
Synthesis, Anticonvulsant, and Antinociceptive Activity of New 3-(2-Chlorophenyl)- and 3-(3-Chlorophenyl)-2,5-dioxo-pyrrolidin-1-yl-acetamides

 , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
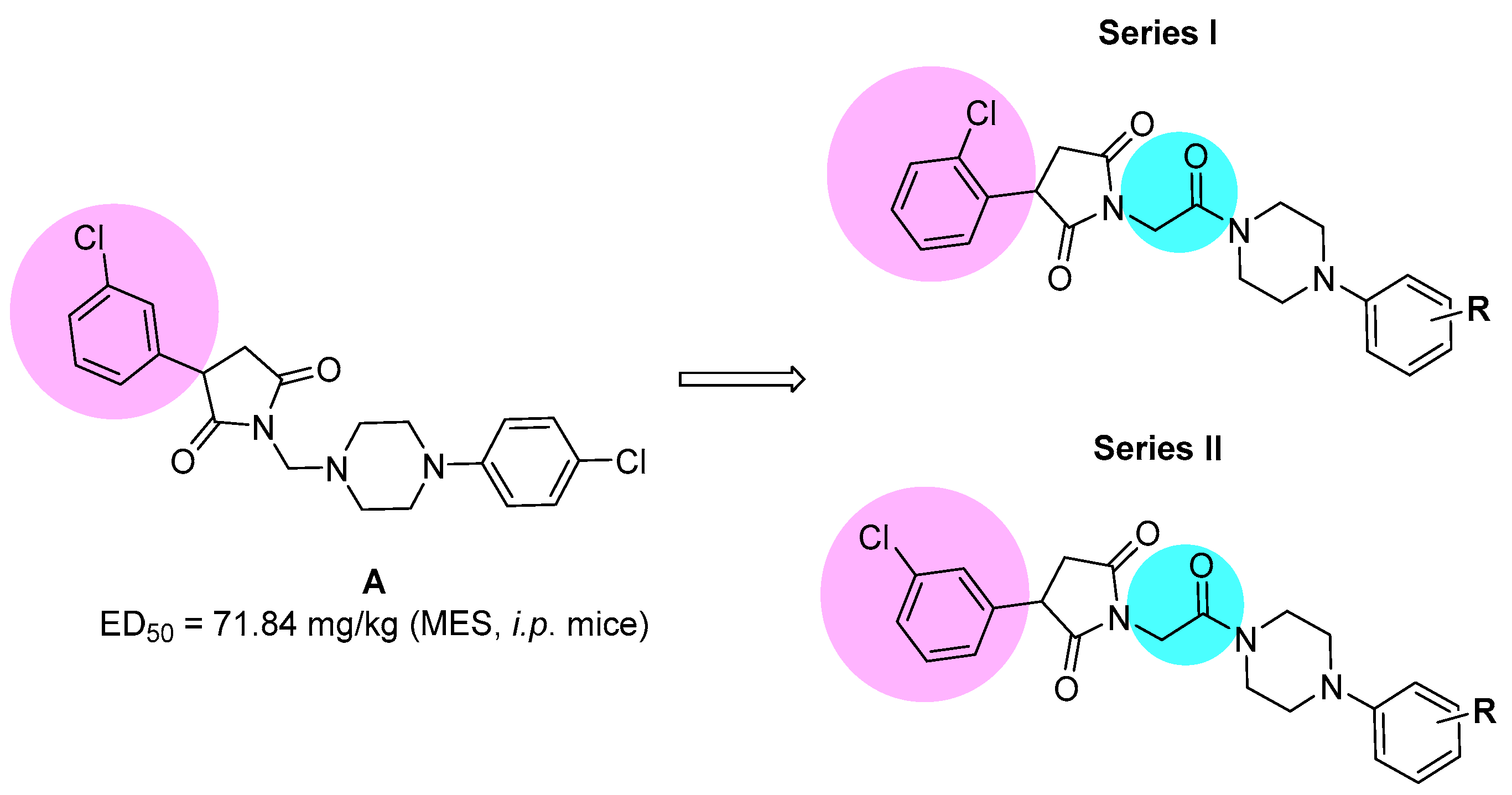
2.1. Synthesis and Drug-Likeness Properties
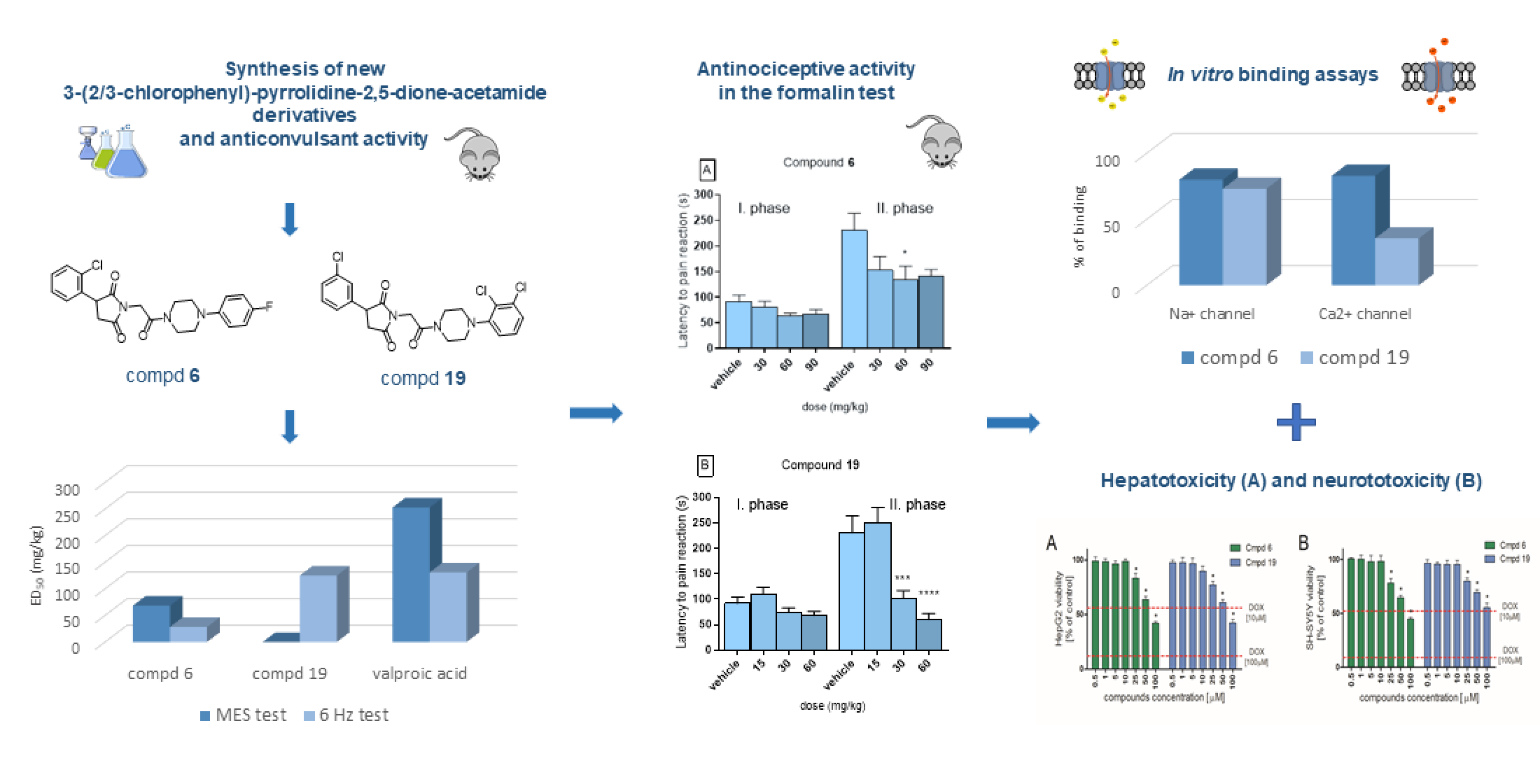
2.2. Anticonvulsant Activity
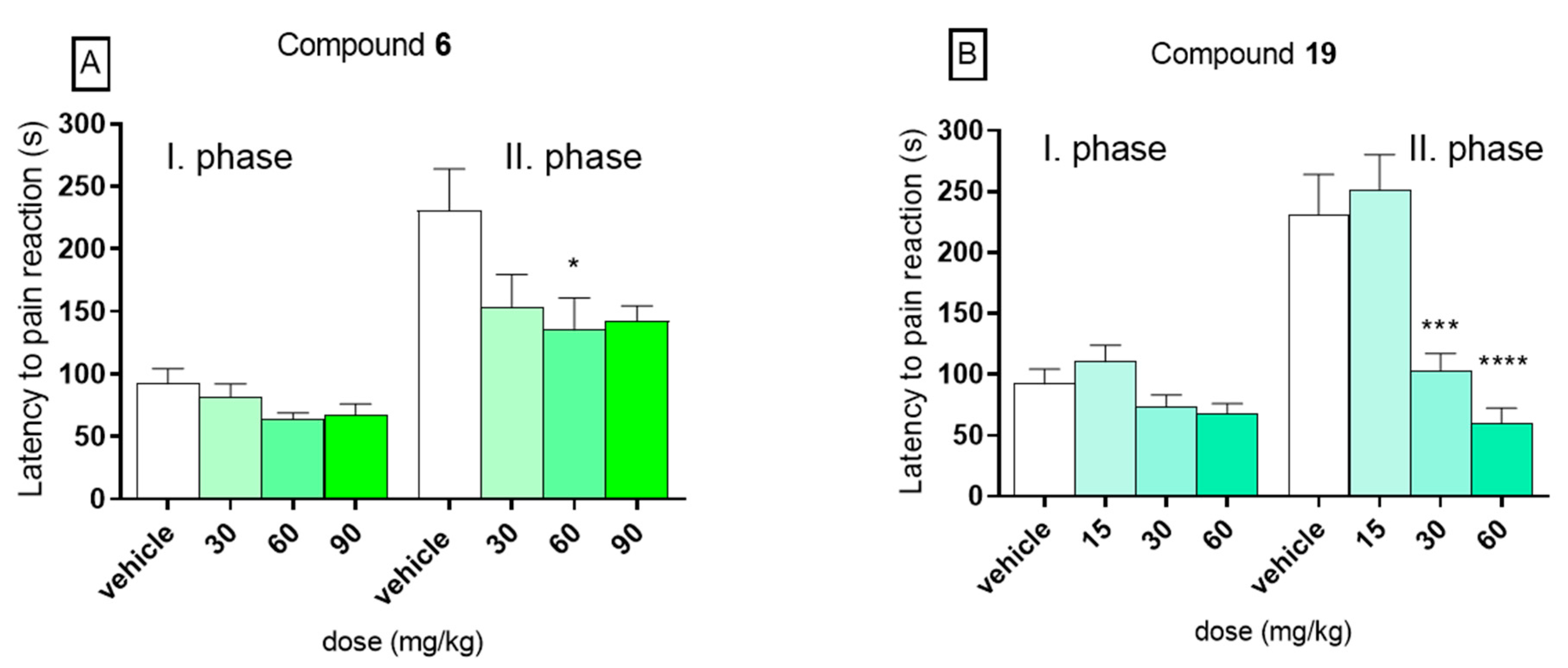
2.3. Antinociceptive Activity in the Formalin Test
2.4. In Vitro Radioligand Binding Studies
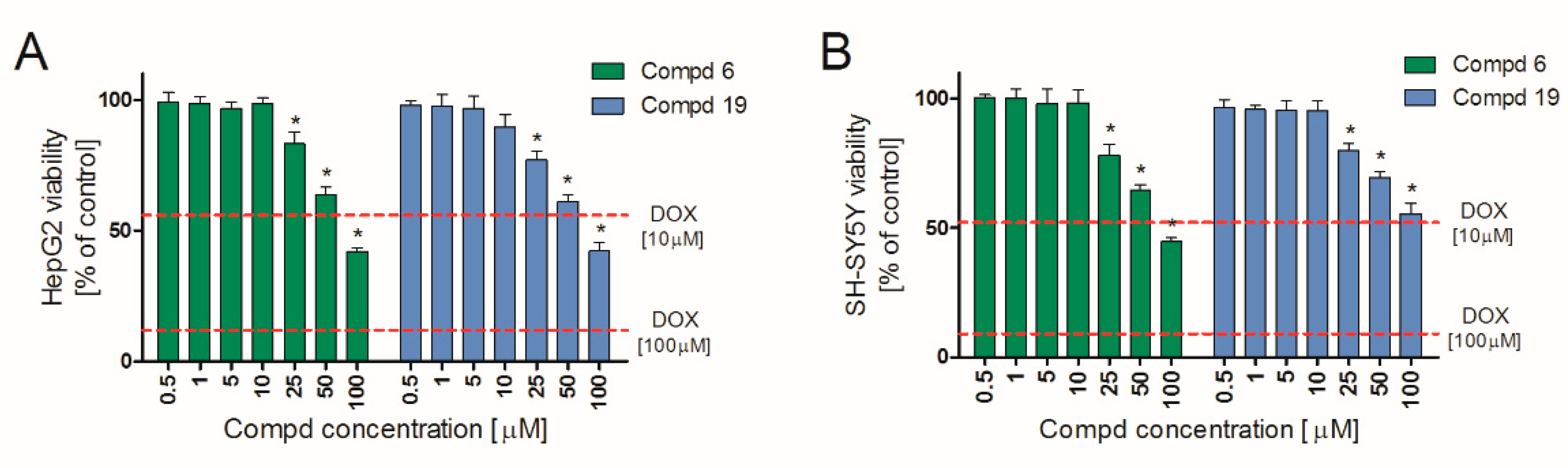
2.5. Hepatotoxicity and Neurotoxicity Study
3. Materials and Methods
3.1. Chemistry
3.1.1. General Remarks
3.1.2. Chemical Synthesis
General Procedure for the Preparation of the 3-(2-chlorophenyl)-(3) and 3-(3-chlorophenyl)-2,5-dioxo-pyrrolidin-1-yl-acetic Acids (4)
General Procedure for the Synthesis of Compounds 5–20
3.2. In Vivo Experiments
3.2.1. Animals
3.2.2. Maximal Electroshock Seizure Test (MES)
3.2.3. The 6 hertz (6 Hz) Psychomotor Seizure Test
3.2.4. PTZ Seizure Test
3.2.5. Neurotoxicity Screening (NT)—Rotarod Test
3.2.6. Median Effective Dose (ED50), Median Toxic Dose (TD50), and Protective Index (PI)
3.2.7. Formalin Test
3.3. In Vitro Experiments
3.3.1. Binding and Functional Assays
3.3.2. In Vitro Hepatotoxicity and Neurotoxicity Assessment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational Classification of Seizure Types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef]
