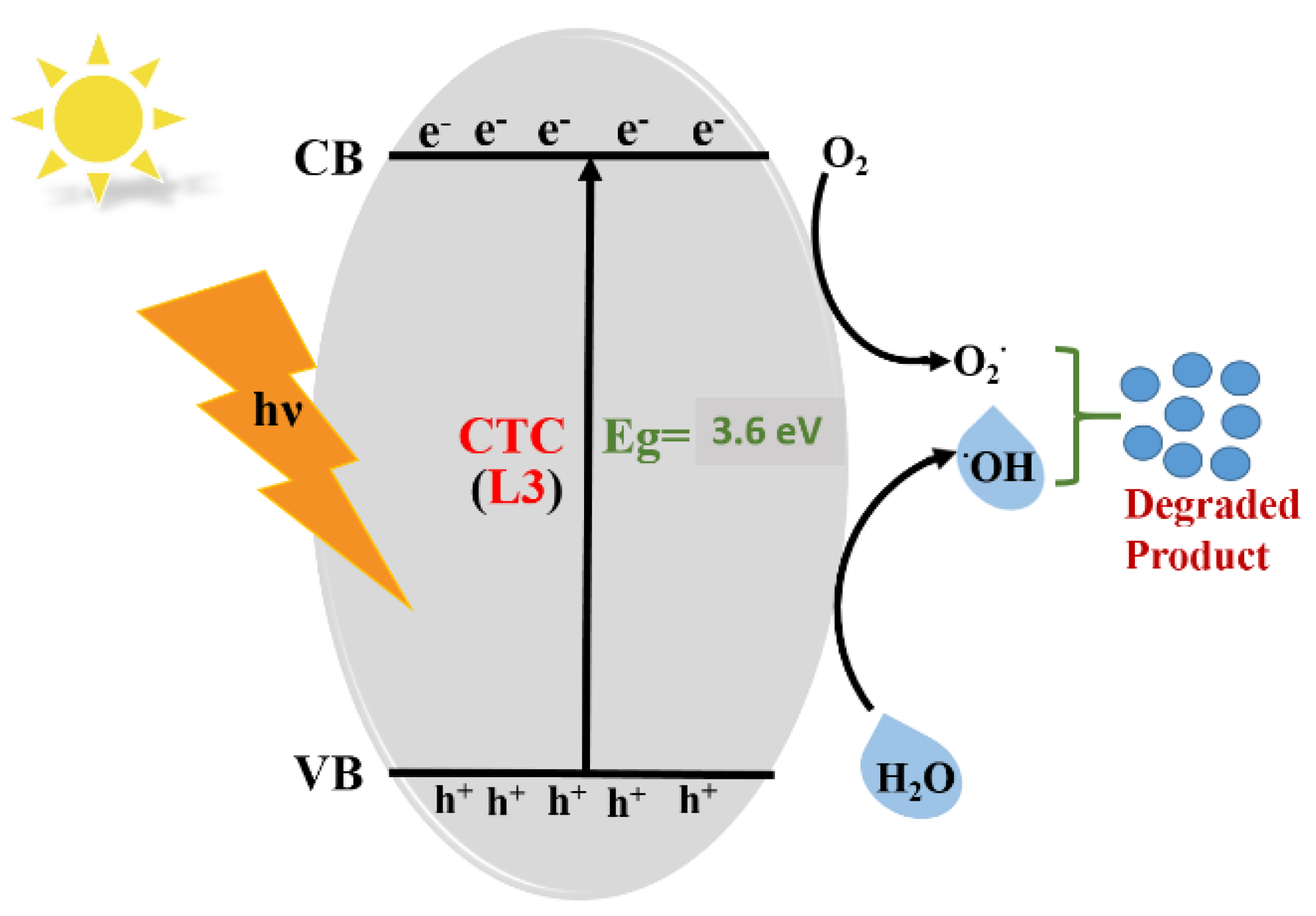
3.6. Mechanism Associated with MB Dye Degradation: Theoretical Approach
3.7. Vibrational Spectral Study
3.7.1. Ring Vibrations
3.7.2. C=O, N–H and O–H Vibrations
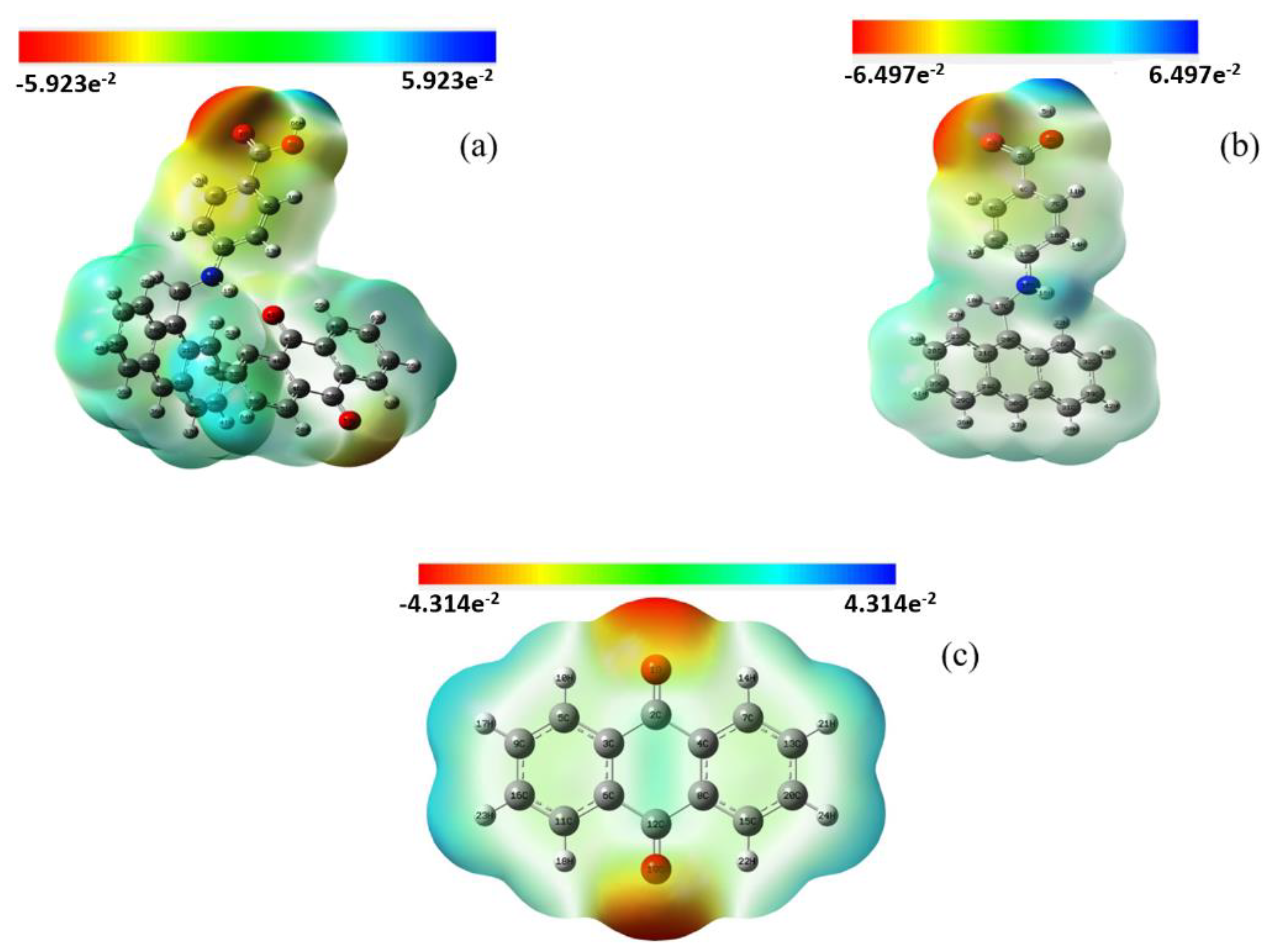
3.8. Molecular Electrostatic Potential (MEP)
3.9. Non-Linear Optical (NLO) Analysis
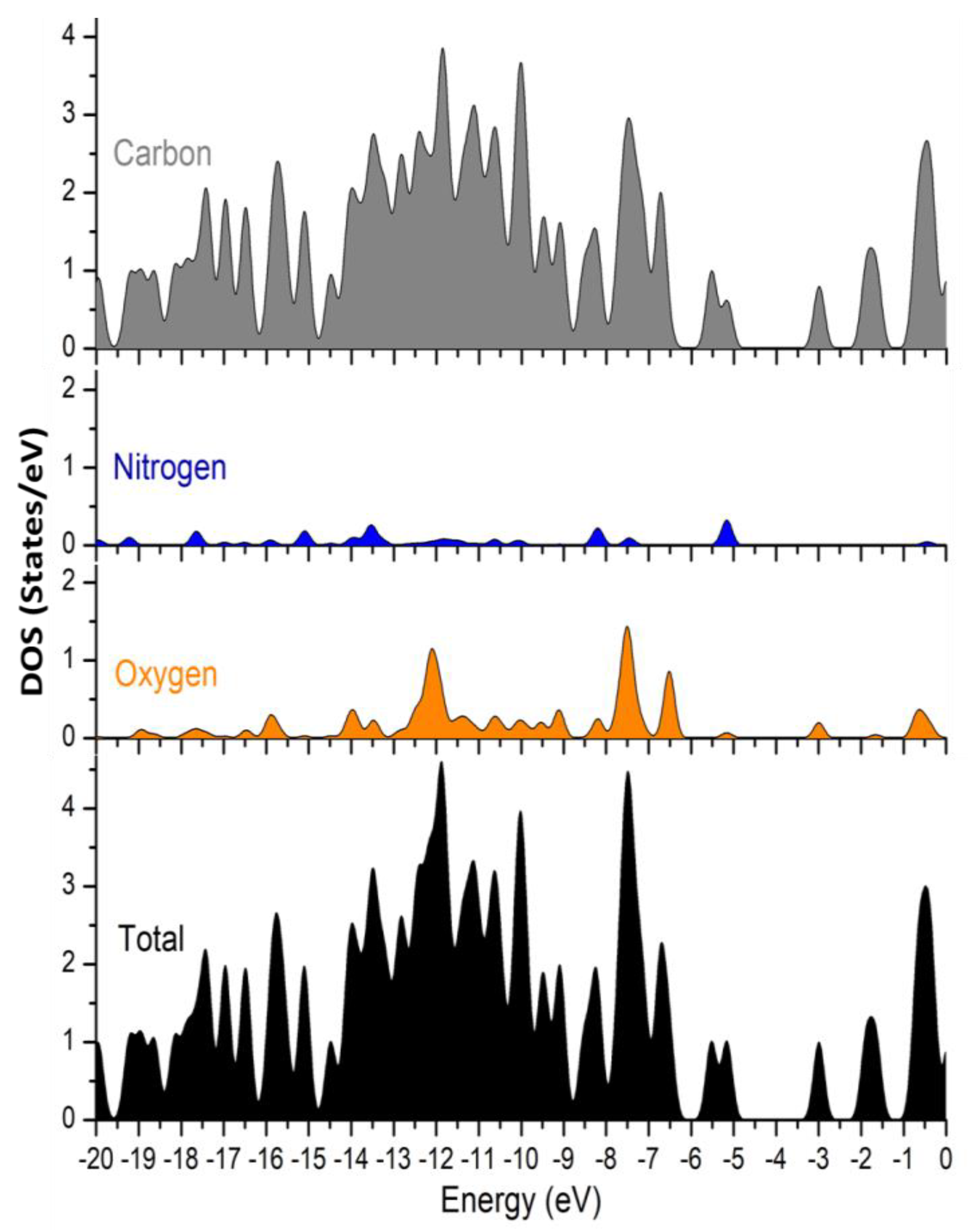
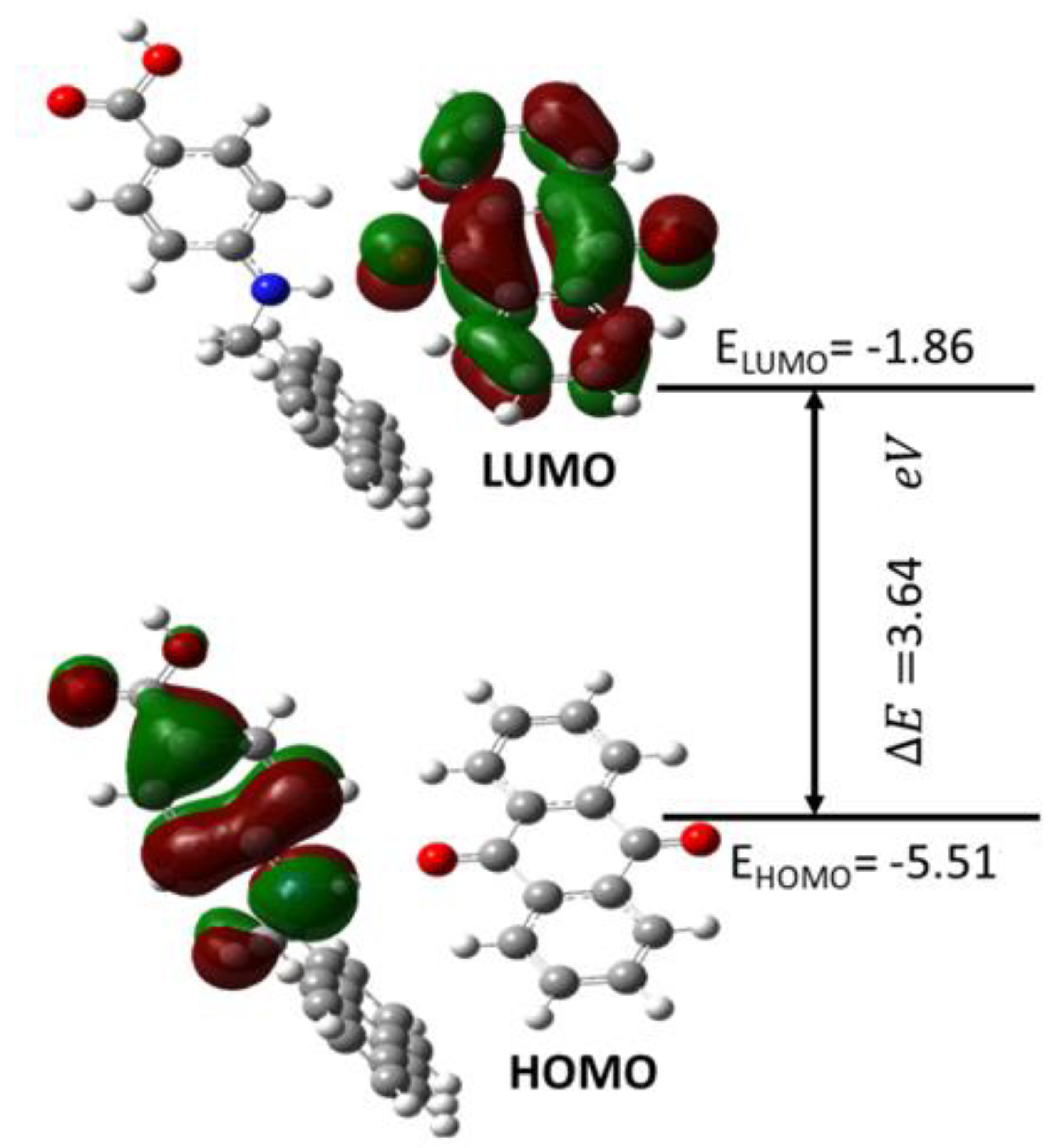
3.10. UV–Vis and Electronic Properties
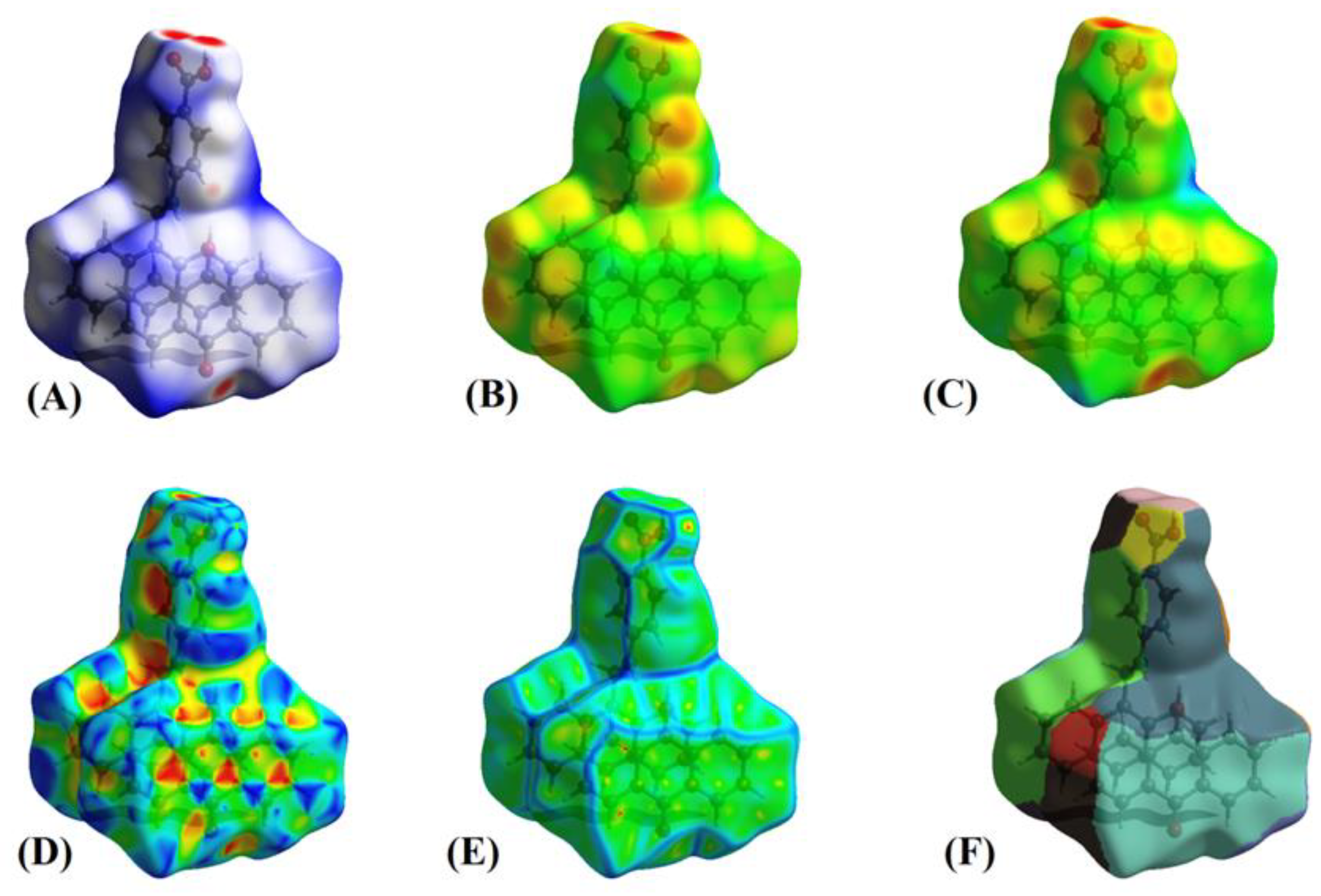
3.11. Hirshfeld Surface Analysis
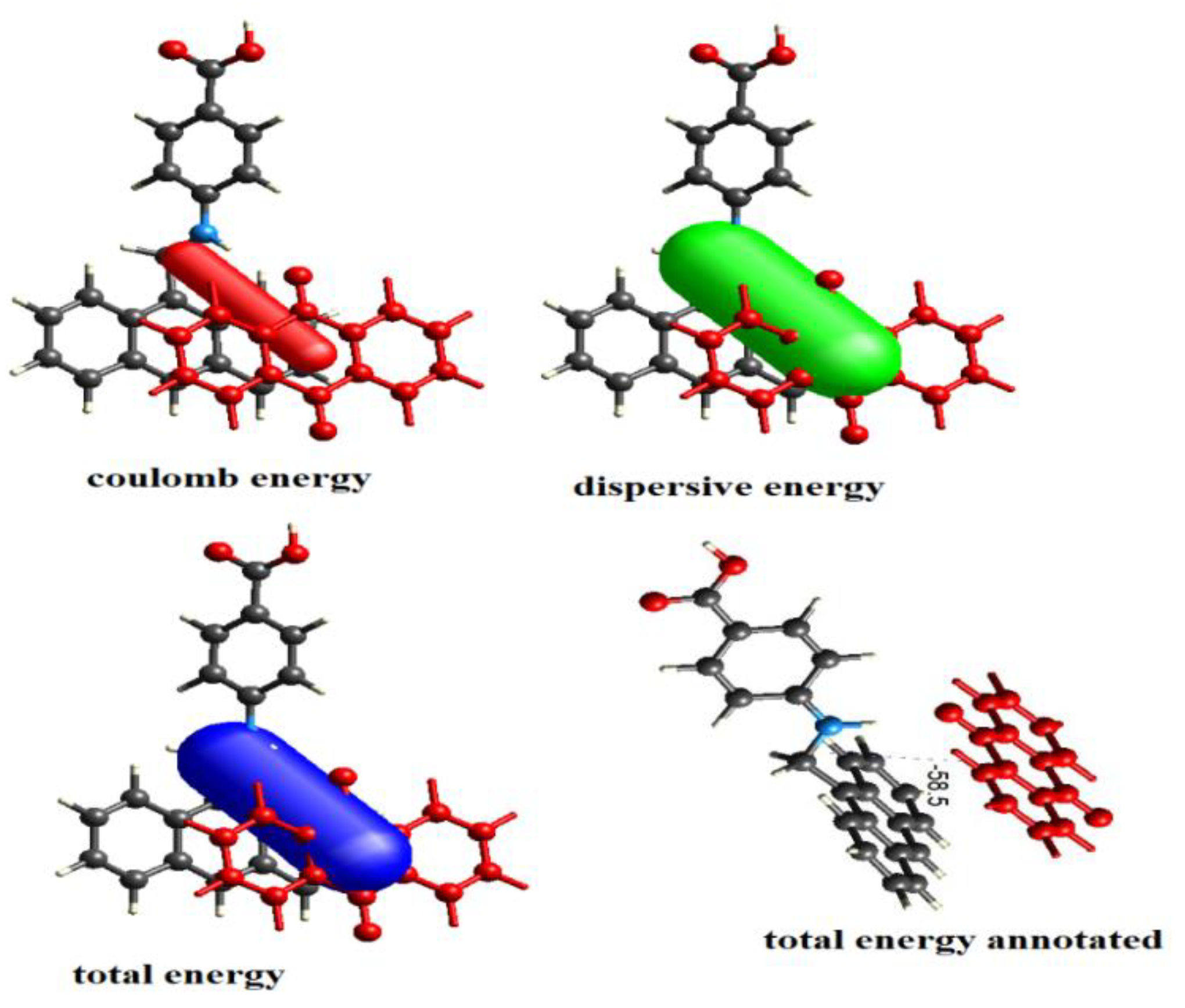
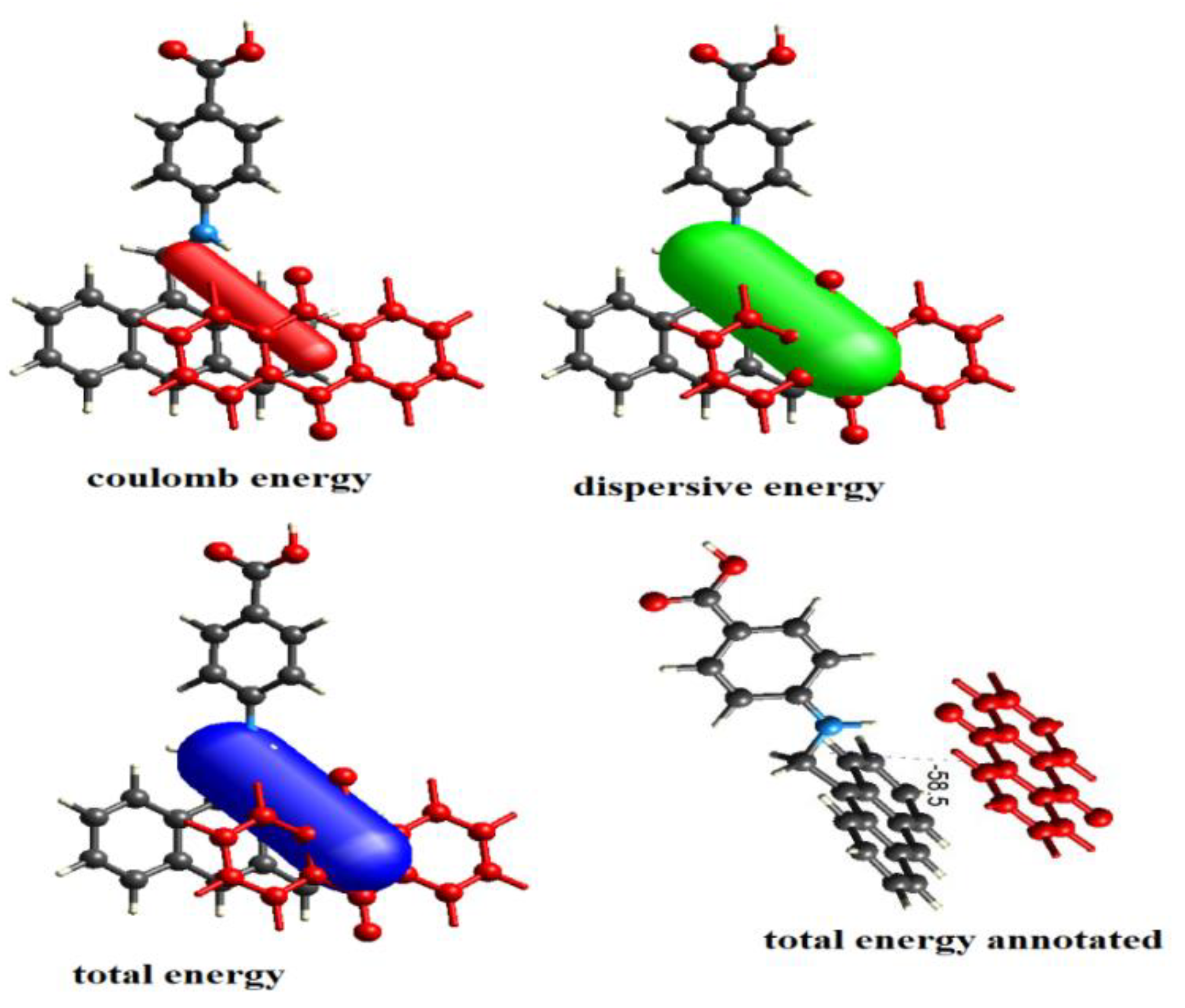
3.12. Energy Framework Calculations
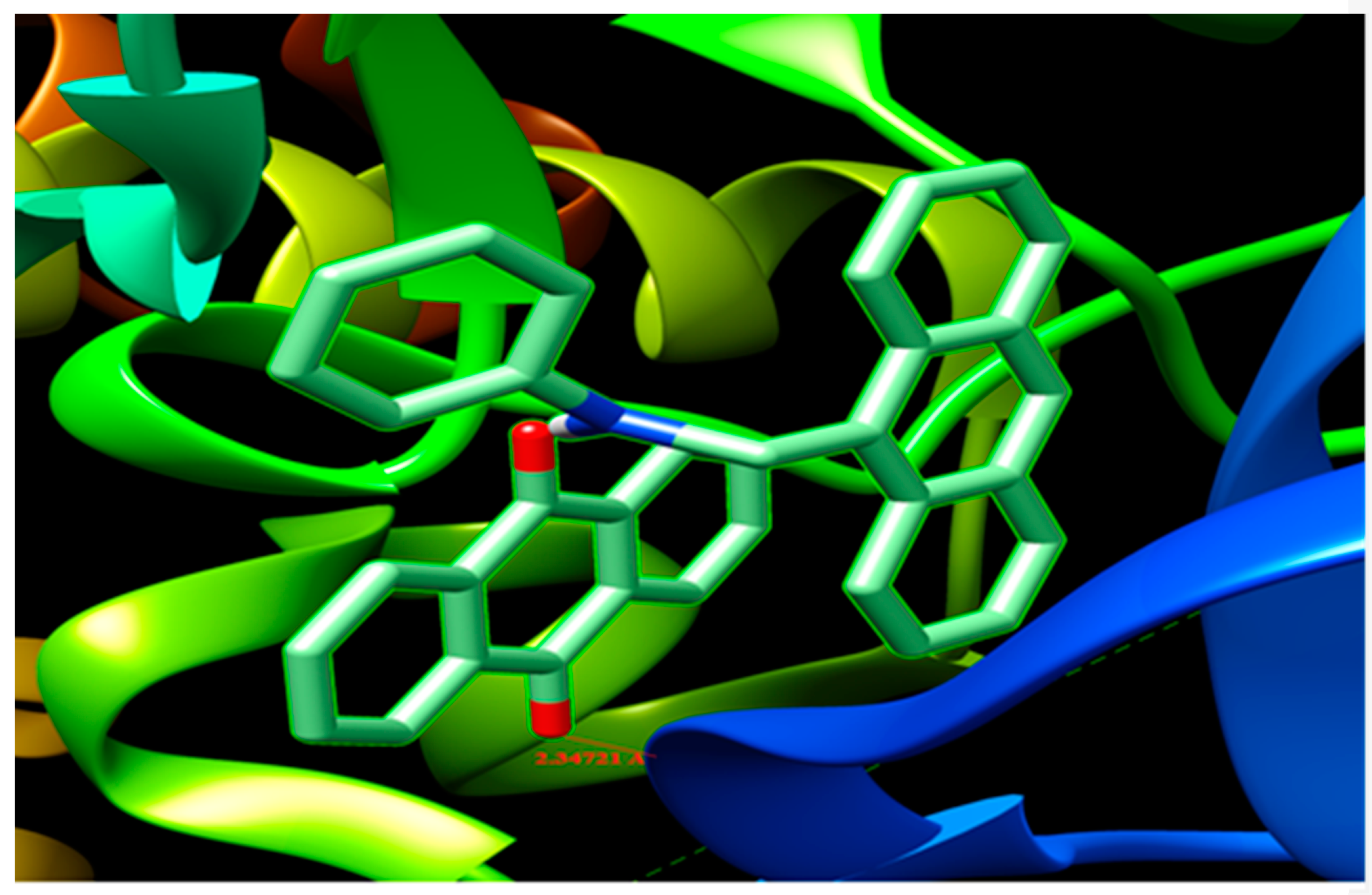
3.13. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Eychmuller, A.; Rogach, A.L. Chemistry and photophysics of thiol-stabilized II-VI semiconductor nanocrystals. J. Pure Appl. Chem. 2000, 72, 179–188. [Google Scholar] [CrossRef]
- Singh, P.; Kumar, P.; Katyal, R.; Dass, S.K.; Prakash, S.; Chandra, R. Synthesis and Electrochemical studies of Charge-Transfer Complexes of Thiazolidine-2,4-dione with Sigma and Pi Acceptors. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 75, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Dabestani, R.; Reska, K.J.; Sigman, M.E. Surface catalyzed electron transfer from polycyclic aromatic hydrocarbons (PAH) to methyl viologen dication: Evidence for ground-state charge transfer complex formation on silica gel. J. Photochem. Photobiol. A 1998, 117, 223–233. [Google Scholar] [CrossRef]
- Dozal, A.; Keyzer, H.; Kin, H.K.; Wang, W.W. Charge transfer complexes of K vitamins with several classes of antimicrobials. Int. J. Antimicrob. Agent 2000, 14, 261–265. [Google Scholar] [CrossRef]
- Mandal, R.; Lahiri, S.C. Interactions of L-amino acids with metronidazole and tinidazole. J. Indian Chem. Soc. 1999, 76, 347–349. [Google Scholar]
- Pandeswaran, M.; Elango, K.P. Spectroscopic studies on the interaction of cimetidine drug with biologically significant sigma- and pi-acceptors. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 75, 1462–1469. [Google Scholar] [CrossRef]
