
Mass Spectrometry for the Monitoring of Lipoprotein Oxidations by Myeloperoxidase in Cardiovascular Diseases

 , ,
, ,
Abstract
:1. The Oxidative Activity of Myeloperoxidase on Lipoproteins in the Context of Atherosclerosis
2. Reconsideration of the Actual Cardiovascular Risk Assessments
2.1. Actual Measurements
2.2. Treatment Targets
3. Upgrading Current Methods with Mass Spectrometry
3.1. Issues with Current Methods
3.2. Comparison between Immunoassay and Mass Spectrometry for Protein Analysis
3.3. Development and Challenges of A Mass Spectrometry Method to Analyze Oxidized Proteins
3.4. Mass Spectrometry Allows Quantitation of Serum ApoA-1 and ApoB-100
4. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Husain, K.; Hernandez, W.; Ansari, R.A.; Ferder, L. Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J. Biol. Chem. 2015, 6, 209–217. [Google Scholar] [CrossRef]
- Libby, P.; Okamoto, Y.; Rocha, V.Z.; Folco, E. Inflammation in Atherosclerosis. Circ. J. 2010, 74, 213–220. [Google Scholar]
- Boudjeltia, K.Z.; Legssyer, I.; Van Antwerpen, P.; Kisoka, R.L.; Babar, S.; Moguilevsky, N.; Delree, P.; Ducobu, J.; Remacle, C.; Vanhaeverbeek, M.; et al. Triggering of inflammatory response by myeloperoxidase-oxidized LDL. Biochem. Cell Biol. 2006, 84, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, R.; Henkels, K.M.; Wrenshall, L.E.; Kanaho, Y.; Paolo, G.D.; Frohman, M.A.; Gomez-Cambronero, J. Oxidized LDL phagocytosis during foam cell formation in atherosclerotic plaques relies on a PLD2–CD36 functional interdependence. J. Leukoc. Biol. 2018, 103, 867–883. [Google Scholar] [CrossRef]
- Bielski, B.H.J.; Allen, A.O. Mechanism of the disproportionation of superoxide radicals. J. Phys. Chem. 1977, 81, 1048–1050. [Google Scholar] [CrossRef]
- Klebanoff, S.J.; Kettle, A.J.; Rosen, H.; Winterbourn, C.C.; Nauseef, W.M. Myeloperoxidase: A front-line defender against phagocytosed microorganisms. J. Leukoc. Biol. 2013, 93, 185–198. [Google Scholar] [CrossRef] [Green Version]
- Sorescu, D.; Weiss, D.; Lassègue, B.; Clempus, R.E.; Szöcs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide Production and Expression of Nox Family Proteins in Human Atherosclerosis. Circulation 2002, 105, 1429. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Horke, S.; Förstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef]
- Douglas, G.; Bendall, J.K.; Crabtree, M.J.; Tatham, A.L.; Carter, E.E.; Hale, A.B.; Channon, K.M. Endothelial-specific Nox2 overexpression increases vascular superoxide and macrophage recruitment in ApoE(−/−) mice. Cardiovasc. Res. 2012, 94, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.W.; Schmitt, M.E. A central role for the endothelial NADPH oxidase in atherosclerosis. FEBS Lett. 2000, 472, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.F.; Qiao, M.; Schröder, K.; Zhao, Q.; Asmis, R. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ. Res. 2010, 106, 1489–1497. [Google Scholar] [CrossRef] [Green Version]
- Tavora, F.R.; Ripple, M.; Li, L.; Burke, A.P. Monocytes and neutrophils expressing myeloperoxidase occur in fibrous caps and thrombi in unstable coronary plaques. BMC Cardiovasc. Disord. 2009, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Aratani, Y. Myeloperoxidase: Its role for host defense, inflammation, and neutrophil function. Arch. Biochem. Biophys. 2018, 640, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Alsahli, M.A.; Rahmani, A.H. Myeloperoxidase as an Active Disease Biomarker: Recent Biochemical and Pathological Perspectives. Med. Sci. 2018, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocatta, T.J.; Pilbrow, A.P.; Cameron, V.A.; Senthilmohan, R.; Frampton, C.M.; Richards, A.M.; Winterbourn, C.C. Plasma Concentrations of Myeloperoxidase Predict Mortality After Myocardial Infarction. J. Am. Coll. Cardiol. 2007, 49, 1993–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franck, T.; Minguet, G.; Delporte, C.; Derochette, S.; Zouaoui Boudjeltia, K.; Van Antwerpen, P.; Gach, O.; Deby-Dupont, G.; Mouithys-Mickalad, A.; Serteyn, D. An immunological method to combine the measurement of active and total myeloperoxidase on the same biological fluid, and its application in finding inhibitors which interact directly with the enzyme. Free Radic. Res. 2015, 49, 790–799. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2017, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Zouaoui Boudjeltia, K.; Moguilevsky, N.; Legssyer, I.; Babar, S.; Guillaume, M.; Delree, P.; Vanhaeverbeek, M.; Brohee, D.; Ducobu, J.; Remacle, C. Oxidation of low density lipoproteins by myeloperoxidase at the surface of endothelial cells: An additional mechanism to subendothelium oxidation. Biochem. Biophys. Res. Commun. 2004, 325, 434–438. [Google Scholar] [CrossRef]
- Holland, J.A.; Ziegler, L.M.; Meyer, J.W. Atherogenic levels of low-density lipoprotein increase hydrogen peroxide generation in cultured human endothelial cells: Possible mechanism of heightened endocytosis. J. Cell. Physiol. 1996, 166, 144–151. [Google Scholar] [CrossRef]
- Tien, M. Myeloperoxidase-Catalyzed Oxidation of Tyrosine. Arch. Biochem. Biophys. 1999, 367, 61–66. [Google Scholar] [CrossRef]
- Finley, E.L.; Dillon, J.; Crouch, R.K.; Schey, K.L. Identification of tryptophan oxidation products in bovine α-crystallin. Protein Sci. 1998, 7, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- Hazen, S.L.; Heinecke, J.W. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Investig. 1997, 99, 2075–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiserich, J.P.; Hristova, M.; Cross, C.E.; Jones, A.D.; Freeman, B.A.; Halliwell, B.; Van Der Vliet, A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature 1998, 391, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Bergt, C.; Reicher, H.; Malle, E.; Sattler, W. Hypochlorite modification of high density lipoprotein: Effects on cholesterol efflux from J774 macrophages. FEBS Lett. 1999, 452, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Bergt, C.; Pennathur, S.; Fu, X.; Byun, J.; O’Brien, K.; McDonald, T.O.; Singh, P.; Anantharamaiah, G.M.; Chait, A.; Brunzell, J.; et al. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc. Natl. Acad. Sci. USA 2004, 101, 13032–13037. [Google Scholar] [CrossRef] [Green Version]
- Oram, J.F. HDL Apolipoproteins and ABCA1. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 720–727. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.-J.; Yin, K.; Fu, Y.-C.; Tang, C.-K. The interaction of ApoA-I and ABCA1 triggers signal transduction pathways to mediate efflux of cellular lipids. Mol. Med. 2012, 18, 149–158. [Google Scholar] [CrossRef]