- Stevens, C.E.; Stafstrom, C. Pharmacotherapy for Focal Seizures in Children and Adolescents. Drugs 2018, 78, 1321–1337. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Brodie, M.J.; Liew, D.; Kwan, P. Treatment Outcomes in Patients With Newly Diagnosed Epilepsy Treated With Established and New Antiepileptic Drugs: A 30-Year Longitudinal Cohort Study. JAMA Neurol. 2018, 75, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, K.S.; West, P.J.; Metcalf, C.S. The Current Approach of the Epilepsy Therapy Screening Program Contract Site for Identifying Improved Therapies for the Treatment of Pharmacoresistant Seizures in Epilepsy. Neuropharmacology 2020, 166, 107811. [Google Scholar] [CrossRef] [PubMed]
- Derry, S.; Bell, R.F.; Straube, S.; Wiffen, P.J.; Aldington, D.; Moore, R.A. Pregabalin for Neuropathic Pain in Adults. Cochrane Database Syst. Rev. 2019, 1, CD007076. [Google Scholar] [CrossRef]
- Rapacz, A.; Obniska, J.; Wiklik-Poudel, B.; Rybka, S.; Sałat, K.; Filipek, B. Anticonvulsant and Antinociceptive Activity of New Amides Derived from 3-Phenyl-2,5-Dioxo-Pyrrolidine-1-Yl-Acetic Acid in Mice. Eur. J. Pharmacol. 2016, 781, 239–249. [Google Scholar] [CrossRef]
- Byrtus, H.; Obniska, J.; Czopek, A.; Kamiński, K.; Pawłowski, M. Synthesis and Anticonvulsant Activity of New N-Mannich Bases Derived from 5-Cyclopropyl-5-Phenyl- and 5-Cyclopropyl-5-(4-Chlorophenyl)-Imidazolidine-2,4-Diones. Bioorg. Med. Chem. 2011, 19, 6149–6156. [Google Scholar] [CrossRef] [PubMed]
- Obniska, J.; Kopytko, M.; Zagórska, A.; Chlebek, I.; Kamiński, K. Synthesis and Anticonvulsant Properties of New Mannich Bases Derived from 3-Aryl-Pyrrolidine-2,5-Diones. Part 1. Archiv. Pharm. 2010, 343, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Long, L. Anticonvulsants. I. An Investigation of N-R-a-R,-a-Phenylsuccinimides. J. Am. Chem. Soc. 1951, 73, 4895–4898. [Google Scholar] [CrossRef]
