
Synthesis of 3-Aryl-3-(Furan-2-yl)Propanoic Acid Derivatives, and Study of Their Antimicrobial Activity

 , , , , , , and
, , , , , , and 
Abstract
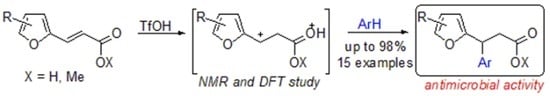
:
1. Introduction
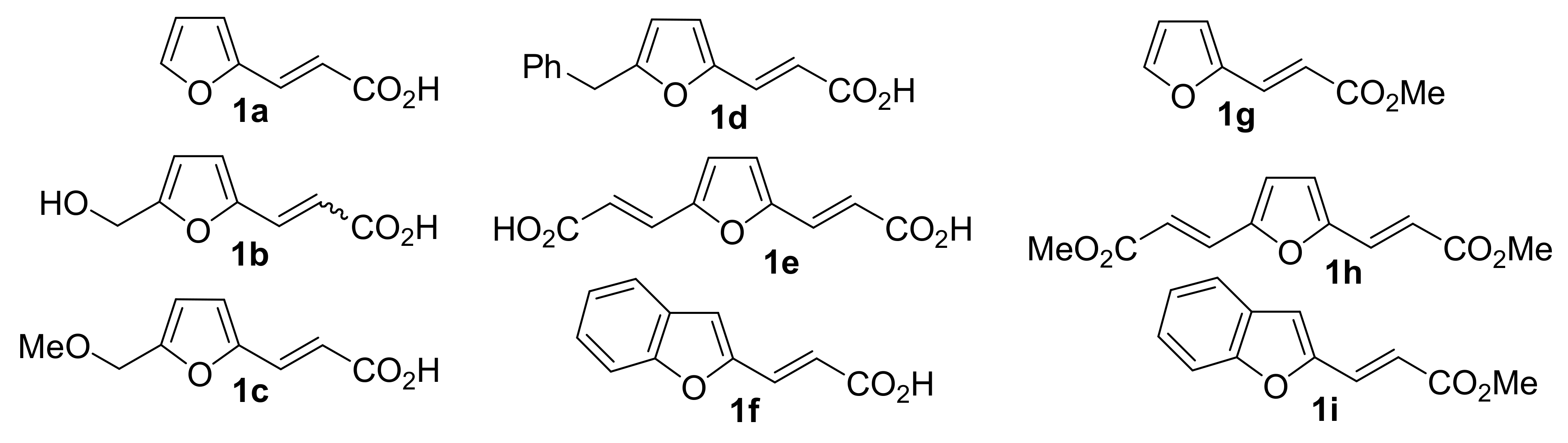
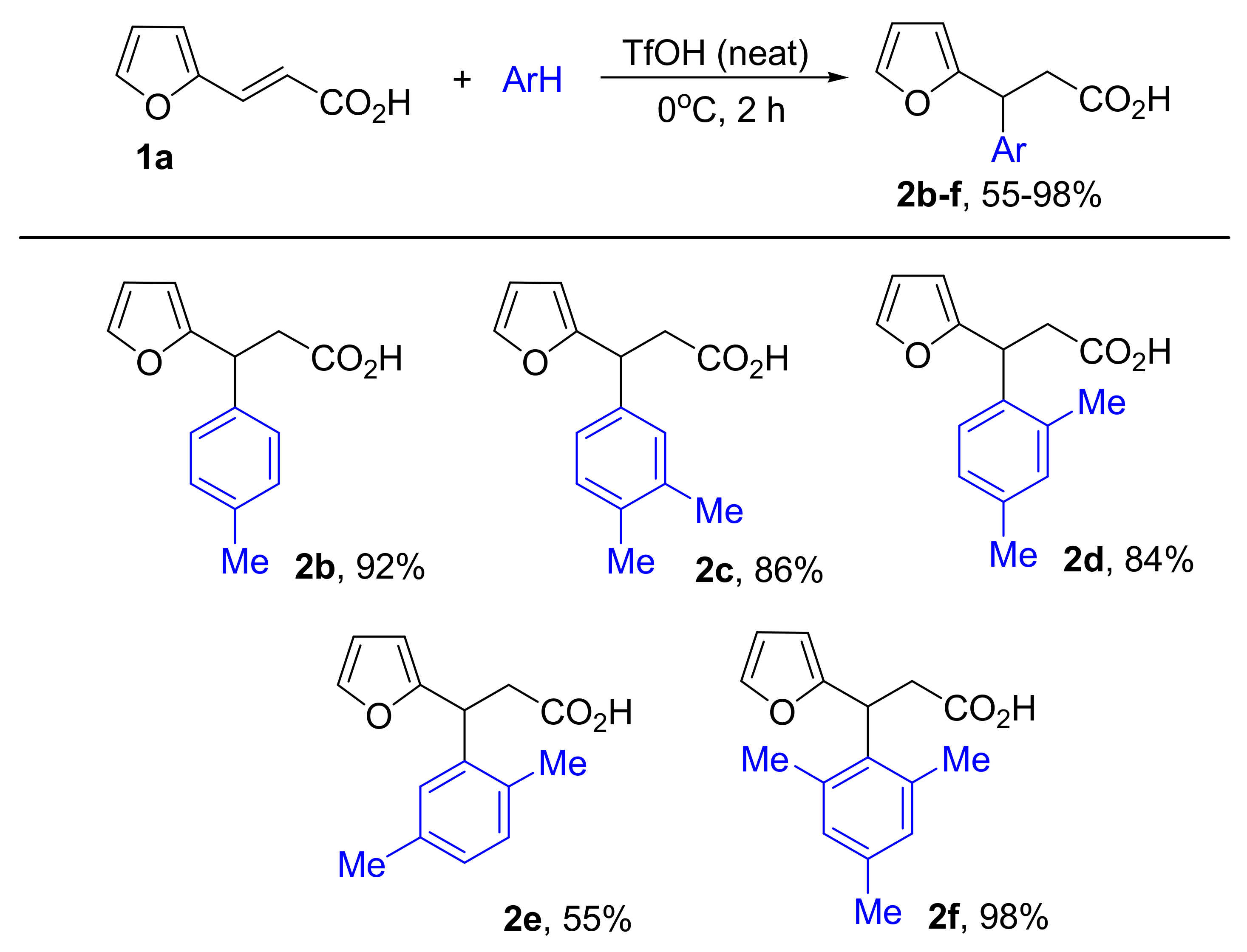
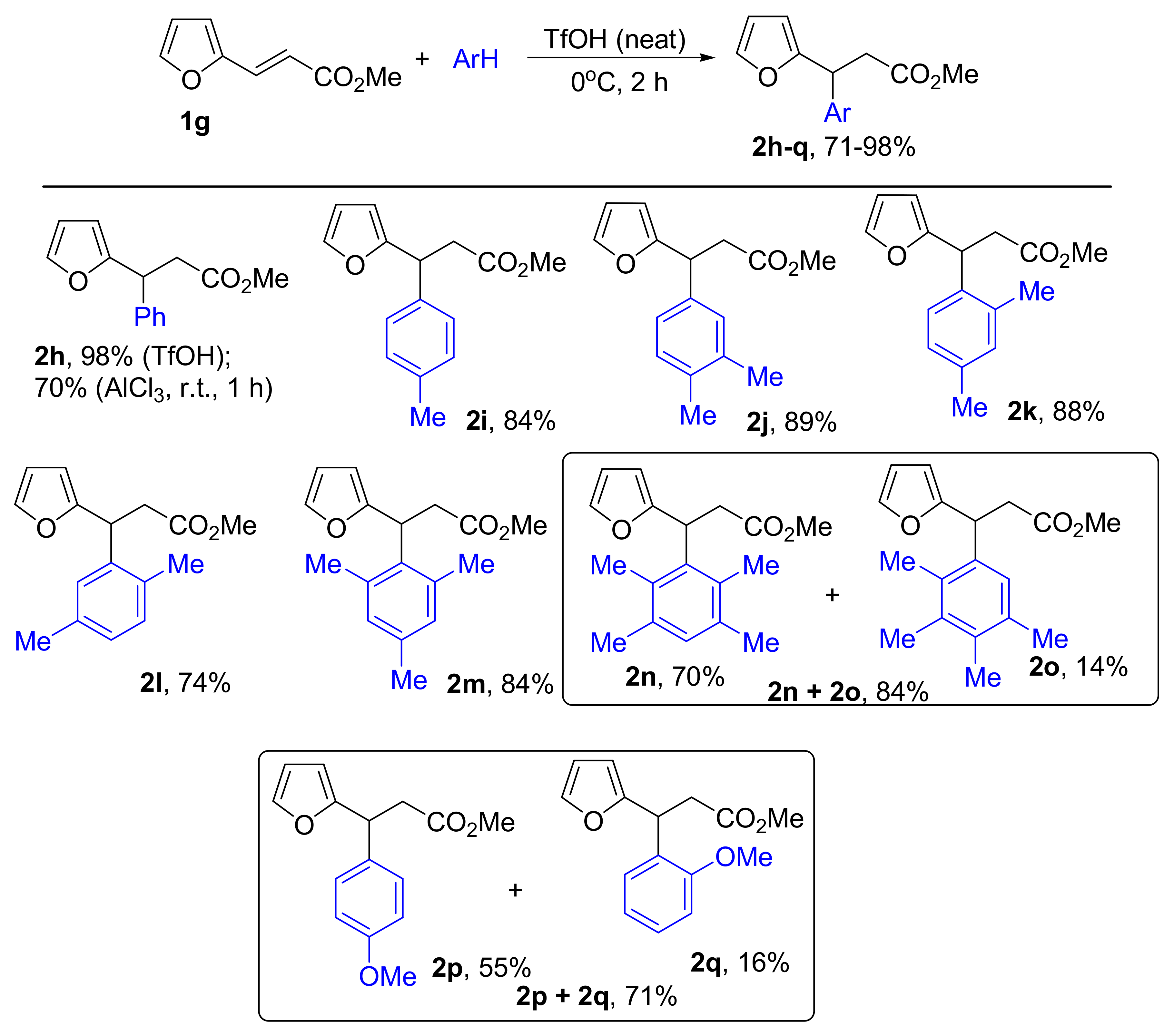
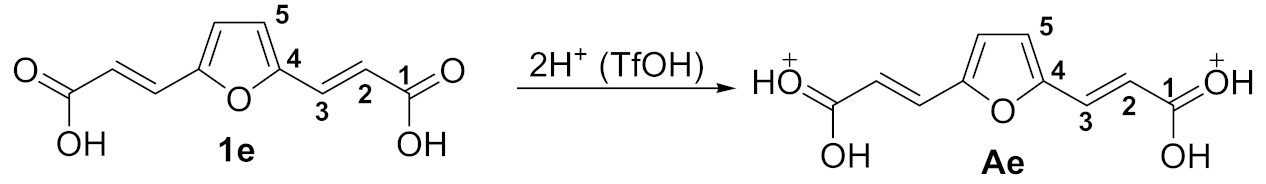
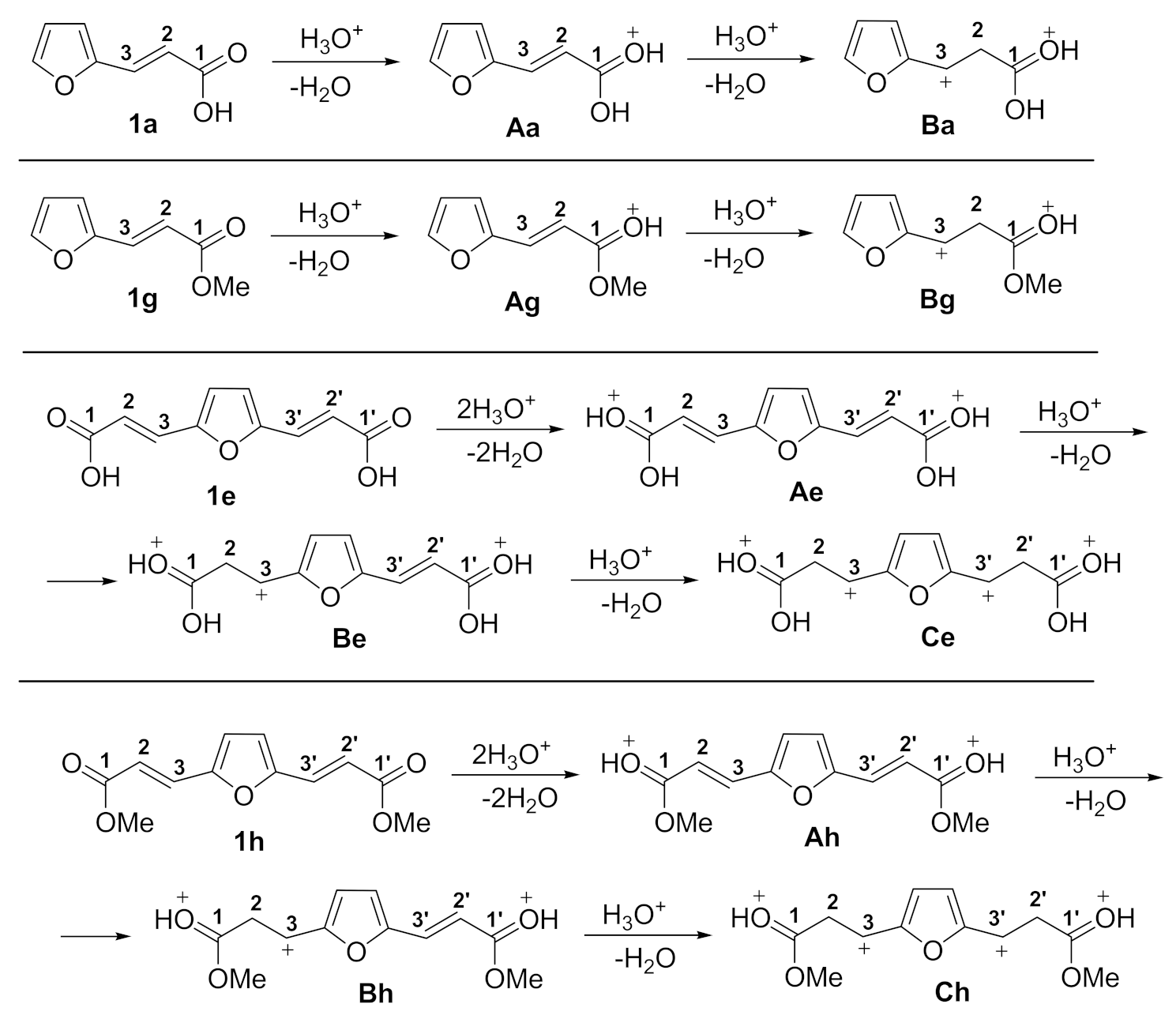
2. Results and Discussion
3. Conclusions
4. Experimental Part
4.1. General Information
4.2. DFT Calculations
4.3. Study of Biological Activity
4.4. Preparation and Characterization of Compounds
4.4.1. General Procedure for Synthesis of 3-(furan-2-yl)propenoic Acids 1a–f from furan-2-carbaldehydes and Malonic Acid
4.4.2. General Procedure for Synthesis of 3-(furan-2-yl)-3-phenylpropanoic Esters 1g-i from 3-(furan-2-yl)propeonic Acids 1a,e,f
4.4.3. General Procedure for Synthesis of 3-(furan-2-yl)-3-phenylpropanoic Acids and Esters 2a-r from Compounds 1 and Arenes in TfOH
4.4.4. General Procedure for Synthesis of 3-(furan-2-yl)-3-phenylpropanoic Acids and Esters 2 from Compounds 1 and Benzene under the Action of AlX3 (X = Cl, Br)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kashparova, V.P.; Chernysheva, D.V.; Klushin, V.A.; Andreeva, V.E.; Kravchenko, O.A.; Smirnova, N.V. Furan monomers and polymers from renewable plant biomass. Russ. Chem. Rev. 2021, 90, 750–784. [Google Scholar] [CrossRef]
- Dutta, S. Valorization of biomass-derived furfurals: Reactivity patterns, synthetic strategies, and applications. Biomass Conv. Bioref. 2021. [CrossRef]
- Chernyshev, V.M.; Kravchenko, O.A.; Ananikov, V.P. Conversion of plant biomass to furan derivatives and sustainable access to the new generation of polymers, functional materials and fuels. Russ. Chem. Rev. 2017, 86, 357–387. [Google Scholar] [CrossRef]