- Peng, D.Q.; Brubaker, G.; Wu, Z.; Zheng, L.; Willard, B.; Kinter, M.; Hazen, S.L.; Smith, J.D. Apolipoprotein A-I tryptophan substitution leads to resistance to myeloperoxidase-mediated loss of function. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2063–2070. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.; Bergt, C.; Fu, X.; Green, P.; Voss, J.C.; Oda, M.N.; Oram, J.F.; Heinecke, J.W. Tyrosine 192 in apolipoprotein A-I is the major site of nitration and chlorination by myeloperoxidase, but only chlorination markedly impairs. J. Biol. Chem. 2005, 280, 5983–5993. [Google Scholar] [CrossRef] [Green Version]
- Bergt, C.; Oettl, K.; Keller, W.; Andreae, F.; Jörg Leis, H.; Malle, E.; Sattler, W. Reagent or Myeloperoxidase-Generated Hypochlorite Affects Discrete Regions in Lipid-Free and Lipid-Associated Human Apolipoprotein A-I. Biochem. J. 2000, 346, 345–354. [Google Scholar] [CrossRef]
- Moran, C.S.; Jose, R.J.; Biros, E.; Golledge, J. The Vascular Biology Unit, Queensland Research Centre for Peripheral Vascular Disease, School of Medicine and Dentistry. Arter. Thromb Vasc Biol 2014, 34, 2609–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergt, C.; Fu, X.; Huq, N.P.; Kao, J.; Heinecke, J.W. Lysine residues direct the chlorination of tyrosines in YXXK motifs of apolipoprotein A-I when hypochlorous acid oxidizes high density lipoprotein. J. Biol. Chem. 2004, 279, 7856–7866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Settle, M.; Brubaker, G.; Schmitt, D.; Hazen, S.L.; Smith, J.D.; Kinter, M. Localization of nitration and chlorination sites on apolipoprotein A-I catalyzed by myeloperoxidase in human atheroma and associated oxidative impairment in. J. Biol. Chem. 2005, 280, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, B.; Oda, M.N.; Bergt, C.; Fu, X.; Green, P.S.; Brot, N.; Oram, J.F.; Heinecke, J.W. Myeloperoxidase impairs ABCA1-dependent cholesterol efflux through methionine oxidation and site-specific tyrosine chlorination of apolipoprotein A-I. J. Biol. Chem. 2006, 281, 9001–9004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Didonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Delporte, C.; Van Antwerpen, P.; Zouaoui Boudjeltia, K.; Noyon, C.; Abts, F.; Metral, F.; Vanhamme, L.; Reye, F.; Rousseau, A.; Vanhaeverbeek, M.; et al. Optimization of apolipoprotein-B-100 sequence coverage by liquid chromatography-tandem mass spectrometry for the future study of its posttranslational modifications. Anal. Biochem. 2011, 411, 129–138. [Google Scholar] [CrossRef]
- Zouaoui Boudjeltia, K.; Roumeguere, T.; Delree, P.; Moguilevsky, N.; Ducobu, J.; Vanhaeverbeek, M.; Wespes, E. Presence of LDL Modified by Myeloperoxidase in the Penis in Patients with Vascular Erectile Dysfunction: A Preliminary Study. Eur. Urol. 2007, 51, 262–269. [Google Scholar] [CrossRef]
- Moguilevsky, N.; Zouaoui Boudjeltia, K.; Babar, S.; Delrée, P.; Legssyer, I.; Carpentier, Y.; Vanhaeverbeek, M.; Ducobu, J. Monoclonal antibodies against LDL progressively oxidized by myeloperoxidase react with ApoB-100 protein moiety and human atherosclerotic lesions. Biochem. Biophys. Res. Commun. 2004, 323, 1223–1228. [Google Scholar] [CrossRef]
- Yang, C.Y.; Gu, Z.W.; Yang, H.X.; Yang, M.; Gotto, A.M.; Smith, C.V. Oxidative modifications of apoB-100 by exposure of low density lipoproteins to HOCL in vitro. Free Radic. Biol. Med. 1997, 23, 82–89. [Google Scholar] [CrossRef]
- Yang, C.; Wang, J.; Krutchinsky, A.N.; Chait, B.T.; Morrisett, J.D.; Smith, C.V. Selective oxidation in vitro by myeloperoxidase of the N-terminal amine in apolipoprotein B-100. J. Lipid Res. 2001, 42, 1891–1896. [Google Scholar] [CrossRef]
- Hamilton, R.T.; Asatryan, L.; Nilsen, J.T.; Isas, J.M.; Gallaher, T.K.; Sawamura, T.; Hsiai, T.K. LDL protein nitration: Implication for LDL protein unfolding. Arch. Biochem. Biophys. 2008, 479, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Cai, Y.; Tarr, M.A. Mapping oxidations of apolipoprotein B-100 in human low-density lipoprotein by liquid chromatography–tandem mass spectrometry. Anal. Biochem. 2010, 404, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Delporte, C.; Boudjeltia, K.Z.; Noyon, C.; Furtmüller, P.G.; Nuyens, V.; Slomianny, M.-C.; Madhoun, P.; Desmet, J.-M.; Raynal, P.; Dufour, D.; et al. Impact of myeloperoxidase-LDL interactions on enzyme activity and subsequent posttranslational oxidative modifications of apoB-100. J. Lipid Res. 2014, 55, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obama, T.; Kato, R.; Masuda, Y.; Takahashi, K.; Aiuchi, T.; Itabe, H. Analysis of modified apolipoprotein B-100 structures formed in oxidized low-density lipoprotein using LC-MS/MS. Proteomics 2007, 7, 2132–2141. [Google Scholar] [CrossRef] [PubMed]
- Catapano, A.L.; Graham, I.; De Backer, G.; Wiklund, O.; Chapman, M.J.; Drexel, H.; Hoes, A.W.; Jennings, C.S.; Landmesser, U.; Pedersen, T.R.; et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias. Eur. Heart J. 2016, 37, 2999–3058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannel, W.B.; Castelli, W.P.; Gordon, T. Cholesterol in the prediction of atherosclerotic disease. New perspectives based on the Framingham study. Ann. Intern. Med. 1979, 90, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C.; Lloyd-Jones, D.M.; Bennett, G.; Coady, S.; D’Agostino, R.B.; Gibbons, R.; Greenland, P.; Lackland, D.T.; Levy, D.; O’Donnell, C.J.; et al. 2013 ACC/AHA Guideline on the Assessment of Cardiovascular Risk. Circulation 2014, 129, S49–S73. [Google Scholar] [CrossRef] [Green Version]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.-T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice. Eur. J. Prev. Cardiol. 2016, 23, NP1–NP96. [Google Scholar]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: A systematic review and meta-analysis. JAMA J. Am. Med. Assoc. 2016, 316, 1289–1297. [Google Scholar] [CrossRef] [Green Version]
- Borén, J.; John Chapman, M.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef] [PubMed]
- Di Angelantonio, E.; Gao, P.; Pennells, L.; Kaptoge, S.; Caslake, M.; Thompson, A.; Butterworth, A.S.; Sarwar, N.; Wormser, D.; Saleheen, D.; et al. Lipid-related markers and cardiovascular disease prediction. JAMA J. Am. Med. Assoc. 2012, 307, 2499–2506. [Google Scholar]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.V.; Asselbergs, F.W.; Palmer, T.M.; Drenos, F.; Lanktree, M.B.; Nelson, C.P.; Dale, C.E.; Padmanabhan, S.; Finan, C.; Swerdlow, D.I.; et al. Mendelian randomization of blood lipids for coronary heart disease. Eur. Heart J. 2015, 36, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonneil, C.; Benedittini, D.; Cecchin, M.; Lascols, S.; Tuil, L.A. Place des Dosages des Apolipoproteines a1 et b Dans le Bilan Lipidique. 2008. Available online: https://www.has-sante.fr/portail/upload/docs/application/pdf/2008-12/texte_court_apolipoa1b.pdf (accessed on 7 April 2019).