- SwissAdme. Available online: http://www.swissadme.ch/ (accessed on 25 August 2020).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings1PII of Original Article: S0169-409X(96)00423-1. The Article Was Originally Published in Advanced Drug Delivery Reviews 23 (1997) 3–25.1. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef]
- Petit, J.; Meurice, N.; Kaiser, C.; Maggiora, G. Softening the Rule of Five—Where to Draw the Line? Bioorg. Med. Chem. 2012, 20, 5343–5351. [Google Scholar] [CrossRef]
- Kamiński, K.; Rapacz, A.; Łuszczki, J.J.; Latacz, G.; Obniska, J.; Kieć-Kononowicz, K.; Filipek, B. Design, Synthesis and Biological Evaluation of New Hybrid Anticonvulsants Derived from N-Benzyl-2-(2,5-Dioxopyrrolidin-1-Yl)Propanamide and 2-(2,5-Dioxopyrrolidin-1-Yl)Butanamide Derivatives. Bioorg. Med. Chem 2015, 23, 2548–2561. [Google Scholar] [CrossRef]
- Dantas, L.L.S.F.R.; Fonseca, A.G.; Pereira, J.R.; Furtado, A.A.; Gomes, P.T.M.; Fernandes-Pedrosa, M.F.; Leite, A.C.L.; Rêgo, M.J.B.M.; Pitta, M.G.R.; Lemos, T.M.M. Anti-Inflammatory and Antinociceptive Effects of the Isatin Derivative (Z)-2-(5-Chloro-2-Oxoindolin-3-Ylidene)-N-Phenyl-Hydrazinecarbothioamide in Mice. J. Med. Biol. Res. 2020, 53, e10204. [Google Scholar] [CrossRef]
- Laughlin, T.M.; Tram, K.V.; Wilcox, G.L.; Birnbaum, A.K. Comparison of Antiepileptic Drugs Tiagabine, Lamotrigine, and Gabapentin in Mouse Models of Acute, Prolonged, and Chronic Nociception. J. Pharmacol. Exp. Ther. 2002, 302, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Chen, X.; Chai, J.; Li, R.; Han, X.; Chen, X.; Liu, S.; Chen, M.; Xu, X. Antipyretic, Anti-Inflammatory and Analgesic Activities of Periplaneta Americana Extract and Underlying Mechanisms. Biomed. Pharm. 2020, 123, 109753. [Google Scholar] [CrossRef] [PubMed]