- Gandini, A.; Lacerda, T.M.; Carvalho, A.J.F.; Trovatti, E. Progress of Polymers from Renewable Resources: Furans, Vegetable Oils, and Polysaccharides. Chem. Rev. 2016, 116, 1637−1669. [Google Scholar] [CrossRef]
- Wang, T.; Nolte, M.W.; Shanks, B.H. Catalytic dehydration of C6 carbohydrates for the production of hydroxymethylfurfural (HMF) as a versatile platform chemical. Green Chem. 2014, 16, 548–572. [Google Scholar] [CrossRef]
- Teong, S.P.; Yia, G.; Zhang, Y. Hydroxymethylfurfural production from bioresources: Past, present and future. Green Chem. 2014, 16, 2015–2026. [Google Scholar] [CrossRef]
- Cai, J.; Li, K.; Wu, S. Recent advances in catalytic conversion of biomass derived 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid. Biomass Bioenergy 2022, 158, 106358. [Google Scholar] [CrossRef]
- Le, H.S.; Said, Z.; Pham, M.T.; Le, T.H.; Veza, I.; Nguyen, V.N.; Deepanraj, B.; Nguyen, L. Production of HMF and DMF biofuel from carbohydrates through catalytic pathways as a sustainable strategy for the future energy sector. Fuel 2022, 324, 124474. [Google Scholar]
- Slak, J.; Pomeroy, D.; Kostyniuk, A.; Grilc, M.; Likozar, B. A review of bio-refining process intensification in catalytic conversion reactions, separations and purifications of hydroxymethylfurfural (HMF) and furfural. Chem. Eng. J. 2022, 429, 132325. [Google Scholar] [CrossRef]
- Makarov, A.S.; Uchuskin, M.G.; Trushkov, I.V. Furan Oxidation Reactions in the Total Synthesis of Natural Products. Synthesis 2018, 50, 3059–3086. [Google Scholar]
- Sidwell, R.W.; Bailey, K.W.; Wong, M.H.; Barnard, D.L.; Smee, D.F. In vitro and in vivo influenza virus-inhibitory effects of viramidine. Antivir. Res. 2005, 68, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Kazakova, A.N.; Vasilyev, A.V. Trifluoromethanesulfonic acid in organic synthesis. Russ. J. Org. Chem. 2017, 53, 485–509. [Google Scholar] [CrossRef]