- Whelton, S.P.; Meeusen, J.W.; Donato, L.J.; Jaffe, A.S.; Saenger, A.; Sokoll, L.J.; Blumenthal, R.S.; Jones, S.R.; Martin, S.S. Evaluating the atherogenic burden of individuals with a Friedewald-estimated low-density lipoprotein cholesterol <70 mg/dL compared with a novel low-density lipoprotein estimation method. J. Clin. Lipidol. 2017, 11, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Reignier, A.; Sacchetto, É.; Hardouin, J.B.; Orsonneau, J.L.; Le Carrer, D.; Delaroche, O.; Bigot-Corbel, E. Évaluation d’une méthode de dosage direct du LDL-cholestérol et de son impact potentiel en termes de prise en charge thérapeutique. Ann. Biol. Clin. 2014, 72, 593–598. [Google Scholar]
- Lin, A.V. Indirect ELISA. Methods Mol. Biol. 2015, 1318, 51–59. [Google Scholar]
- Langlois, M.R.; Chapman, M.J.; Cobbaert, C.; Mora, S.; Remaley, A.T.; Ros, E.; Watts, G.F.; Borén, J.; Baum, H.; Bruckert, E.; et al. Quantifying Atherogenic Lipoproteins: Current and Future Challenges in the Era of Personalized Medicine and Very Low Concentrations of LDL Cholesterol. A Consensus Statement from EAS and EFLM. Clin. Chem. 2018, 64, 1006–1033. [Google Scholar] [CrossRef] [Green Version]
- van den Berg, M.J.; Visseren, F.L.J.; de Borst, G.J.; Kappelle, L.J.; Nathoe, H.M.; van der Graaf, Y.; van Petersen, R.; van Dinther, B.G.F.; Algra, A.; van der Graaf, Y.; et al. Low-Density Lipoprotein Cholesterol, Non–High-Density Lipoprotein Cholesterol, Triglycerides, and Apolipoprotein B and Cardiovascular Risk in Patients With Manifest Arterial Disease. Am. J. Cardiol. 2016, 118, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sniderman, A.D.; Islam, S.; Yusuf, S.; McQueen, M.J. Discordance analysis of Apolipoprotein B and non-high density lipoprotein cholesterol as markers of cardiovascular risk in the INTERHEART study. Atherosclerosis 2012, 225, 444–449. [Google Scholar] [CrossRef]
- Packard, R.R.S.; Libby, P. Inflammation in Atherosclerosis: From Vascular Biology to Biomarker Discovery and Risk Prediction. Clin. Chem. 2007, 54, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Sniderman, A.D.; Robinson, J.G. ApoB in clinical care: Pro and Con. Atherosclerosis 2019, 282, 169–175. [Google Scholar] [CrossRef]
- Ho, E.; Karimi Galougahi, K.; Liu, C.-C.; Bhindi, R.; Figtree, G.A. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biol. 2013, 1, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Trpkovic, A.; Resanovic, I.; Stanimirovic, J.; Radak, D.; Mousa, S.A.; Cenic-Milosevic, D.; Jevremovic, D.; Isenovic, E.R. Oxidized low-density lipoprotein as a biomarker of cardiovascular diseases. Crit. Rev. Clin. Lab. Sci. 2015, 52, 70–85. [Google Scholar] [CrossRef]
- Wen, Y.; Leake, D.S. Low density lipoprotein undergoes oxidation within lysosomes in cells. Circ. Res. 2007, 100, 1337–1343. [Google Scholar] [CrossRef] [Green Version]
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized LDL: Diversity, Patterns of Recognition, and Pathophysiology. Antioxid. Redox Signal. 2010, 13, 39–75. [Google Scholar] [CrossRef] [Green Version]
- Liao, F.; Berliner, J.A.; Mehrabian, M.; Navab, M.; Demer, L.L.; Lusis, A.J.; Fogelman, A.M. Minimally modified low density lipoprotein is biologically active in vivo in mice. J. Clin. Investig. 1991, 87, 2253–2257. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Willett, W.C.; Rifai, N.; Shai, I.; Manson, J.E.; Rimm, E.B. Is Plasma Oxidized Low-Density Lipoprotein, Measured With the Widely Used Antibody 4E6, an Independent Predictor of Coronary Heart Disease Among U.S. Men and Women? J. Am. Coll. Cardiol. 2006, 48, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Boudjeltia, K.Z.; Delporte, C.; Van Antwerpen, P.; Franck, T.; Serteyn, D.; Moguilevsky, N.; Raes, M.; Vanhamme, L.; Vanhaeverbeek, M.; Van Meerhaeghe, A.; et al. Myeloperoxidase-dependent LDL modifications in bloodstream are mainly predicted by angiotensin II, adiponectin, and myeloperoxidase activity: A cross-sectional study in men. Mediators Inflamm. 2013, 2013, 750742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Yancey, P.G.; Tao, H.; Borja, M.S.; Smith, L.E.; Kon, V.; Davies, S.S.; Linton, M.F. Reactive dicarbonyl scavenging effectively reduces mpo-mediated oxidation of hdl and restores pon1 activity. Nutrients 2020, 12, 1937. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, Y.; Shi, J.; Gao, M.; Liu, Y.; Cong, Y.; Li, Y.; Wang, Y.; Yu, M.; Lu, Y.; et al. Entire Peroxidation Reaction System of Myeloperoxidase Correlates with Progressive Low-Density Lipoprotein Modifications via Reactive Aldehydes in Atherosclerotic Patients with Hypertension. Cell. Physiol. Biochem. 2018, 50, 1245–1254. [Google Scholar] [CrossRef]
- Sodhi, K.; Bracero, L.; Feyh, A.; Nichols, A.; Srikanthan, K.; Latif, T.; Preston, D.; Shapiro, J.I.; Elitsur, Y. Role of Serum Biomarkers in Early Detection of Non-Alcoholic Steatohepatitis and Fibrosis in West Virginian Children. J. Clin. Cell. Immunol. 2016, 7, 393. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.-Q.; Wu, Z.; Brubaker, G.; Zheng, L.; Settle, M.; Gross, E.; Kinter, M.; Hazen, S.L.; Smith, J.D. Tyrosine Modification Is Not Required for Myeloperoxidase-induced Loss of Apolipoprotein A-I Functional Activities. J. Biol. Chem. 2005, 280, 33775–33784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobbaert, C.; Boerma, G.; Lindemans, J. Evaluation of the Cholestech LDX desktop analyser for cholesterol, HDL-cholesterol, and triacylglycerols in heparinized venous blood. Clin. Chem. Lab. Med. 1994, 32, 391–394. [Google Scholar]
- Warnick, G.R.; Nauck, M.; Rifai, N. Evolution of Methods for Measurement of HDL-Cholesterol: From Ultracentrifugation to Homogeneous Assays. Clin. Chem. 2001, 43, 1809–1810. [Google Scholar] [CrossRef] [Green Version]
- Eugui, J.; Logroño, J.; Ruiz, R.; Zugaza, C.; Mirabel, J.; Martínez, C. Immunoturbidimetry of serum apolipoproteins A-I and B on the Cobas Bio centrifugal analyzer: Method validation and reference values. Clin. Biochem. 1994, 27, 310–315. [Google Scholar] [CrossRef]
- Cobbaert, C.; Zwang, L.; Ceriotti, F.; Modenese, A.; Cremer, P.; Herrmann, W.; Hoss, G.; Jarausch, J.; Tü, R.; Mä, W.; et al. Reference Standardization and Triglyceride Interference of a New Homogeneous HDL-Cholesterol Assay Compared with a Former Chemical Precipitation Assay Lipids and Lipoproteins. 1998. Available online: http://clinchem.aaccjnls.org/content/clinchem/44/4/779.full.pdf (accessed on 8 January 2019).
- Nauck, M.; Graziani, M.S.; Bruton, D.; Cobbaert, C.; Cole, T.G.; Lefevre, F.; Riesen, W.; Bachorik, P.S.; Rifai, N. Analytical and Clinical Performance of a Detergent-based Homogeneous LDL-Cholesterol Assay: A Multicenter Evaluation. 2000. Available online: http://clinchem.aaccjnls.org/content/clinchem/46/4/506.full.pdf (accessed on 8 January 2019).