- Haverkate, L.A.; Zbiri, M.; Johnson, M.R.; Carter, E.; Kotlewski, A.; Picken, S.; Mulder, F.M.; Kearley, G.J. Electronic and vibronic properties of a discotic liquid-crystal and its charge transfer complex. J. Chem. Phys. 2014, 140, 014903–014908. [Google Scholar] [CrossRef] [Green Version]
- Moulton, B.; Zaworotko, M.J. From Molecules to Crystal Engineering: Supramolecular Isomerism and Polymorphism in Network Solids. Chem. Rev. 2001, 101, 1629–1658. [Google Scholar] [CrossRef]
- Hakiri, R.; Ameur, I.; Abid, S.; Derbel, N. Synthesis, X-ray structural, Hirshfeld surface analysis, FTIR, MEP and NBO analysis using DFT study of a 4-chlorobenzylammonium nitrate (C7ClH9N)+(NO3)−. J. Mol. Struct. 2018, 1164, 486–492. [Google Scholar] [CrossRef]
- Ahmed, A.; Faizi, M.S.H.; Ahmad, A.; Ahmad, M.; Fritsky, I.O. Crystal structure and Hirshfeld surface analysis of 4-{[(anthracen-9-yl)methyl]amino}benzoic acid. Acta Cryst. 2019, 76, 62–65. [Google Scholar]
- Obali, A.Y.; Ucan, H.I. Aromatic Chromophore-Tethered Schiff Base Ligands and Their Iron(III)/Chromium(III) Salen and Saloph Capped Complexes. J. Fluoresc. 2012, 22, 1357–1370. [Google Scholar] [CrossRef] [PubMed]
- Monks, T.J.; Hanzlik, R.P.; Cohen, G.M.; Ross, D.; Graham, D.G. Quinone chemistry and toxicity. Toxicol Appl Pharmacol. 1992, 112, 2–16. [Google Scholar] [CrossRef]
- Wraight, C.A. Proton and electron transfer in the acceptor quinone complex of photosynthetic reaction centers from rhodobacter sphaeroides. Front. Biosci. 2004, 9, 309–337. [Google Scholar] [CrossRef] [PubMed]
- Morton, R.A. Biochemistry of Quinones; Academic Press: New York, NY, USA, 1965. [Google Scholar]
- Balraj, C.; Satheshkumar, A.; Ganesh, K.; Elango, K.P. Charge transfer complexes of quinones in aqueous medium: Spectroscopic and theoretical studies on interaction of cimetidine with novel substituted 1,4-benzoquinones and its application in colorimetric sensing of anions. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 114, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Rauf, M.A.; Meetani, M.A.; Khaleel, A.; Ahmed, A. Photocatalytic degradation of Methylene Blue using a mixed catalyst and product analysis by LC/MS. J. Chem. Eng. 2010, 157, 373–378. [Google Scholar] [CrossRef]