- Rosland, J.H.; Tjølsen, A.; Maehle, B.; Hole, K. The Formalin Test in Mice: Effect of Formalin Concentration. Pain 1990, 42, 235–242. [Google Scholar] [CrossRef]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic Pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef]
- Beyreuther, B.K.; Freitag, J.; Heers, C.; Krebsfänger, N.; Scharfenecker, U.; Stöhr, T. Lacosamide: A Review of Preclinical Properties. CNS Drug Rev. 2007, 13, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Rapacz, A.; Głuch-Lutwin, M.; Mordyl, B.; Filipek, B.; Abram, M.; Kamiński, K. Evaluation of Anticonvulsant and Analgesic Activity of New Hybrid Compounds Derived from N-Phenyl-2-(2,5-Dioxopyrrolidin-1-Yl)-Propanamides and -Butanamides. Epilepsy Res. 2018, 143, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Rapacz, A.; Obniska, J.; Koczurkiewicz, P.; Wójcik-Pszczoła, K.; Siwek, A.; Gryboś, A.; Rybka, S.; Karcz, A.; Pękala, E.; Filipek, B. Antiallodynic and Antihyperalgesic Activity of New 3,3-Diphenyl-Propionamides with Anticonvulsant Activity in Models of Pain in Mice. Eur. J. Pharm. 2018, 821, 39–48. [Google Scholar] [CrossRef]
- Soldatow, V.Y.; Lecluyse, E.L.; Griffith, L.G.; Rusyn, I. In Vitro Models for Liver Toxicity Testing. Toxicol. Res. 2013, 2, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, D.J.; Capela, J.P.; de Lourdes Bastos, M.; Carvalho, F. In Vitro Models for Neurotoxicology Research. Toxicol. Res. 2015, 4, 801–842. [Google Scholar] [CrossRef]