- Jungner, I.; Marcovina, S.M.; Walldius, G.; Holme, I.; Kolar, W.; Steiner, E. Apolipoprotein B and A-I values in 147576 Swedish males and females, standardized according to the World Health Organization-International Federation of Clinical Chemistry First International Reference Materials. Clin. Chem. 1998, 44, 1641–1649. [Google Scholar] [CrossRef]
- Hoofnagle, A.N.; Wener, M.H. The fundamental flaws of immunoassays and potential solutions using tandem mass spectrometry. J. Immunol. Methods 2009, 347, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Geromanos, S.J.; Vissers, J.P.C.; Silva, J.C.; Dorschel, C.A.; Li, G.-Z.; Gorenstein, M.V.; Bateman, R.H.; Langridge, J.I. The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data dependant LC-MS/MS. Proteomics 2009, 9, 1683–1695. [Google Scholar] [CrossRef]
- Rogier, E.; Plucinski, M.; Lucchi, N.; Mace, K.; Chang, M.; Lemoine, J.F.; Candrinho, B.; Colborn, J.; Dimbu, R.; Fortes, F.; et al. Bead-based immunoassay allows sub-picogram detection of histidine-rich protein 2 from Plasmodium falciparum and estimates reliability of malaria rapid diagnostic tests. PLoS ONE 2017, 12, e0172139. [Google Scholar] [CrossRef] [Green Version]
- Rösch, A.; Beck, B.; Hollender, J.; Singer, H. Picogram per liter quantification of pyrethroid and organophosphate insecticides in surface waters: A result of large enrichment with liquid-liquid extraction and gas chromatography coupled to mass spectrometry using atmospheric pressure chemical ionization. Anal. Bioanal. Chem. 2019, 411, 3151–3164. [Google Scholar] [PubMed]
- Elpa, D.P.; Prabhu, G.R.D.; Wu, S.P.; Tay, K.S.; Urban, P.L. Automation of mass spectrometric detection of analytes and related workflows: A review. Talanta 2019, 208, 120304. [Google Scholar] [CrossRef] [PubMed]
- Güzel, C.; Govorukhina, N.I.; Stingl, C.; Dekker, L.J.M.; Boichenko, A.; van der Zee, A.G.J.; Bischoff, R.P.H.; Luider, T.M. Comparison of Targeted Mass Spectrometry Techniques with an Immunoassay: A Case Study for HSP90α. PROTEOMICS-Clin. Appl. 2018, 12, 1700107. [Google Scholar] [CrossRef] [PubMed]
- Smit, N.P.M.; Ruhaak, L.R.; Romijn, F.P.H.T.M.; Pieterse, M.M.; van der Burgt, Y.E.M.; Cobbaert, C.M. The Time Has Come for Quantitative Protein Mass Spectrometry Tests That Target Unmet Clinical Needs. J. Am. Soc. Mass Spectrom. 2021, 32, 636–647. [Google Scholar] [CrossRef]