- Rahman, Q.I.; Ali, A.; Ahmad, N.; Lohani, M.B.; Mehta, S.K.; Muddassir, M. Synthesis and characterization of CuO rods for enhanced visible light driven dye degradation. J. Nanosci. Nanotechnol. 2020, 20, 7716–7723. [Google Scholar] [CrossRef]
- Hoffmann, M.R.; Martin, S.T.; Choi, W.; Bahnemann, D.W. Environmental Applications of Semiconductor Photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar]
- Shakoor, S.; Nasar, A. Removal of methylene blue dye from artificially contaminated water using citrus limetta peel waste as a very low cost adsorbent. Taiwan Inst. Chem. Eng. 2006, 66, 154–163. [Google Scholar] [CrossRef]
- Maruthapandi, M.; Luong, J.H.T.; Gedanken, A. Kinetic, isotherm and mechanism studies of organic dye adsorption on poly(4,4′-oxybisbenzenamine) and copolymer of poly(4,4′-oxybisbenzenamine-pyrrole) macro-nanoparticles synthesized by multifunctional carbon dots. New J. Chem. 2018, 43, 1926–1935. [Google Scholar] [CrossRef]
- Rahman, Q.I.; Ahmad, M.; Misra, S.K.; Lohani, M.J. Hexagonal ZnO nanorods assembled flowers for photocatalytic dye degradation: Growth, structural and optical properties. Superlattices Microstruct. 2014, 64, 495–506. [Google Scholar] [CrossRef]
- Ahmed, A.; Ali, A.; Ahmed, M.; Parida, K.N.; Ahmad, M.; Ahmad, A. Construction and topological studies of a three dimensional (3D) coordination polymer showing selective adsorption of aromatic hazardous dyes. J. Sep. Purif. Technol. 2021, 265, 118482–118493. [Google Scholar] [CrossRef]
- Khan, I.M.; Alam, K.; Alam, M.J.; Ahmad, M. Spectrophotometric and photocatalytic studies of H-bonded charge transfer complex of oxalic acid with imidazole: Single crystal XRD, experimental and DFT/TD-DFT studies. New J. Chem. 2019, 43, 9039–9051. [Google Scholar] [CrossRef]
- Zhiyong, Y.; Keppner, H.; Laub, D.; Mielczarski, E.; Mielczarski, J.; Kiwi-Minsker, L.; Renken, A.; Kiwi, J. Photocatalytic discoloration of Methyl Orange on innovative parylene–TiO2 flexible thin films under simulated sunlight. Appl. Catal. B Environ. 2008, 179, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Rahman, Q.I.; Hasan, H.; Ali, A.; Mehta, S.K.; Raja, M.A.; Ahmad, N.; Khan, A.R.; Muddassir, M. Synthesis and Characterizations of Nitrogen (N) Doped Strontium Titanate (SrTiO3) Nanoparticles for Enhanced Visible Light Driven Photocatalytic Degradation. J. Nanosci. Nanotechnol. 2020, 20, 6475–6481. [Google Scholar] [CrossRef] [PubMed]
