


Adsorption and Electropolymerization of p-Aminophenol Reduces Reproducibility of Electrochemical Immunoassays
 , , , , and
, , , , and 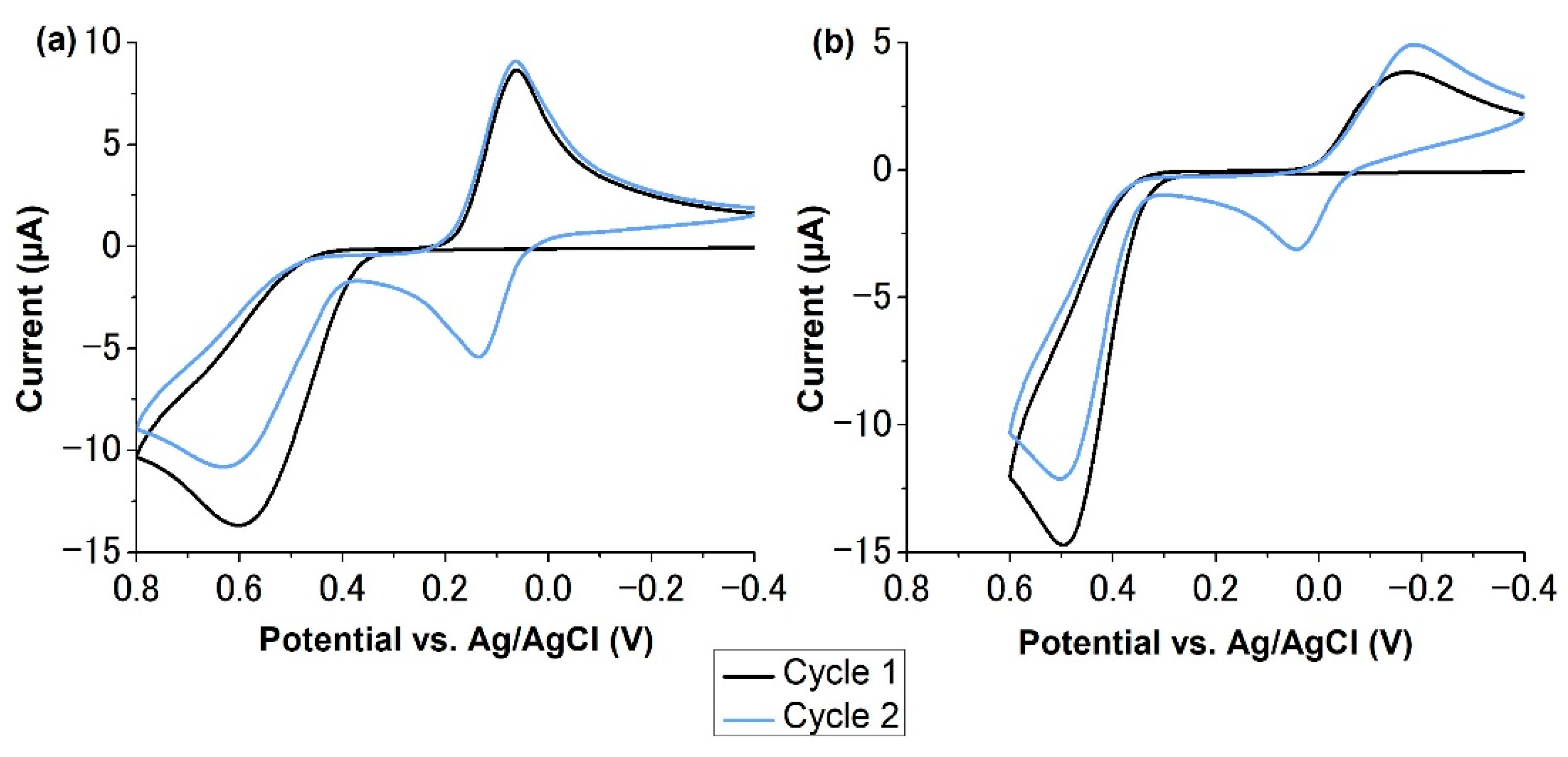{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Electrochemical Characterization of p-Aminophenyl Phosphate
2.2. The Effects of Sonication on p-Aminophenyl Phosphate
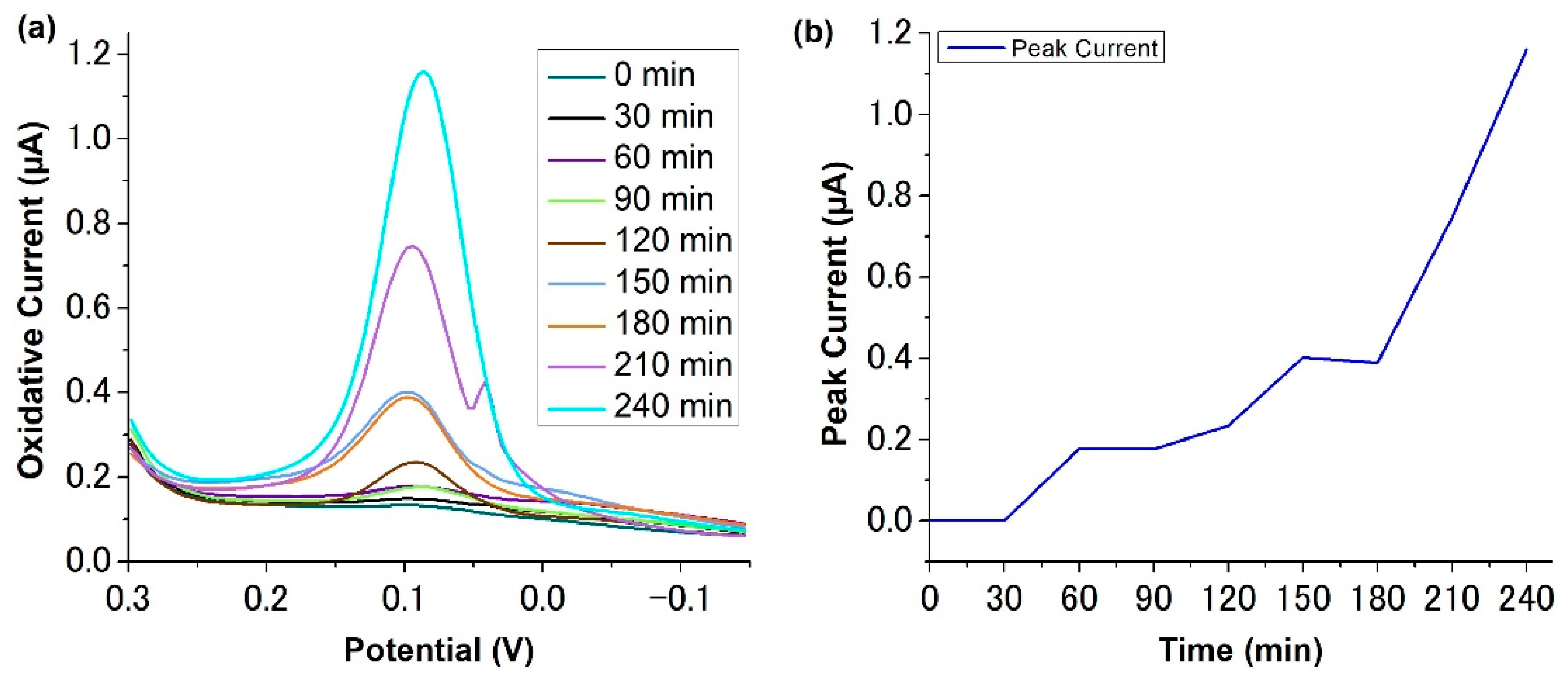
2.3. Adsorption of p-Aminophenol
2.4. Electropolymerization of p-Aminophenol
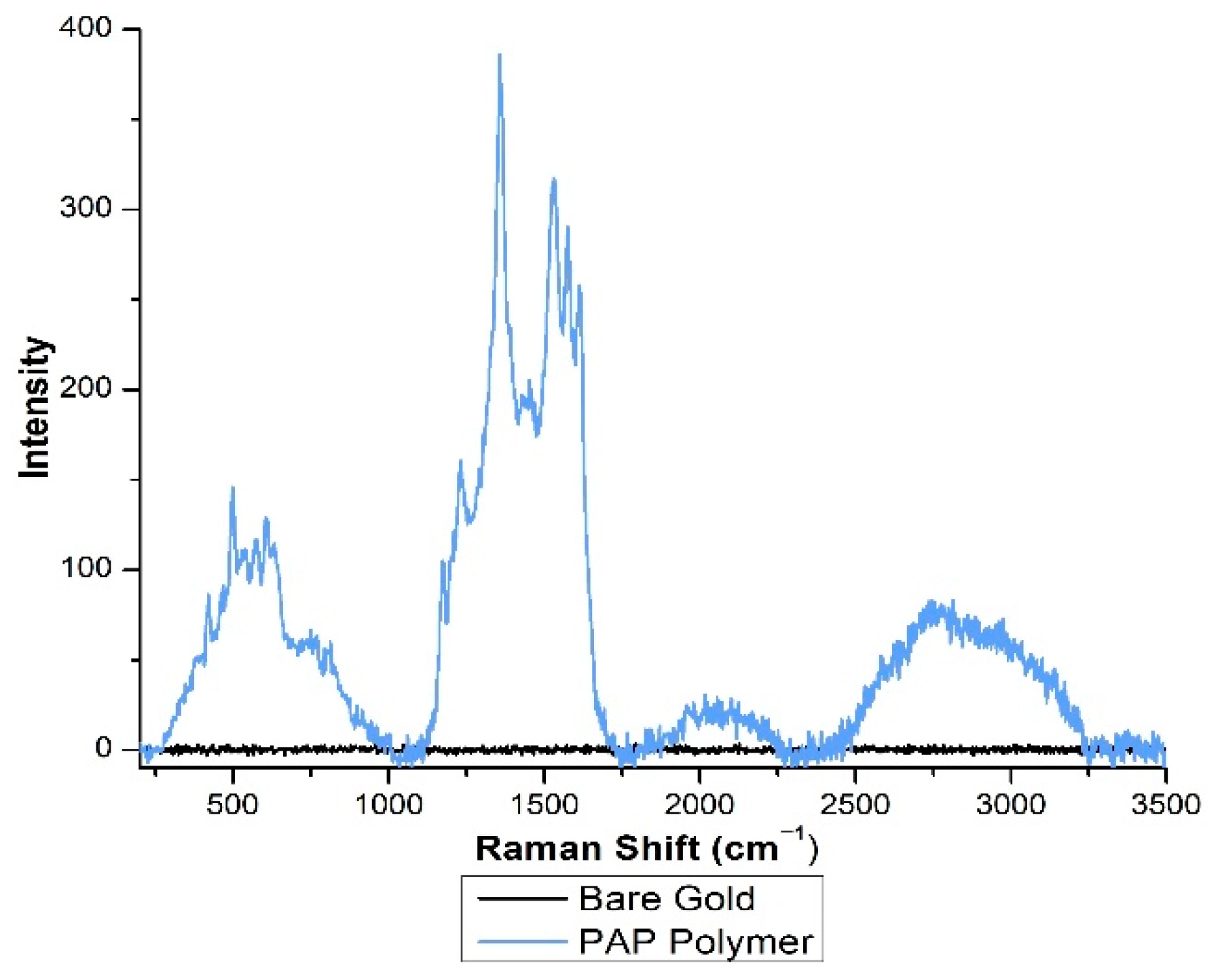
2.5. Spectral Analysis of Adsorbed PAP and PAP Polymer
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Instrumentation
4.3. Analytical Procedures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Weston, M.; Nash, C.; Fritsch, I. Redox-Magnetohydrodynamic Microfluidics Without Channels and Compatible with Electrochemical Detection Under Immunoassay Conditions. Anal. Chem. 2010, 82, 7068–7072. [Google Scholar] [CrossRef]
- Pemberton, R.; Hart, J.; Stoddard, P.; Foulkes, J. A comparison of 1-naphthyl phosphate and 4 aminophenyl phosphate as enzyme substrates for use with a screen-printed amperometric immunosensor for progesterone in cows’ milk. Biosens. Bioelectron. 1999, 14, 495–503. [Google Scholar] [CrossRef]
- Fanjul-Bolado, P.; Hernández-Santos, D.; Lamas-Ardisana, P.; Martín-Pernía, A.; Costa-García, A. Electrochemical characterization of screen-printed and conventional carbon paste electrodes Electrochim. Acta 2008, 53, 3635–3642. [Google Scholar]