- Löscher, W.; Hönack, D.; Fassbender, C.P.; Nolting, B. The Role of Technical, Biological and Pharmacological Factors in the Laboratory Evaluation of Anticonvulsant Drugs. III. Pentylenetetrazole Seizure Models. Epilepsy Res. 1991, 8, 171–189. [Google Scholar] [CrossRef]
- Florek-Luszczki, M.; Wlaz, A.; Kondrat-Wrobel, M.W.; Tutka, P.; Luszczki, J.J. Effects of WIN 55,212-2 (a Non-Selective Cannabinoid CB1 and CB 2 Receptor Agonist) on the Protective Action of Various Classical Antiepileptic Drugs in the Mouse 6 Hz Psychomotor Seizure Model. J. Neural Transm. 2014, 121, 707–715. [Google Scholar] [CrossRef]
- Leclercq, K.; Kaminski, R.M. Genetic Background of Mice Strongly Influences Treatment Resistance in the 6 Hz Seizure Model. Epilepsia 2015, 56, 310–318. [Google Scholar] [CrossRef]
- Löscher, W.; Nolting, B. The Role of Technical, Biological and Pharmacological Factors in the Laboratory Evaluation of Anticonvulsant Drugs. IV. Protective Indices. Epilepsy Res. 1991, 9, 1–10. [Google Scholar] [CrossRef]
- Brown, G. 3H-Batrachotoxinin-A Benzoate Binding to Voltage-Sensitive Sodium Channels: Inhibition by the Channel Blockers Tetrodotoxin and Saxitoxin. J. Neurosci. 1986, 6, 2064–2070. [Google Scholar] [CrossRef]
- Gould, R.J.; Murphy, K.M.; Snyder, S.H. [3H]Nitrendipine-Labeled Calcium Channels Discriminate Inorganic Calcium Agonists and Antagonists. Proc. Natl. Acad. Sci. USA 1982, 79, 3656–3660. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | Lipinski Rules | Veber Rules | ||||
|---|---|---|---|---|---|---|
| MW ≤ 500 | Logp ≤ 5 | NHD ≤ 5 a | NHA ≤ 10 b | NBR ≤ 10 c | TPSA ≤ 140 d | |
| 5 | 429.87 | 2.74 | 0 | 4 | 5 | 60.93 |
| 6 | 429.87 | 2.73 | 0 | 4 | 5 | 60.93 |
| 7 | 479.88 | 3.47 | 0 | 6 | 6 | 60.93 |