- Rahman, Q.I.; Ahmad, M.; Mehta, S.K. Hydrothermal synthesis of Cr-doped SrTiO3 nanoparticles for rhodamine-B dye degradation under visible light illumination. Colloid Polym. Sci. 2017, 295, 933–937. [Google Scholar] [CrossRef]
- Rahman, Q.I.; Ahmad, M.; Misra, S.K.; Lohani, M. Effective photocatalytic degradation of rhodamine B dye by ZnO nanoparticles. Mater. Lett. 2013, 91, 170–174. [Google Scholar] [CrossRef]
- Avansi, W., Jr.; de Mendonça, V.R.; Lopes, O.F.; Ribeiro, C. Vanadium pentoxide 1-D nanostructures applied to dye removal from aqueous systems by coupling adsorption and visible-light photodegradation. RSC Adv. 2015, 5, 12000–12006. [Google Scholar] [CrossRef]
- Mansour, A.M.; Bakry, E.M.; Abdel-Ghani, N.T. Photocatalytic degradation of methylene blue with copper(II) oxide synthesized by thermal decomposition of Flubendazole complexes. J. Photochim. Photobiol. A 2016, 327, 21–24. [Google Scholar] [CrossRef]
- Neto, J.O.M.; Bellato, C.R.; Souza, C.H.F.; Silva, R.C.; Rocha, P.A. Synthesis, Characterization and Enhanced Photocatalytic Activity of Iron Oxide/Carbon Nanotube/Ag-doped TiO2 Nanocomposites. J. Braz. Chem. Soc. 2017, 28, 2301–2312. [Google Scholar]
- Hong, Y.; Li, J.C.; Fan, W.; Yan, X.; Yan, M.; Shi, W. In-situ synthesis of direct solid-state Z-scheme V2O5/g-C3N4 heterojunctions with enhanced visible light efficiency in photocatalytic degradation of pollutants. Appl. Catal. B Environ. 2016, 180, 663–673. [Google Scholar] [CrossRef]
- Wang, X.L.; Luan, J.; Lin, H.Y.; Lu, Q.L.; Le, M.; Liu, G.C.; Shao, J.Y. Metal(II)–Organic Coordination Polymers Modulated by Two Isomeric Semirigid Bis-Pyridyl–Bis-Amide Ligands: Structures, Fluorescent Sensing Behavior, and Selective Photocatalysis. ChemPlusChem 2014, 79, 1691–1702. [Google Scholar] [CrossRef]