- Fda. Cder Bioanalytical Method Validation Guidance for Industry Biopharmaceutics Bioanalytical Method Validation Guidance for Industry Biopharmaceutics Contains Nonbinding Recommendations. 2018. Available online: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htmand/orhttp://www.fda.gov/AnimalVeterinary/GuidanceComplianceEnforcement/GuidanceforIndustry/default.htm (accessed on 5 August 2021).
- Van Den Broek, I.; Nouta, J.; Razavi, M.; Yip, R.; Bladergroen, M.R.; Romijn, F.P.H.T.M.; Smit, N.P.M.; Drews, O.; Paape, R.; Suckau, D.; et al. Quantification of serum apolipoproteins A-I and B-100 in clinical samples using an automated SISCAPA-MALDI-TOF-MS workflow. Methods 2015, 81, 74–85. [Google Scholar] [CrossRef]
- Van De Merbel, N.C. Protein quantification by LC-MS: A decade of progress through the pages of Bioanalysis. Bioanalysis 2019, 11, 629–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christians, U.; Klepacki, J.; Shokati, T.; Klawitter, J.; Klawitter, J. Mass Spectrometry-Based Multiplexing for the Analysis of Biomarkers in Drug Development and Clinical Diagnostics- How Much is too Much? Microchem. J. 2012, 105, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Smit, N.P.M.; Romijn, F.P.H.T.M.; van den Broek, I.; Drijfhout, J.W.; Haex, M.; van der Laarse, A.; van der Burgt, Y.E.M.; Cobbaert, C.M. Metrological traceability in mass spectrometry-based targeted protein quantitation: A proof-of-principle study for serum apolipoproteins A-I and B100. J. Proteom. 2014, 109, 143–161. [Google Scholar] [CrossRef]
- Verrastro, I.; Pasha, S.; Jensen, K.T.; Pitt, A.R.; Spickett, C.M.; Breitenbach, M.; Eckl, P. Mass Spectrometry-Based Methods for Identifying Oxidized Proteins in Disease: Advances and Challenges. Biomolecules 2015, 5, 378–411. [Google Scholar] [CrossRef] [Green Version]
- Egeland, S.V.; Reubsaet, L.; Halvorsen, T.G. The pros and cons of increased trypsin-to-protein ratio in targeted protein analysis. J. Pharm. Biomed. Anal. 2016, 123, 155–161. [Google Scholar] [CrossRef]
- Šlechtová, T.; Gilar, M.; Kalíková, K.; Tesařová, E. Insight into Trypsin Miscleavage: Comparison of Kinetic Constants of Problematic Peptide Sequences. Anal. Chem. 2015, 87, 7636–7643. [Google Scholar] [CrossRef]