- Vasilyev, A.V. Superelectrophilic activation of alkynes, alkenes, and allenes. Adv. Org. Synt. 2018, 8, 81–120. [Google Scholar]
- Kompanec, M.O.; Kusch, O.V.; Litvinov, Y.E.; Pliekhov, O.L.; Novikova, K.V.; Novokhatko, A.O.; Shendrick, A.N.; Vasilyev, A.V.; Opieda, L.O. Oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran with molecular oxygen in the presence of N-hydroxyphthalimide. Cat. Commun. 2014, 57, 60–63. [Google Scholar] [CrossRef]
- Ryabukhin, D.S.; Zakusilo, D.N.; Kompanets, M.O.; Tarakanov, A.A.; Boyarskaya, I.A.; Artamonova, T.O.; Khohodorkovskiy, M.A.; Opeida, I.O.; Vasilyev, A.V. Superelectrophilic activation of 5-hydroxymethylfurfural and 2,5-diformylfuran: Organic synthesis based on biomass-derived products. Beilstein J. Org. Chem. 2016, 12, 2125–2135. [Google Scholar] [CrossRef] [Green Version]
- Van Zandvoort, I.; Wang, Y.; Rasrendra, C.B.; van Eck, E.R.H.; Bruijnincx, P.C.A.; Heeres, H.J.; Weckhuysen, B.M. Formation, Molecular Structure, and Morphology of Humins in Biomass Conversion: Influence of Feedstock and Processing Conditions. ChemSusChem 2013, 6, 1745–1758. [Google Scholar] [CrossRef]
- Hoang, T.M.; van Eck, E.R.H.; Bula, W.; Gardeniers, J.G.E.; Lefferts, L.; Seshan, K. Humin based by-products from biomass processing as a potential carbonaceous source for synthesis gas production. Green Chem. 2015, 17, 959–972. [Google Scholar] [CrossRef]
- Parr, R.; Szentpaly, L.V.; Liu, K.J. Computational Analysis of Theacrine, a Purported Nootropic and Energy-Enhancing Nutritional Supplement. Am. Chem. Soc. 1999, 121, 1922−1924. [Google Scholar]
- Koltunov, K.Y.; Repinskaya, I. Interaction of phenols and their derivatives with aromatic compounds in the presence of acidic agents XIV. Protonation of α,β-enones in the HF-SbF5-SO2FCl superacid system. Russ. J. Org. Chem. 1994, 30, 82–85, (Translated from ZhOrKh, 1994, 30, 90–93). [Google Scholar]