- Jha, A.; Mote, K.R.; Chandra, S.; Madhu, P.K.; Dasgupta, J. Photoactive Anthraquinone-Based Host–Guest Assembly for Long-Lived Charge Separation. J. Phys. Chem. C 2021, 125, 10891–10900. [Google Scholar] [CrossRef]
- Zheng, Q.; Kuwabara, H.; Potscavage, W.J., Jr.; Huang, S.; Hatae, Y.; Shibata, T.; Adachi, C. Anthraquinone-Based Intramolecular Charge-Transfer Compounds: Computational Molecular Design, Thermally Activated Delayed Fluorescence, and Highly Efficient Red Electroluminescence. J. Am. Chem. Soc. 2014, 136, 18070–18081. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Nie, Z.H.; Zhao, L.; Lu, L.; Cheng, F.; Chen, X.; Tan, G.; Liu, Q.Q.; Wang, J.; Chauhan, R.; et al. Efficient degradation of dyes in water by two Ag-based coordination polymers containing 1,3-bis(3,5-dicarboxylphenoxy)benzene and N-donor linkers. Polyhedron 2021, 207, 115362–115369. [Google Scholar] [CrossRef]
- Murry, J.S.; Sen, K. Molecular Electrostatic Potential Concepts and Applications; Elsevier: Amsterdam, The Netherlands, 1996. [Google Scholar]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Karabacak, M.; Kose, E.; Atac, A.; Cipiloglu, M.A.; Kurt, M. Molecular structure investigation and spectroscopic studies on 2,3-difluorophenylboronic acid: A combined experimental and theoretical analysis. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 97, 892–908. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Tenderholt, A.L.; Langer, K.M. A library for package-independent computational chemistry algorithms. J. Comput. Inside Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Xavier, S.; Periandy, S. Spectroscopic (FT-IR, FT-Raman, UV and NMR) investigation on 1-phenyl-2-nitropropene by quantum computational calculations. Spectrochim, Acta Part A Mol. Biomol. Spectrosc. 2015, 149, 216–230. [Google Scholar] [CrossRef]
- Tahenti, M.; Gatfaoui, S.; Issaoui, N.; Roisnel, T.; Marouani, H. A tetrachlorocobaltate(II) salt with 2-amino-5-picolinium: Synthesis, theoretical and experimental characterization. J. Mol. Struct. 2020, 1207, 127781–127792. [Google Scholar] [CrossRef]
- Mleh, C.B.; Brandan, S.A.; Issaoui, N.; Roisnel, T.; Marouani, H. Synthesis, molecular structure, vibrational and theoretical studies of a new non-centrosymmetric organic sulphate with promising NLO properties. J. Mol. Struct. 2018, 1171, 771–785. [Google Scholar]