- Messina, G.; Panini, N.; Martinez, N.; Raba, J. Microfluidic immunosensor design for the quantification of interleukin-6 in human serum samples. Anal. Biochem. 2008, 380, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Lunte, C.; Halsall, H.; Heineman, W. p-aminophenyl phosphate: An improved substrate for electrochemical enzyme immunoassay. Anal. Chim. Acta 1988, 214, 187–195. [Google Scholar] [CrossRef]
- Niwa, O.; Xu, Y.; Halsall, H.; Heineman, W. Small-Volume Voltammetric Detection of 4-Aminophenol with Interdigitated Array Electrodes and Its Application to Electrochemical Enzyme Immunoassay. Anal. Chem. 1993, 65, 1559–1563. [Google Scholar] [CrossRef]
- Yin, H.; Ma, Q.; Zhou, Y.; Ai, S.; Zhu, L. Electrochemical behavior and voltammetric determination of 4-aminophenol based on graphene–chitosan composite film modified glassy carbon electrode. Electrochim. Acta 2010, 55, 7102–7108. [Google Scholar] [CrossRef]
- Wang, J.; Jin, B.; Cheng, L. Investigation on redox mechanism of p-aminophenol in non-aqueous media by FT-IR spectroelectrochemistry. Electrochim. Acta 2013, 91, 152–157. [Google Scholar] [CrossRef]
- Taj, S.; Ahmed, M.; Sankarapapavinasam, S. Poly(para-aminophenol): A new soluble, electroactive conducting polymer. J. Electroanal. Chem. 1992, 338, 347–352. [Google Scholar] [CrossRef]