- Walspurger, S.; Vasilyev, A.V.; Sommer, J.; Pale, P. Chemistry of 3-arylindenones: Behavior in superacids and photodimerization. Tetrahedron 2005, 61, 3559–3564. [Google Scholar] [CrossRef]
- Koltunov, K.Y.; Walspurger, S.; Sommer, J. Superacidic Activation of α,β-Unsaturated Amides and Their Electrophilic Reactions. Eur. J. Org. Chem. 2004, 2004, 4039–4047. [Google Scholar] [CrossRef]
- Koltunov, K.Y.; Walspurger, S.; Sommer, J. Friedel–Crafts alkylation of benzene with α,β-unsaturated amides. Tetrahedron Lett. 2004, 45, 3547–3549. [Google Scholar] [CrossRef]
- Rendy, R.; Zhang, Y.; McElrea, A.; Gomez, A.; Klumpp, D.A. Superacid-Catalyzed Reactions of Cinnamic Acids and the Role of Superelectrophiles1. J. Org. Chem. 2004, 69, 2340–2347. [Google Scholar] [CrossRef]
- Stadler, D.; Goeppert, A.; Rasul, G.; Olah, G.A.; Prakash, G.K.S.; Bach, T.J. Chiral Benzylic Carbocations: Low-Temperature NMR Studies and Theoretical Calculations. Org. Chem. 2009, 74, 312–318. [Google Scholar] [CrossRef]
- Allen, J.M.; Johnston, K.M.; Jones, J.F.; Shotter, R.G. Friedel-crafts cyclisation—VI1: Polyphosphoric acid-catalysed reactions of crotonophenones and chalcones. Tetrahedron 1977, 33, 2083–2087. [Google Scholar] [CrossRef]
- Suzuki, T.; Ohwada, T.; Shudo, K.J. Superacid-Catalyzed Electrocyclization of 1-Phenyl-2-propen-1-ones to 1-Indanones. Kinetic and Theoretical Studies of Electrocyclization of Oxonium−Carbenium Dications. Am. Chem. Soc. 1997, 119, 6774–6780. [Google Scholar]
- Prakash, G.K.S.; Yan, P.; Török, B.; Olah, G.A. Superacidic Trifluoromethanesulfonic Acid-Induced Cycli-Acyalkylation of Aromatics. Catal. Lett. 2003, 87, 109–112. [Google Scholar] [CrossRef]