- Turner, M.J.; Mckinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer 17; The University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- Jayatilaka, D.; Grimwood, D.J. Tonto: A Fortran Based Object-Oriented System for Quantum Chemistry and Crystallography. In International Computational Science ICCS 2003, Melbourne, Australia; St. Petersburg, Russia, 2–4 June 2003; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- International Tables for X-ray Crystallography, Vol. III; Kynoch Press: Birmingham, UK, 1952.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Blatov, V.A.; Shevchenko, A.P.; Proserpio, D.M. Applied topological analysis of crystal structures with the program package ToposPro, Cryst. Growth Des. 2014, 14, 3576–3586. [Google Scholar] [CrossRef]
- O’Keeffe, M.; Peskov, M.A.; Ramsden, S.J.; Yaghi, O.M. The reticular chemistry structure resource (RCSR) database of, and symbols for, crystal nets. Acc. Chem. Res. 2008, 41, 1782–1789. [Google Scholar] [CrossRef]
- Alexandrov, E.V.; Blatov, V.A.; Kochetkov, A.V.; Proserpio, D.M. Underlying nets in three-periodic coordination polymers: Topology, taxonomy and prediction from a computer-aided analysis of the Cambridge Structural Database. CrystEngComm 2011, 13, 3947–3958. [Google Scholar] [CrossRef]
- Shevchenko, A.P.; Blatov, V.A. Simplify to understand: How to elucidate crystal structures? Struct. Chem. 2021, 32, 507–519. [Google Scholar] [CrossRef]
- Alexandrov, E.V.; Shevchenko, A.P.; Blatov, V.A. Topological Databases: Why Do We Need Them for Design of Coordination Polymers? Cryst. Growth Des. 2019, 19, 2604–2614. [Google Scholar] [CrossRef]
- Petersson, G.A.; Al-Laham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennet, A.; Tensfeld, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Origin 8.0; OriginLab Corp.: Northampton, MA, USA, 2021.