- Schwarz, J.; Oelßner, W.; Kaden, H.; Schumer, F.; Hennig, H. Voltammetric and spectroelectrochemical studies on 4-aminophenol at gold electrodes in aqueous and organic media. Electrochim. Acta 2003, 48, 2479–2486. [Google Scholar] [CrossRef]
- Salavagione, H.; Arias, J.; Garcés, P.; Morallón, E.; Barbero, C.; Vázquez, J. Spectroelectrochemical study of the oxidation of aminophenols on platinum electrode in acid medium. J. Electroanal. Chem. 2004, 565, 375–383. [Google Scholar] [CrossRef]
- Vieira, S.; Ferreira, L.; Franco, D.; Afonso, A.; Conçalves, R.; Brito-Madurro, A.; Madurro, J. Electrochemical Modification of Graphite Electrodes with Poly(4-aminophenol). Macromol. Symp. 2006, 245–246, 236–242. [Google Scholar] [CrossRef]
- Menezes, H.; Maia, G. Films formed by the electrooxidation of p-aminophenol (p-APh) in aqueous medium: What do they look like? J. Electroanal. Chem. 2006, 586, 39–48. [Google Scholar] [CrossRef]
- Chandrashekar, B.; Swamy, B.; Pandurangachar, M.; Sathisha, T.; Sherigara, B. Electrochemical Investigation of 4-Aminophenol at CTAB Modified Carbon Paste Electrode: A Cyclic Voltammetric Technique. Anal. Bioanal. Electrochem. 2011, 3, 227–232. [Google Scholar]
- Heras, A.; Avila, J.; Ruiz, J.; García-Blanco, F. A contribution to the study of the electrochemical oxidation of p-aminophenol on a mercury electrode. Electrochim. Acta 1984, 29, 541–545. [Google Scholar] [CrossRef]