- Wu, G.; Yang, Q.; Long, M.; Guo, L.; Li, B.; Meng, Y.; Zhang, A.; Wang, H.; Liu, S.; Zou, L.J. Evaluation of agar dilution and broth microdilution methods to determine the disinfectant susceptibility. J. Antibiot. 2015, 68, 661–665. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; He, X.; Meng, Y.; Guo, L.; Long, M.; Yu, H.; Li, B.; Fan, L.; Liu, S.; Wang, H.; et al. Superacidic Trifluoromethanesulfonic Acid-Induced Cycli-Acyalkylation of Aromatics. Microb. Drug Resist. 2016, 22, 80–87. [Google Scholar] [CrossRef]
- Gus’kova, T.A.; Durnev, A.D.; Reikhart, D.V.; Chernyavtseva, A.P.; Robb, J.R.; Cheeseman, G.; Scalmani, V.; Barone, B.; Mennucci, G.A.; Petersson, H.; et al. Fox in Gaussian 09, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Peng, Y.; Song, G. Combined microwave and ultrasound accelerated Knoevenagel–Doebner reaction in aqueous media: A green route to 3-aryl acrylic acids. Green Chem. 2003, 5, 704–706. [Google Scholar] [CrossRef]
- Mndzhoyan, O.L.; Avetisyan, S.A.; Azaryan, L.V. Synthesis of 3-(5-Hydroxymethylfuran-2-yl)propenoic acid. Sint. Geterotsiklikh Soedin. 1981, 12, 47–49. (In Russian) [Google Scholar]
- Serum, E.M.; Selvakumar, S.; Zimmermann, N.; Sibi, M.P. Valorization of 2,5-furandicarboxylic acid. Diels–Alder reactions with benzyne. Green Chem. 2018, 20, 1448–1454. [Google Scholar]
- Shindo, M.; Makigawa, S.; Kodama, K.; Sugiyama, H.; Matsumoto, T.; Iwataa, N.; Wasanoc, A.; Kano, M.T.; Morita, Y.; Fujii, Y. Design and chemical synthesis of root gravitropism inhibitors: Bridged analogues of ku-76 have more potent activity. Phytochemistry 2020, 179, 112508. [Google Scholar] [CrossRef]
- Lasseuguette, E.; Gandini, A.; Timpe, H.-J. Photoreactive furan derivatives. J. Photochem. Photobiol. A Chem. 2005, 174, 222–228. [Google Scholar] [CrossRef]