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta. 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. Cryst. Eng. Comm. 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Wang, W.; Ling, Y.; Yang, L.J.; Liu, Q.L.; Luo, Y.H.; Sun, B.W. Crystals of 4-(2-benzimidazole)-1,2,4-triazole and its hydrate: Preparations, crystal structure and Hirshfeld surfaces analysis. Res. Chem. Intermed. 2016, 42, 3157–3168. [Google Scholar] [CrossRef]
- Safin, D.A.; Robeyns, K.; Garcia, Y. 1,2,4-Triazole-based molecular switches: Crystal structures, Hirshfeld surface analysis and optical properties. Cryst. Eng. Comm. 2016, 18, 7284–7296. [Google Scholar] [CrossRef]
- Roeges, N.P.G. A Guide to the Complete Interpretation of the Infrared Spectra of Organic Structures; Wiley: New York, NY, USA, 1994. [Google Scholar]
- Rekik, N.; Issaoui, N.; Ghalla, H.; Oujia, B.; Wojcik, M.J. Infrared spectral density of H-bonds within the strong anharmonic coupling theory: Indirect relaxation effect. J. Mol. Struct. 2007, 844–845, 21–31. [Google Scholar] [CrossRef]
- Ganeshvar, P.S.; Kanagaraj, M.; Gunasekaran, S.; Gnanasambandan, T. Experimental and theoretical investigation of Gabapentin by Density functional theory. Int. J. Sci. Eng. Res. 2016, 7, 8–15. [Google Scholar]

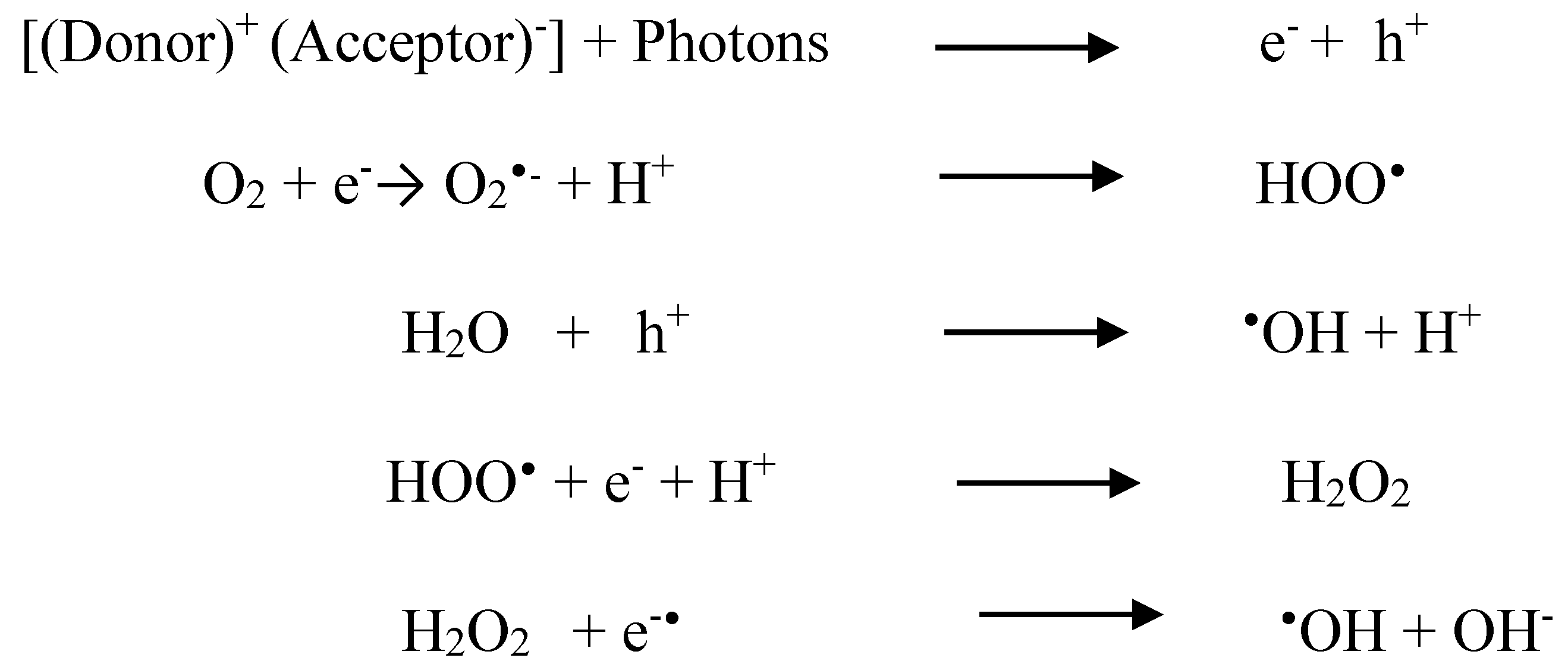

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCDC Number | 2102211 |
|---|---|
| Empirical formula | C29H21NO3 |
| Formula weight | 431.49 |
| Temperature/K | 100(2) |
| Crystal system | Triclinic |
| Space group | P-1 |
| a/Å | 8.1901(7) |
| b/Å | 12.4958(10) |
| c/Å | 12.5930(11) |
| α/° | 64.784(2) |
| β/° | 82.636(2) |
| γ/° | 77.491(2) |
| Volume/Å3 | 1137.36(17) |
| Z | 2 |
| ρcalcg/cm3 | 1.2599 |
| μ/mm−1 | 0.082 |
| F(000) | 452.2 |
| Crystal size/mm3 | 0.48 × 0.27 × 0.15 |
| Radiation | Mo Kα (λ = 0.71073) |
| 2Θ range for data collection/° | 6.08 to 50.1 |
| Index ranges | −10 ≤ h ≤ 10, −16 ≤ k ≤ 16, −16 ≤ l ≤ 16 |
| Reflections collected | 16,550 |
| Independent reflections | 4008 [Rint = 0.0399, Rsigma = 0.0471] |
| Data/restraints/parameters | 4008/1/302 |
| Goodness-of-fit on F2 | 1.063 |
| Final R indexes [I> = 2σ (I)] | R1 = 0.0512, wR2 = 0.1153 |
| Final R indexes [all data] | R1 = 0.0629, wR2 = 0.1215 |
| Largest diff. peak/hole/e Å−3 | 0.32/−0.28 |
| Solvent | TD/DFT |
|---|---|
| λmax (nm) | Band gap (eV) |
| Experimental | |
| Ethanol (L3) | |