| 8 | 446.33 | 2.94 | 0 | 3 | 5 | 60.93 |
| 9 | 446.33 | 2.95 | 0 | 3 | 5 | 60.93 |
| 10 | 446.33 | 2.93 | 0 | 3 | 5 | 60.93 |
| 11 | 480.77 | 3.47 | 0 | 3 | 5 | 60.93 |
| 12 | 480.77 | 3.48 | 0 | 3 | 5 | 60.93 |
| 13 | 429.87 | 2.75 | 0 | 4 | 5 | 60.93 |
| 14 | 429.87 | 2.76 | 0 | 4 | 5 | 60.93 |
| 15 | 479.88 | 3.45 | 0 | 6 | 6 | 60.93 |
| 16 | 446.33 | 3.06 | 0 | 3 | 5 | 60.93 |
| 17 | 446.33 | 2.96 | 0 | 3 | 5 | 60.93 |
| 18 | 446.33 | 2.97 | 0 | 3 | 5 | 60.93 |
| 19 | 480.77 | 3.59 | 0 | 3 | 5 | 60.93 |
| 20 | 480.77 | 3.55 | 0 | 3 | 5 | 60.93 |

| Compd | R1 | R2 | Intraperitoneal Administration in Mice | ||
|---|---|---|---|---|---|
| MES a | 6 Hz b | NT c | |||
| 5 | 2-Cl | 2-F | 1/4 | 1/4 | 0/4 |
| 6 | 2-Cl | 4-F | 4/4 | 3/4 | 0/4 |
| 7 | 2-Cl | 3-CF3 | 0/4 | 2/4 | 1/4 |
| 8 | 2-Cl | 2-Cl | 0/4 | 0/4 | 0/4 |
| 9 | 2-Cl | 3-Cl | 0/4 | 0/4 | 0/4 |
| 10 | 2-Cl | 4-Cl | 1/4 | 1/4 | 0/4 |
| 11 | 2-Cl | 2,3-diCl | 0/4 | 1/4 | 1/4 |
| 12 | 2-Cl | 3,4-diCl | 0/4 | 0/4 | 0/4 |
| 13 | 3-Cl | 2-F | 1/4 | 0/4 | 0/4 |
| 14 | 3-Cl | 4-F | 1/4 | 1/4 | 0/4 |
| 15 | 3-Cl | 3-CF3 | 1/4 | 2/4 | 0/4 |
| 16 | 3-Cl | 2-Cl | 0/4 | nt | 0/4 |
| 17 | 3-Cl | 3-Cl | 1/4 | nt | 0/4 |
| 18 | 3-Cl | 4-Cl | 1/4 | nt | 0/4 |
| 19 | 3-Cl | 2,3-diCl | 0/4 | 2/4 | 0/4 |
| 20 | 3-Cl | 3,4-diCl | 0/4 | nt | 0/4 |
| Compd | TPE (h) a | ED50 MES (mg/kg) b | ED50 6 Hz (mg/kg) c | TD50 (mg/kg) d | PI (TD50/ED50) e |
|---|---|---|---|---|---|
| 6 | 0.5 | 68.30 (60.31–77.35) | 28.20 (16.86–47.16) | >300 | >4.39 (MES) >10.64 (6 Hz) |
| 19 | 0.5 | – | 124.93 (91.13–191.27) | >300 | >2.4 (6 Hz) |
| ETXf | 0.25 | – | 171.74 (141.43–208.52) | 440.81 (409.01–475.07) | 2.57 (6 Hz) |