- Park, E.; Park, J.; Cheon, C.H. Remarkable Differences in Reactivity between Cyanide and N-Heterocyclic Carbenes in Ring-Closing Reactions of 4-(2-Formylphenoxy)but-2-Enoate Derivatives. Bull. Korean Chem. Soc. 2021, 42, 483–485. [Google Scholar] [CrossRef]
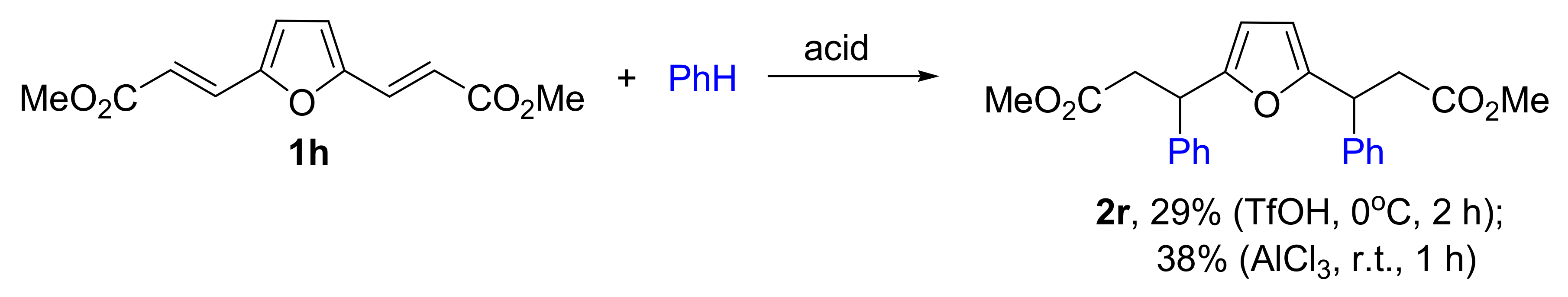
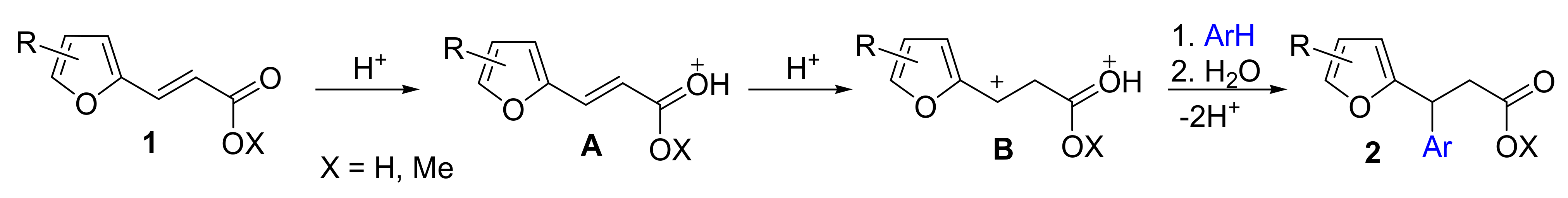
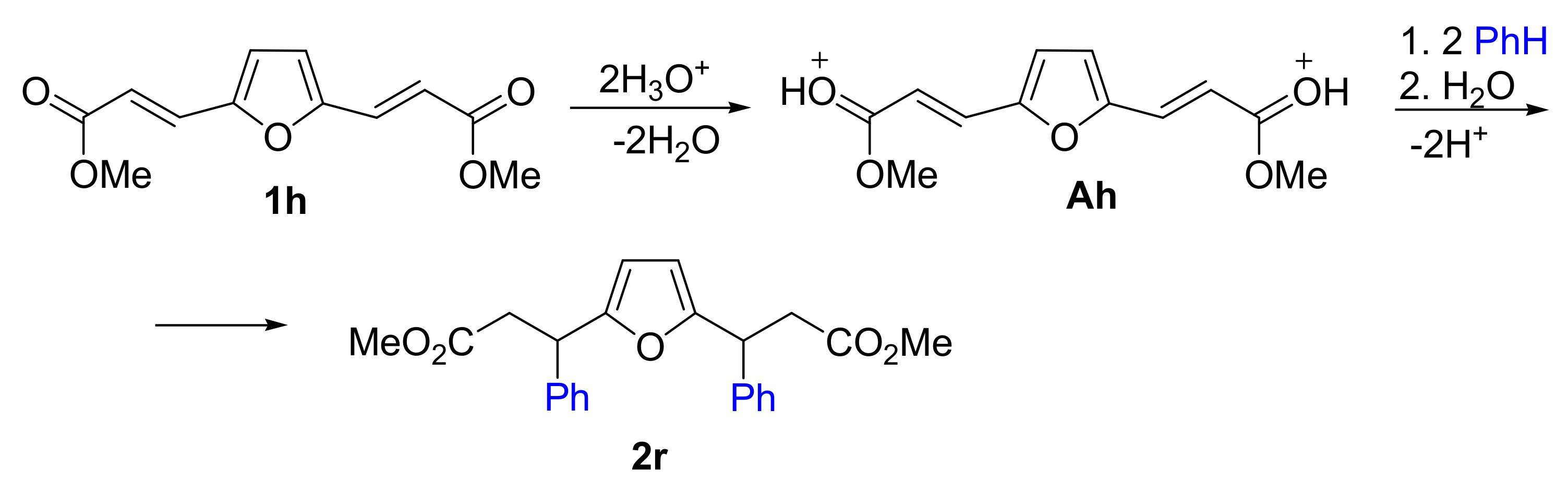

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
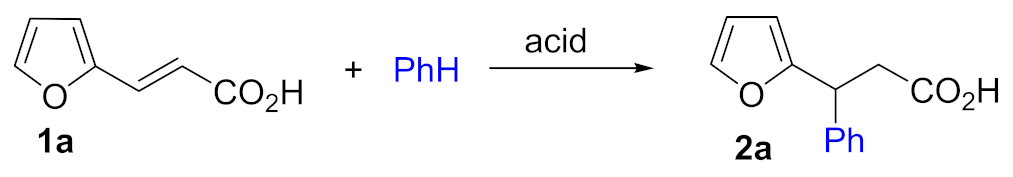 | ||||
| Entry | Reaction Conditions | Yield of 2a, % | ||
| Acid (Equiv.) | Temperature, °C | Time, h | ||
| 1 | CF3CO2H (19) | r.t. | 1 | no reaction |
| 2 | H2SO4 (52) | r.t. | 1 | oligomerization |
| 3 | FeBr3 (5.5) | r.t. | 1 | oligomerization |
| 4 | AlCl3 (5.5) | r.t. | 1 | 65 |
| 5 | AlCl3 (5.5) | r.t. | 4 | 47 |
| 6 | AlBr3 (5.5) | r.t. | 1 | 52 |
| 7 | TfOH (16) | r.t. | 1 | 22 |
| 8 | TfOH (16) | 0 | 0.25 | 28 |
| 9 | TfOH (16) | 0 | 1 | 32 |
| 10 | TfOH (16) | 0 | 2 | 33 |
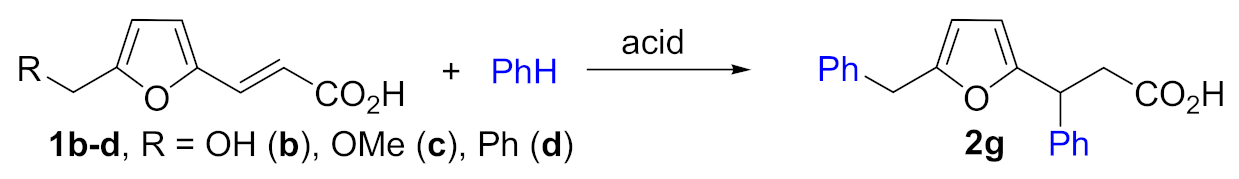 | |||||
| Entry | Starting Compound 1b-d | Reaction Conditions | Yield of 2g, % | ||
| Acid (Equiv.) | Temperature, °C | Time, h | |||
| 1 | 1b | H2SO4 (55) | r.t. | 1 | oligomerization |
| 2 | 1b | AlCl3 (5.5) | r.t. | 1 | 44 |
| 3 | 1b | AlCl3 (5.5) | r.t. | 4 | 17 |
| 4 | 1b | TfOH (19) | r.t. | 1 | 37 |
| 5 | 1b | TfOH (19) | 0 | 0.25 | 43 |
| 6 | 1b | TfOH (19) | 0 | 1 | 37 |
| 7 | 1c | AlCl3 (5.5) | r.t. | 1 | 36 |
| 8 | 1c | TfOH (19) | 0 | 0.25 | 39 |
| 9 | 1c | TfOH (19) | 0 | 1 | 46 |
| 10 | 1d | AlCl3 (5.5) | r.t. | 1 | 75 |
| 11 | 1d | TfOH (19) | 0 | 0.25 | 60 |
| 12 | 1d | TfOH (19) | 0 | 1 | 63 |
| Starting Compound | Retention Time, min | Peak Area, Arb. Units | Elemental Composition | RDB a | [M+H]+ m/z (Found) | [M+H]+ m/z (Calcul.) | Δ, ppm |
|---|---|---|---|---|---|---|---|
| 1a | 10.0 | 472 | C13H14O5 | 7 | 251.0914 | 251.0914 | 0 |
| 10.9 | 1400 | C19H18O7 | 11 | 359.1124 | 359.1125 | −0.4 | |