| 252 | 4.92 |
| Ethanol (L2) | |
| 243.5 | 5.09 |
| Calculated | |
| Ethanol | |
| 262 | 4.73 |
| 241 | 5.14 |
| Energy Model | k_ele | k_pol | k_disp | k_rep |
|---|---|---|---|---|
| B3LYP/6-311G(d,p) electron densities | 1.057 | 0.740 | 0.871 | 0.618 |
| Electron Density | E_ele | E_pol | E_dis | E_rep |
| B3LYP/6-31G(d,p) | −25.9 | −5.3 | −71.2 | 56.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, A.; Fatima, A.; Shakya, S.; Rahman, Q.I.; Ahmad, M.; Javed, S.; AlSalem, H.S.; Ahmad, A. Crystal Structure, Topology, DFT and Hirshfeld Surface Analysis of a Novel Charge Transfer Complex (L3) of Anthraquinone and 4-{[(anthracen-9-yl)meth-yl] amino}-benzoic Acid (L2) Exhibiting Photocatalytic Properties: An Experimental and Theoretical Approach. Molecules 2022, 27, 1724. https://doi.org/10.3390/molecules27051724
Ahmed A, Fatima A, Shakya S, Rahman QI, Ahmad M, Javed S, AlSalem HS, Ahmad A. Crystal Structure, Topology, DFT and Hirshfeld Surface Analysis of a Novel Charge Transfer Complex (L3) of Anthraquinone and 4-{[(anthracen-9-yl)meth-yl] amino}-benzoic Acid (L2) Exhibiting Photocatalytic Properties: An Experimental and Theoretical Approach. Molecules. 2022; 27(5):1724. https://doi.org/10.3390/molecules27051724
Chicago/Turabian StyleAhmed, Adeeba, Aysha Fatima, Sonam Shakya, Qazi Inamur Rahman, Musheer Ahmad, Saleem Javed, Huda Salem AlSalem, and Aiman Ahmad. 2022. "Crystal Structure, Topology, DFT and Hirshfeld Surface Analysis of a Novel Charge Transfer Complex (L3) of Anthraquinone and 4-{[(anthracen-9-yl)meth-yl] amino}-benzoic Acid (L2) Exhibiting Photocatalytic Properties: An Experimental and Theoretical Approach" Molecules 27, no. 5: 1724. https://doi.org/10.3390/molecules27051724
APA StyleAhmed, A., Fatima, A., Shakya, S., Rahman, Q. I., Ahmad, M., Javed, S., AlSalem, H. S., & Ahmad, A. (2022). Crystal Structure, Topology, DFT and Hirshfeld Surface Analysis of a Novel Charge Transfer Complex (L3) of Anthraquinone and 4-{[(anthracen-9-yl)meth-yl] amino}-benzoic Acid (L2) Exhibiting Photocatalytic Properties: An Experimental and Theoretical Approach. Molecules, 27(5), 1724. https://doi.org/10.3390/molecules27051724