| VPAf | 0.5 | 252.74 (220.10–290.22) | 130.64 (117.61–145.19) | 430.77 (407.92–454.90) | 1.70 (MES) 3.30 (6 Hz) |
| Compound | Na+ Channel (Site 2) * | L-type Ca2+ (Dihydropyridine Site, Antagonist Radioligand) * |
|---|---|---|
| % Inhibition of Control Specific Binding | ||
| 6 | 80.0 | 82.9 |
| 19 | 73.1 | 35.5 |
| PHE | 53.9 | 57.8 |
| CBZ a | 17.4 | 2.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Góra, M.; Czopek, A.; Rapacz, A.; Gębska, A.; Wójcik-Pszczoła, K.; Pękala, E.; Kamiński, K. Synthesis, Anticonvulsant, and Antinociceptive Activity of New 3-(2-Chlorophenyl)- and 3-(3-Chlorophenyl)-2,5-dioxo-pyrrolidin-1-yl-acetamides. Molecules 2021, 26, 1564. https://doi.org/10.3390/molecules26061564
Góra M, Czopek A, Rapacz A, Gębska A, Wójcik-Pszczoła K, Pękala E, Kamiński K. Synthesis, Anticonvulsant, and Antinociceptive Activity of New 3-(2-Chlorophenyl)- and 3-(3-Chlorophenyl)-2,5-dioxo-pyrrolidin-1-yl-acetamides. Molecules. 2021; 26(6):1564. https://doi.org/10.3390/molecules26061564
Chicago/Turabian StyleGóra, Małgorzata, Anna Czopek, Anna Rapacz, Anna Gębska, Katarzyna Wójcik-Pszczoła, Elżbieta Pękala, and Krzysztof Kamiński. 2021. "Synthesis, Anticonvulsant, and Antinociceptive Activity of New 3-(2-Chlorophenyl)- and 3-(3-Chlorophenyl)-2,5-dioxo-pyrrolidin-1-yl-acetamides" Molecules 26, no. 6: 1564. https://doi.org/10.3390/molecules26061564
APA StyleGóra, M., Czopek, A., Rapacz, A., Gębska, A., Wójcik-Pszczoła, K., Pękala, E., & Kamiński, K. (2021). Synthesis, Anticonvulsant, and Antinociceptive Activity of New 3-(2-Chlorophenyl)- and 3-(3-Chlorophenyl)-2,5-dioxo-pyrrolidin-1-yl-acetamides. Molecules, 26(6), 1564. https://doi.org/10.3390/molecules26061564