| 11.6 | 1550 | C20H22O9 | 10 | 407.1335 | 407.1337 | −0.4 | |
| 12.4 | 900 | C23H26O10 | 11 | 463.1591 | 463.1599 | −1.7 | |
| 1b | 17.7 | 907 | C28H26O7 | 16 | 475.1744 | 475.1751 | −1.5 |
| 18.3 | 410 | C20H20O4 | 11 | 325.1436 | 325.1434 | 0.5 | |
| 19.8 | 712 | C24H30O6 | 10 | 415.2117 | 415.2115 | 0.4 | |
| 23.4 | 1220 | C28H28O5 | 15 | 445.2008 | 445.2010 | −0.3 |
 | ||||||
| Compound/Cation | 13C NMR, δ, ppm | |||||
| C1 | C2 | C3 | C4 | C5 | ||
| Acid 1e in CDCl3 | 166.9 | 129.7 | 117.3 | 151.8 | 118.1 | |
| Cation Ae in TfOH | 182.2 | 128.5 | 110.1 | 155.8 | 145.0 | |
| Δδ = δAe − δ1e | 15.3 | 1.2 | 7.2 | 4.0 | 26.9 | |
 | ||||||
| Compound/Cation | 13C NMR, δ, ppm | |||||
| C1 | C2 | C3 | C4 | C5 | C6 | |
| Ester 1h in CDCl3 | 166.2 | 129.4 | 116.7 | 151.8 | 115.9 | 51.0 |
| Cation Ah in TfOH | 181.5 | 128.0 | 110.3 | 155.6 | 143.6 | 62.7 |
| Δδ = δAh − δ1h | 15.3 | 1.4 | 6.4 | 3.8 | 27.7 | 11.7 |
 | |||||||||
| Entry | Species | EHOMO, eV | ELUMO, eV | ω, a eV | q(C1), b e | q(C3), b e | k(C1)LUMO, c % | k(C3)LUMO, c % | ΔG, d kJ/mol |
| 1 | 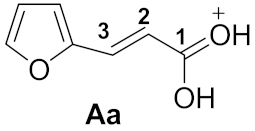 | −6.85 | −3.28 | 3.6 | 0.83 | −0.08 | 22.0 | 27.1 | 1a→Aa −69.1 |
| 2 | 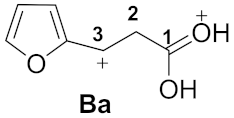 | −9.17 | −4.63 | 5.2 | 0.94 | 0.02 | 6.9 | 30.6 | Aa→Ba 26.0 |
| 3 | 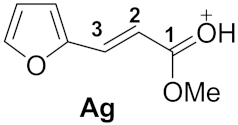 | −6.86 | −3.34 | 3.7 | 0.83 | −0.08 | 23.2 | 26.0 | 1g→Ag −39.9 |
| 4 | 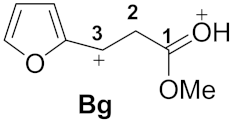 | −9.18 | −4.63 | 5.3 | 0.95 | 0.02 | 13.2 | 27.0 | Ag→Bg 30.8 |
| 5 | 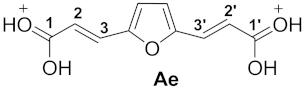 | −7.02 | −4.06 | 5.2 | 0.86 | −0.19 | 11.8 | 11.6 | 1e→Ae −117.3 |
| 6 | 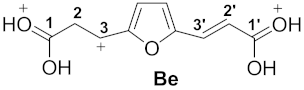 | −8.82 | −5.25 | 6.9 | C1 0.89 C1′ 0.94 | C3 −0.17 C3′ 0.01 | C1 4.3 C1′ 6.2 | C3 4.0 C3′ 19.1 | Ae→Be 53.4 |
| 7 |  | −10.61 | −7.28 | 12.0 | 0.94 | 0.24 | 2.2 | 21.0 | Be→Ce 222 |