- de Souza, J.; Zanoni, M.; Oliveira-Brett, A. Genotoxic permanent hair dye precursors p-aminophenol and p-toluenediamine electrochemical oxidation mechanisms and evaluation in biological fluids. J. Electroanal. Chem. 2020, 857, 113509. [Google Scholar] [CrossRef]
- Michalska, A.; Maksymiuk, K. On the pH Influence on Electrochemical Properties of Poly(pyrrole) and Poly(N-methylpyrrole). Electroanalysis 1998, 10, 177–180. [Google Scholar] [CrossRef]
- Su, W.; Wang, S.; Cheng, S. Electrochemically pretreated screen-printed carbon electrodes for the simultaneous determination of aminophenol isomers. J. Electroanal. Chem. 2011, 651, 166–172. [Google Scholar] [CrossRef]
- Sun, W.; Jiao, K.; Zhang, S.; Zhang, C.; Zhang, Z. Electrochemical detection for horseradish peroxidase-based enzyme immunoassay using p-aminophenol as substrate and its application in detection of plant virus. Anal. Chim. Acta 2001, 434, 43–50. [Google Scholar] [CrossRef]
- Shende, C.; Smith, W.; Brouillette, C.; Farquharson, S. Drug Stability Analysis by Raman Spectroscopy. Pharmaceutics 2014, 6, 651–662. [Google Scholar] [CrossRef]
- Sun, Q.; Tripath, N.; Schuler, R. Time-Resolved Resonance Raman Spectroscopy of p-Aminophenol Radical Cation in Aqueous Solution. J. Phys. Chem. 1990, 94, 6273–6277. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundaments and Applications, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Joschek, H.; Miller, S. Photooxidation of Phenol, Cresols, and Dihydroxybenzenes1,2. J. Am. Chem. Soc. 1966, 88, 3273–3281. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buckey, G.; Owens, O.E.; Gabriel, A.W.; Downing, C.M.; Calhoun, M.C.; Cliffel, D.E. Adsorption and Electropolymerization of p-Aminophenol Reduces Reproducibility of Electrochemical Immunoassays. Molecules 2022, 27, 6046. https://doi.org/10.3390/molecules27186046
Buckey G, Owens OE, Gabriel AW, Downing CM, Calhoun MC, Cliffel DE. Adsorption and Electropolymerization of p-Aminophenol Reduces Reproducibility of Electrochemical Immunoassays. Molecules. 2022; 27(18):6046. https://doi.org/10.3390/molecules27186046
Chicago/Turabian StyleBuckey, Grace, Olivia E. Owens, Ainslee W. Gabriel, Claudia M. Downing, Margaret C. Calhoun, and David E. Cliffel. 2022. "Adsorption and Electropolymerization of p-Aminophenol Reduces Reproducibility of Electrochemical Immunoassays" Molecules 27, no. 18: 6046. https://doi.org/10.3390/molecules27186046
APA StyleBuckey, G., Owens, O. E., Gabriel, A. W., Downing, C. M., Calhoun, M. C., & Cliffel, D. E. (2022). Adsorption and Electropolymerization of p-Aminophenol Reduces Reproducibility of Electrochemical Immunoassays. Molecules, 27(18), 6046. https://doi.org/10.3390/molecules27186046