- Nouri-Nigjeh, E.; Zhang, M.; Ji, T.; Yu, H.; An, B.; Duan, X.; Balthasar, J.; Johnson, R.W.; Qu, J. Effects of calibration approaches on the accuracy for LC-MS targeted quantification of therapeutic protein. Anal. Chem. 2014, 86, 3575–3584. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.; Štys, D. Noise and baseline filtration in mass spectrometry. In Proceedings of the International Work-Conference on Bioinformatics and Biomedical Engineering, IWBBIO 2015, Granada, Spain, 15–17 April 2015; Springer: Berlin/Heidelberg, Germany, 2015; Volume 9044, pp. 418–425. [Google Scholar]
- Van Den Broek, I.; Romijn, F.P.H.T.M.; Nouta, J.; Van Der Laarse, A.; Drijfhout, J.W.; Smit, N.P.M.; Van Der Burgt, Y.E.M.; Cobbaert, C.M. Automated Multiplex LC-MS/MS Assay for Quantifying Serum Apolipoproteins A-I, B, C-I, C-II, C-III, and E with Qualitative Apolipoprotein E Phenotyping. Clin. Chem. 2015, 62, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Hoofnagle, A.; Hochstrasser, D.; Brede, C.; Glueckmann, M.; Cocho, J.A.; Ceglarek, U.; Lenz, C.; Vialaret, J.; Scherl, A.; et al. Quantitative Clinical Chemistry Proteomics (qCCP) using mass spectrometry: General characteristics and application. Clin. Chem. Lab. Med. 2013, 51, 919–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceglarek, U.; Dittrich, J.; Becker, S.; Baumann, F.; Kortz, L.; Thiery, J. Quantification of seven apolipoproteins in human plasma by proteotypic peptides using fast LC-MS/MS. Proteom. Clin. Appl. 2013, 7, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Kay, R.G.; Gregory, B.; Grace, P.B.; Pleasance, S. The application of ultra-performance liquid chromatography/tandem mass spectrometry to the detection and quantitation of apolipoproteins in human serum. Rapid Commun. Mass Spectrom. 2007, 21, 2585–2593. [Google Scholar] [CrossRef] [PubMed]
- Contois, J.H.; Wu, A.H.B.; Li, Z.; Feroze, A.H.; Grunenberger, F.; Haller, J.; Degroot, L.; Lammi-Keefe, C.J. Distribution of serum apolipoproteins A-I and B and lipoprotein(a) in European elderly: The SENECA study. Clin. Chim. Acta 2000, 295, 1–12. [Google Scholar] [CrossRef]
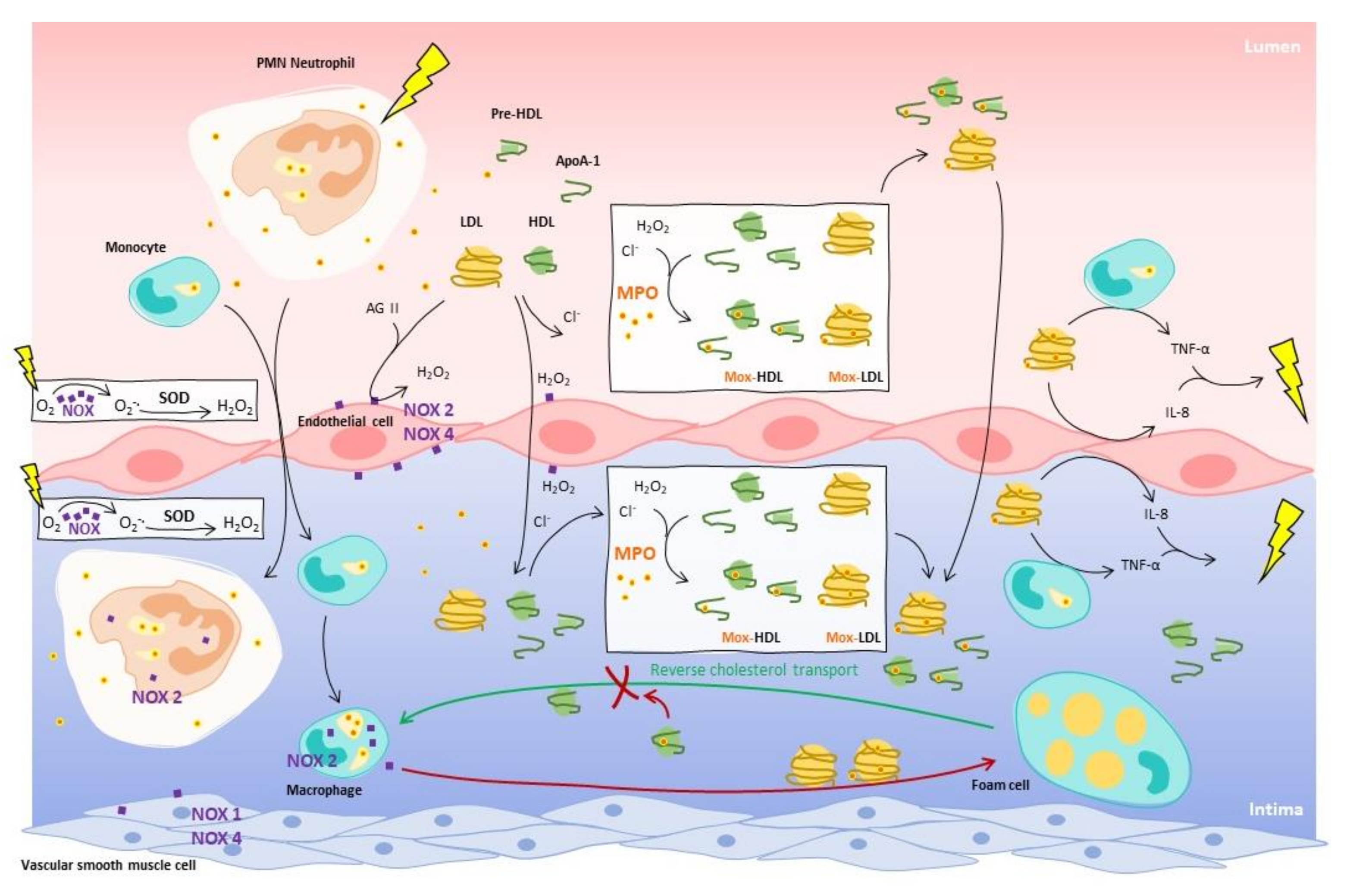
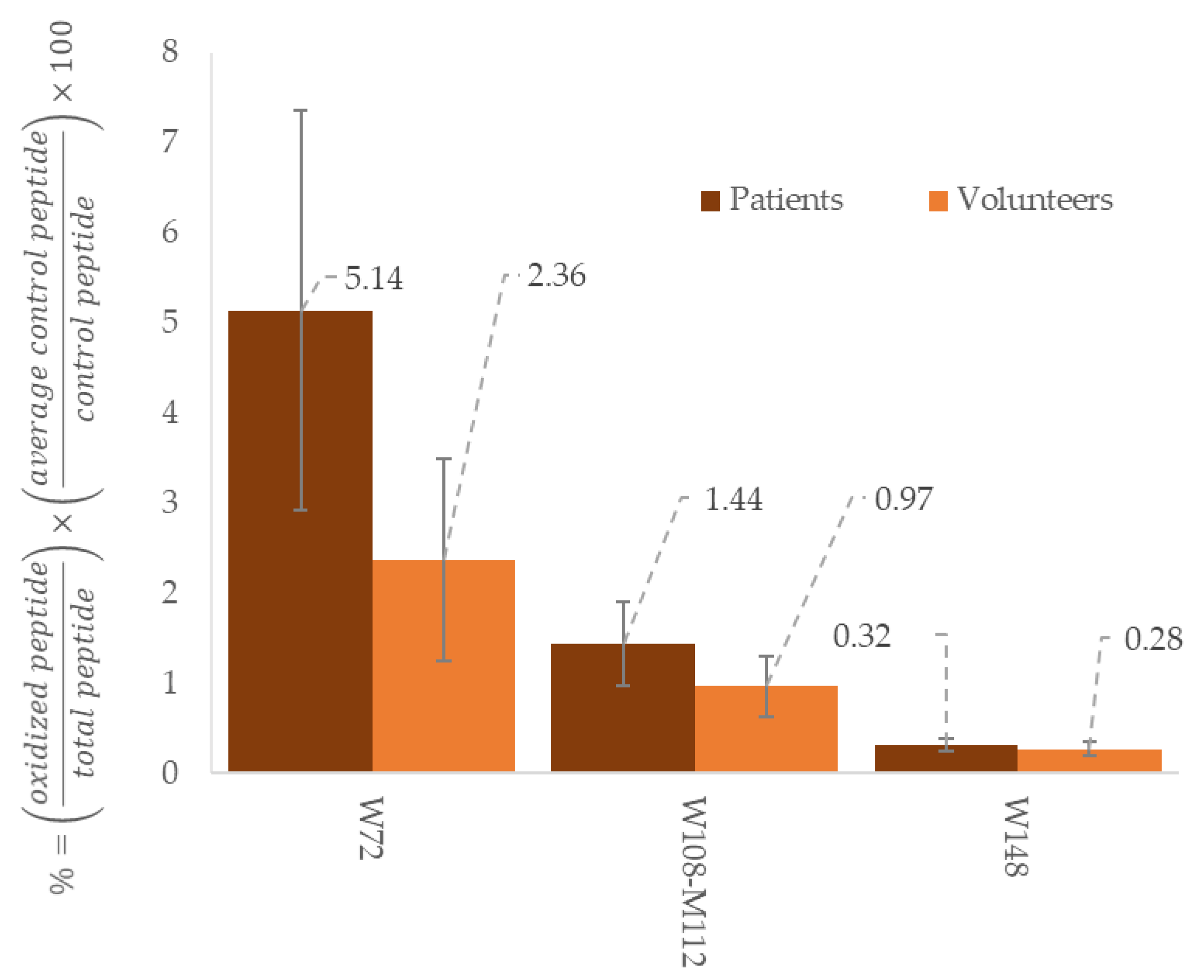
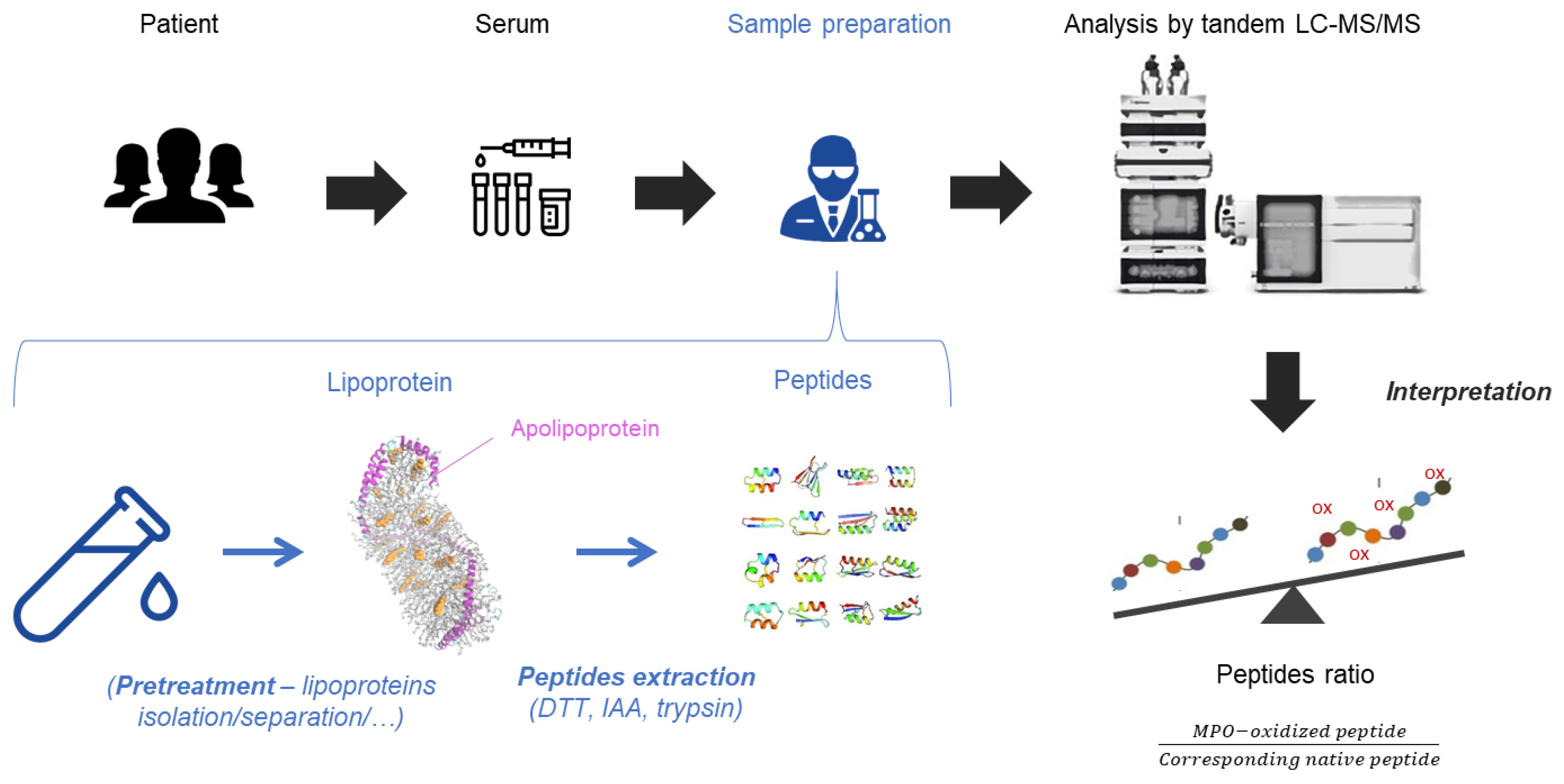

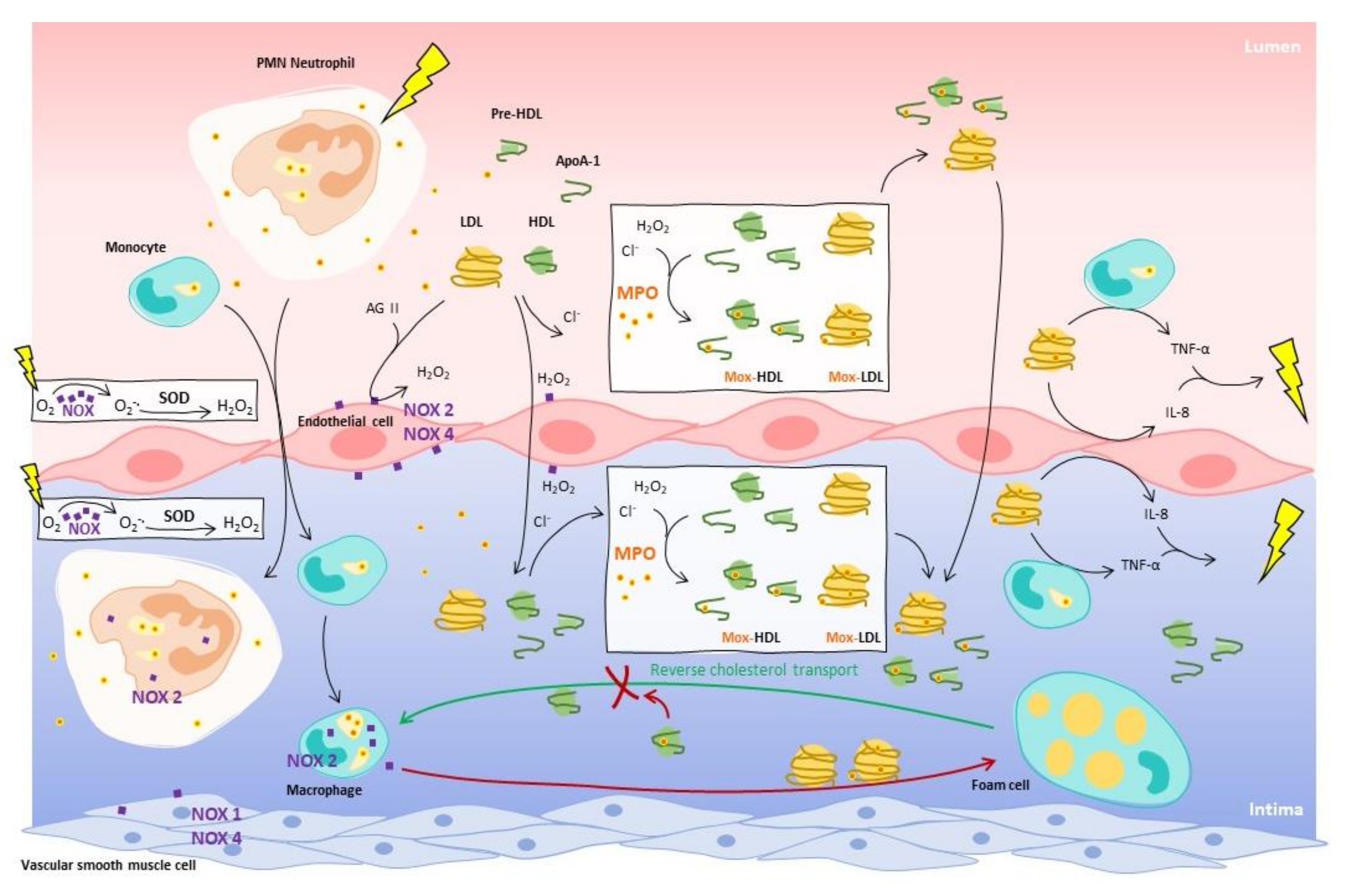{kind=link}
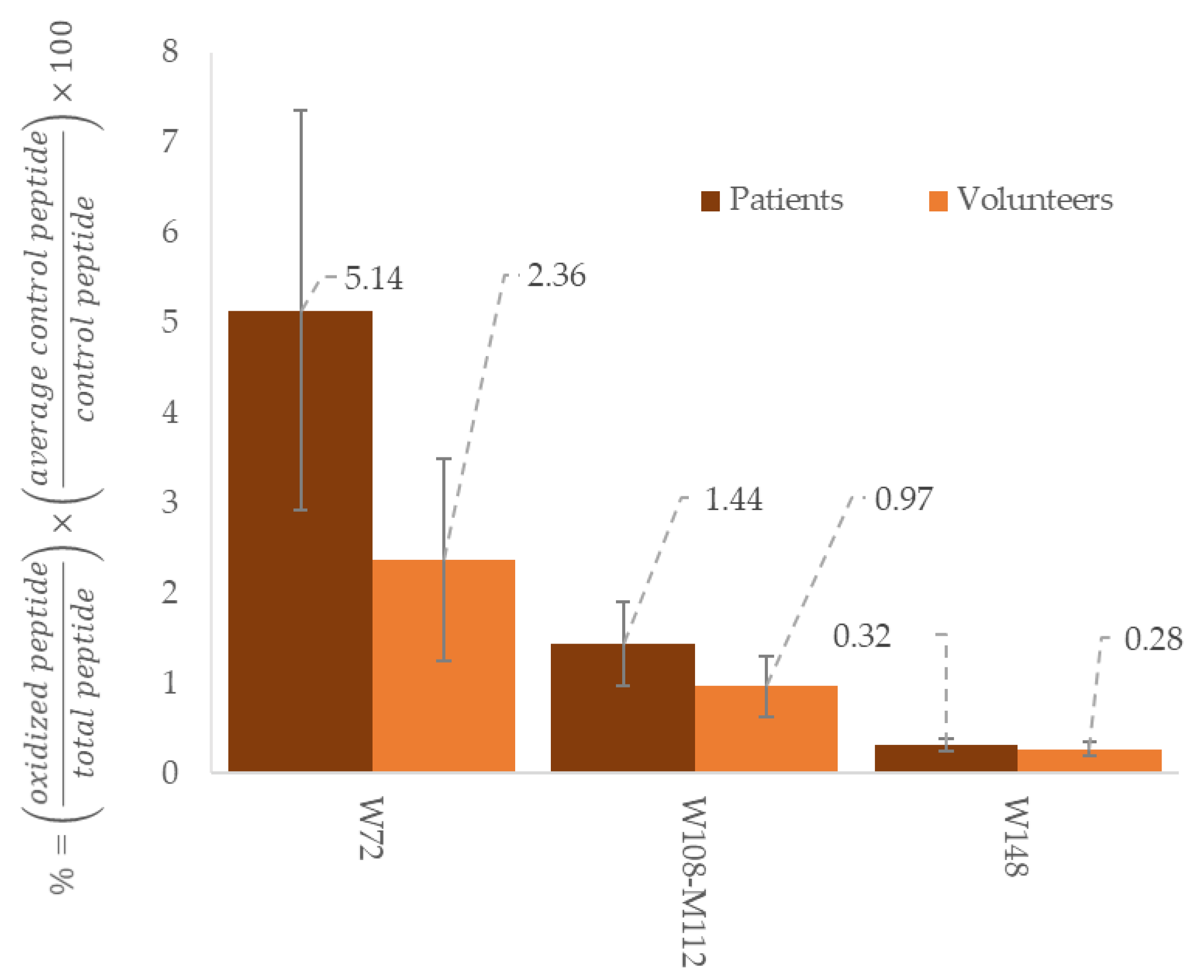{kind=link}
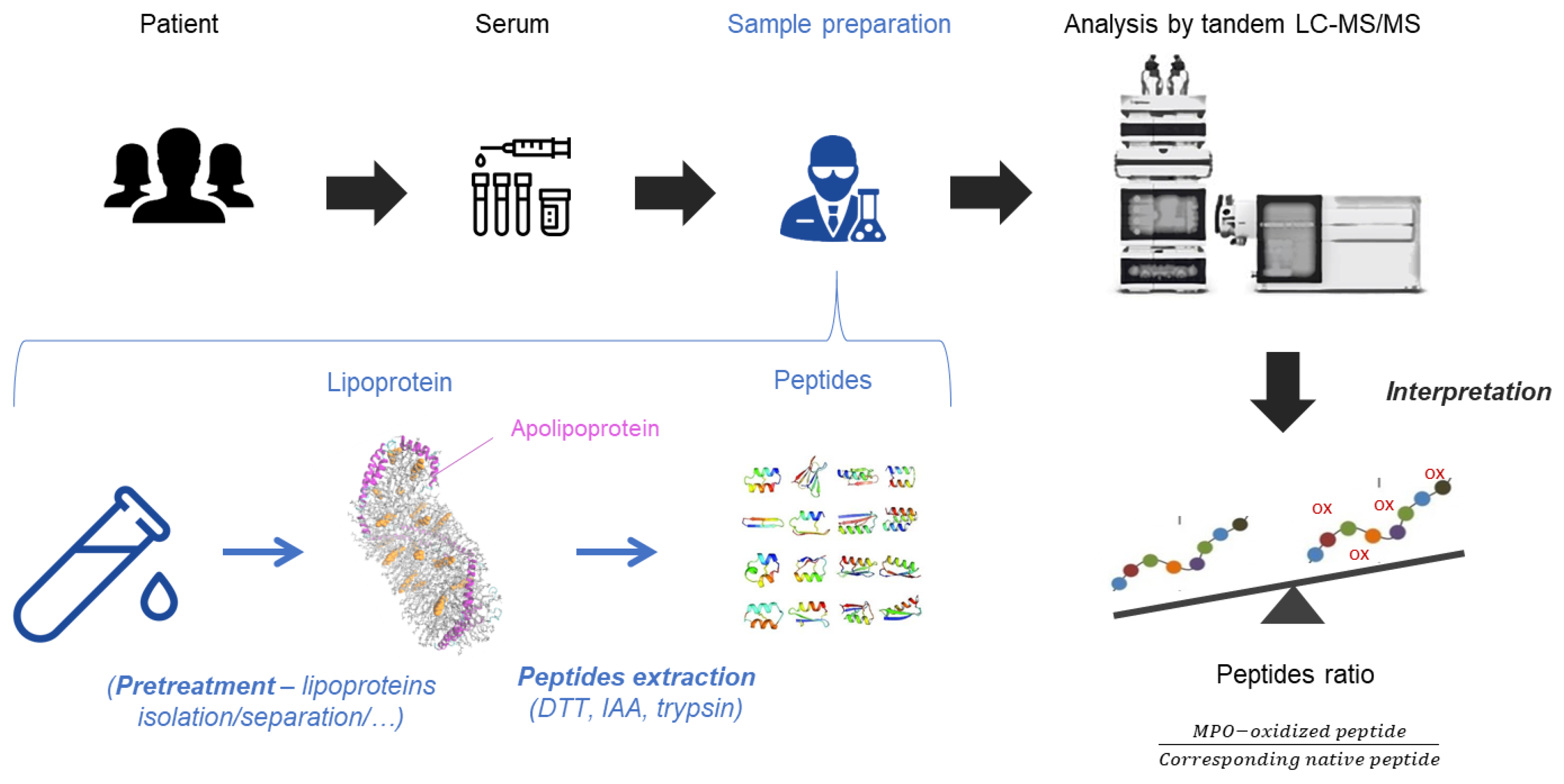{kind=link}