| 8 | 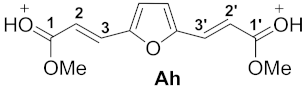 | −7.03 | −4.13 | 5.4 | 0.88 | −0.19 | 12.0 | 11.4 | 1h→Ah −54.6 |
| 9 | 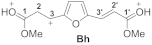 | −8.82 | −5.27 | 7.0 | C1 0.91 C1′ 0.95 | C3 −0.18 C3′ 0.03 | C1 5.2 C1′ 6.5 | C3 4.6 C3′ 19.5 | Ah→Bh 60.7 |
| 10 |  | −10.62 | −7.28 | 12.0 | 0.95 | 0.24 | 11.0 | 10.8 | Bh→Ch 208 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalyaev, M.V.; Ryabukhin, D.S.; Borisova, M.A.; Ivanov, A.Y.; Boyarskaya, I.A.; Borovkova, K.E.; Nikiforova, L.R.; Salmova, J.V.; Ul’yanovskii, N.V.; Kosyakov, D.S.; et al. Synthesis of 3-Aryl-3-(Furan-2-yl)Propanoic Acid Derivatives, and Study of Their Antimicrobial Activity. Molecules 2022, 27, 4612. https://doi.org/10.3390/molecules27144612
Kalyaev MV, Ryabukhin DS, Borisova MA, Ivanov AY, Boyarskaya IA, Borovkova KE, Nikiforova LR, Salmova JV, Ul’yanovskii NV, Kosyakov DS, et al. Synthesis of 3-Aryl-3-(Furan-2-yl)Propanoic Acid Derivatives, and Study of Their Antimicrobial Activity. Molecules. 2022; 27(14):4612. https://doi.org/10.3390/molecules27144612
Chicago/Turabian StyleKalyaev, Mikhail V., Dmitry S. Ryabukhin, Marina A. Borisova, Alexander Yu. Ivanov, Irina A. Boyarskaya, Kristina E. Borovkova, Lia R. Nikiforova, Julia V. Salmova, Nikolay V. Ul’yanovskii, Dmitry S. Kosyakov, and et al. 2022. "Synthesis of 3-Aryl-3-(Furan-2-yl)Propanoic Acid Derivatives, and Study of Their Antimicrobial Activity" Molecules 27, no. 14: 4612. https://doi.org/10.3390/molecules27144612
APA StyleKalyaev, M. V., Ryabukhin, D. S., Borisova, M. A., Ivanov, A. Y., Boyarskaya, I. A., Borovkova, K. E., Nikiforova, L. R., Salmova, J. V., Ul’yanovskii, N. V., Kosyakov, D. S., & Vasilyev, A. V. (2022). Synthesis of 3-Aryl-3-(Furan-2-yl)Propanoic Acid Derivatives, and Study of Their Antimicrobial Activity. Molecules, 27(14), 4612. https://doi.org/10.3390/molecules27144612