| Modifications | Modified Residues | Tested Condition | Reference |
|---|---|---|---|
| Chlorophenylalanine | Phe 57, Phe 71 | ApoA-1/HDL with MPO/H2O2/Cl− | [30] |
| Methionine sulfoxide | Met 86, Met 112, Met 148 | ApoA-1/HDL with MPO/H2O2/Cl− | [30] |
| Met 48, Met 112 | ApoA-1 isolated from human atheroma tissue | [31] | |
| Chlorotyrosine | Tyr 192, Tyr 236, Tyr 29, Tyr 18, Tyr 100, Tyr 115, Tyr 166 | HDL with HOCl | [32] |
| Tyr 192 | HDL with MPO/H2O2/Cl− or HOCl | [29] | |
| Tyr 192, Tyr 166 | HDL with MPO/H2O2 (<50 μM)/Cl− | [33] | |
| Tyr 192, Tyr 166, Tyr 29, Tyr 236 | HDL with MPO/H2O2 (<100 μM)/Cl− or 100 μM HOCl | [33] | |
| Tyr 166 | ApoA-1 in vivo | [33] | |
| Nitrotyrosine | Tyr 192, Tyr 18, Tyr 29, Tyr 236, Tyr 100, Tyr 115, Tyr 166 | ApoA-1 with MPO/H2O2/NO2− or ONOO− | [29] |
| Tyr 18, Tyr 29, Tyr 236, Tyr 100 | HDL with MPO/H2O2/NO2− | [29] | |
| Tyr 192, Tyr 18, Tyr 29, Tyr 236, Tyr 115, Tyr 166 | HDL with ONOO− | [29] | |
| Tyr 192, Tyr 166 | HDL with MPO/H2O2 (<50 μM)/NO2− | [33] | |
| Tyr 192, Tyr 166, Tyr 29, Tyr 236 | HDL with MPO/H2O2 (<100 μM)/NO2− | [33] | |
| Tyr 166, Tyr 18, Tyr 236 | HDL with 100 μM peroxynitrite | [33] | |
| Tyr 192, Tyr 166 | ApoA-1 in vivo | ||
| Nitrotyrosine and methionine sulfoxide | Met 112 and Tyr 115 (single peptide) | ApoA-1 with MPO/H2O2/NO2− or ONOO− | [29] |
| Chlorotyrosine and methionine sulfoxide | Tyr 192 | ApoA-1 with MPO/H2O2/Cl− | [34] |
| Monohydroxytryptophan | Trp 8, Trp 50, Trp 72, Trp 108, | ApoA-1 isolated from human | [31] |
| Trp 72 | [35] | ||
| Dihydroxytryptophan | Trp 108 | [31] | |
| 2-aminoadipic acid | Lysine | [31] |
| Modification | Tested Condition | Modified Residues | Reference |
|---|---|---|---|
| oxCys oxLys oxTrp oxMet | LDL oxidized by HOCl | Cys61/185/234/451/4190/3734/3890 Lys120 Trp1114/1210/1893/3567 Met3569 | [39] |
| oxMet4 | ApoB-100 in vitro by MPO | Met4 | [40] |
| oxTy oxPhe | LDL in vivo | Tyr 103/413/666/2524/3490/3791/4088 Phe 3965 | [41] |
| oxTy oxTrp | Hydroxyl radical and peroxynitrite | Tyr 583 and Trp 2524 | [42] |
| oxTy | Hydroxyl radical and HOCl | Tyr 144, Tyr 276, Tyr 4451 and Tyr 4509 | |
| oxTy | Hydroxyl radical and peroxynitrite and HOCl | Tyr 3295 | |
| oxTyr oxTrp | HOCl | Tyr 3139 and Trp 3153 | |
| oxTrp | Trp 4369 | ||
| oxMet oxTrp | Patients and volunteers | Met 4/4192 Trp 1114/3536 | [43] |
| oxMet oxTrp Cl-Tyr dioxTrp | Patients only | Met 2499 Trp 2894/3606 Tyr 76/102/125/749 Trp 4369 | |
| oxHis oxTrp oxLys | Patients | H2245, H2253, H3960 W1114 Lys293 | [44] |
| Peptide Sequence | Modification | RT (min) | Precursor Ion m/z | Product Ion 1 | Product Ion 2 | ||
|---|---|---|---|---|---|---|---|
| m/z | Frag | m/z | Frag | ||||
| 46LLDNWDSVTSTFSK59 | W50 | 10.7 | 806.90 | 199.18 | a2+ | 1271.59 | y11+ |
| 46LLDNWDSVTSTFSK59 | W50 Ox | 9.93 | 814.89 | 199.18 | 1287.59 | ||
| 62EQLGPVTQEFWDNLEK77 | W72 | 11 | 966.97 | 258.11 | b2+ | 838.42 | y14++ |
| 62EQLGPVTQEFWDNLEK77 | W72 Ox | 11.4 | 974.97 | 258.11 | 846.42 | ||
| 108WQEEMELYR117 | W108 M112 | 9.2 | 642.29 | 969.43 | y7+ | 315.15 | b2+ |
| 108WQEEMELYR117 | W108 M112 Ox | 8.12 | 650.29 | 985.43 | 315.15 | ||
| 108WQEEMELYR117 | W108 Ox M112 | 8.12 | 650.29 | 969.43 | 338.18 | y2+ | |
| 140LSPLGEEMR149 | M148 | 8.5 | 516.26 | 416.20 | y7++ | 621.26 | y5+ |
| 140LSPLGEEMR149 | M148 Ox | 7.37 | 524.26 | 424.20 | 573.26 | y5-64+ | |
| 216QGLLPVLESFK226 | Control 1 | 11.9 | 615.86 | 819.46 | y7+ | ||
| 216QGLLPVLESFK226 | Control 1 labelled | 12 | 619.36 | 826.48 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coremans, C.; Delporte, C.; Cotton, F.; Van De Borne, P.; Boudjeltia, K.Z.; Van Antwerpen, P. Mass Spectrometry for the Monitoring of Lipoprotein Oxidations by Myeloperoxidase in Cardiovascular Diseases. Molecules 2021, 26, 5264. https://doi.org/10.3390/molecules26175264
Coremans C, Delporte C, Cotton F, Van De Borne P, Boudjeltia KZ, Van Antwerpen P. Mass Spectrometry for the Monitoring of Lipoprotein Oxidations by Myeloperoxidase in Cardiovascular Diseases. Molecules. 2021; 26(17):5264. https://doi.org/10.3390/molecules26175264
Chicago/Turabian StyleCoremans, Catherine, Cédric Delporte, Frédéric Cotton, Phillipe Van De Borne, Karim Zouaoui Boudjeltia, and Pierre Van Antwerpen. 2021. "Mass Spectrometry for the Monitoring of Lipoprotein Oxidations by Myeloperoxidase in Cardiovascular Diseases" Molecules 26, no. 17: 5264. https://doi.org/10.3390/molecules26175264
APA StyleCoremans, C., Delporte, C., Cotton, F., Van De Borne, P., Boudjeltia, K. Z., & Van Antwerpen, P. (2021). Mass Spectrometry for the Monitoring of Lipoprotein Oxidations by Myeloperoxidase in Cardiovascular Diseases. Molecules, 26(17), 5264. https://doi.org/10.3390/molecules26175264