
Mechanism of Guanosine Triphosphate Hydrolysis by the Visual Proteins Arl3-RP2: Free Energy Reaction Profiles Computed with Ab Initio Type QM/MM Potentials


Abstract
:
1. Introduction
2. Models and Methods
3. Results
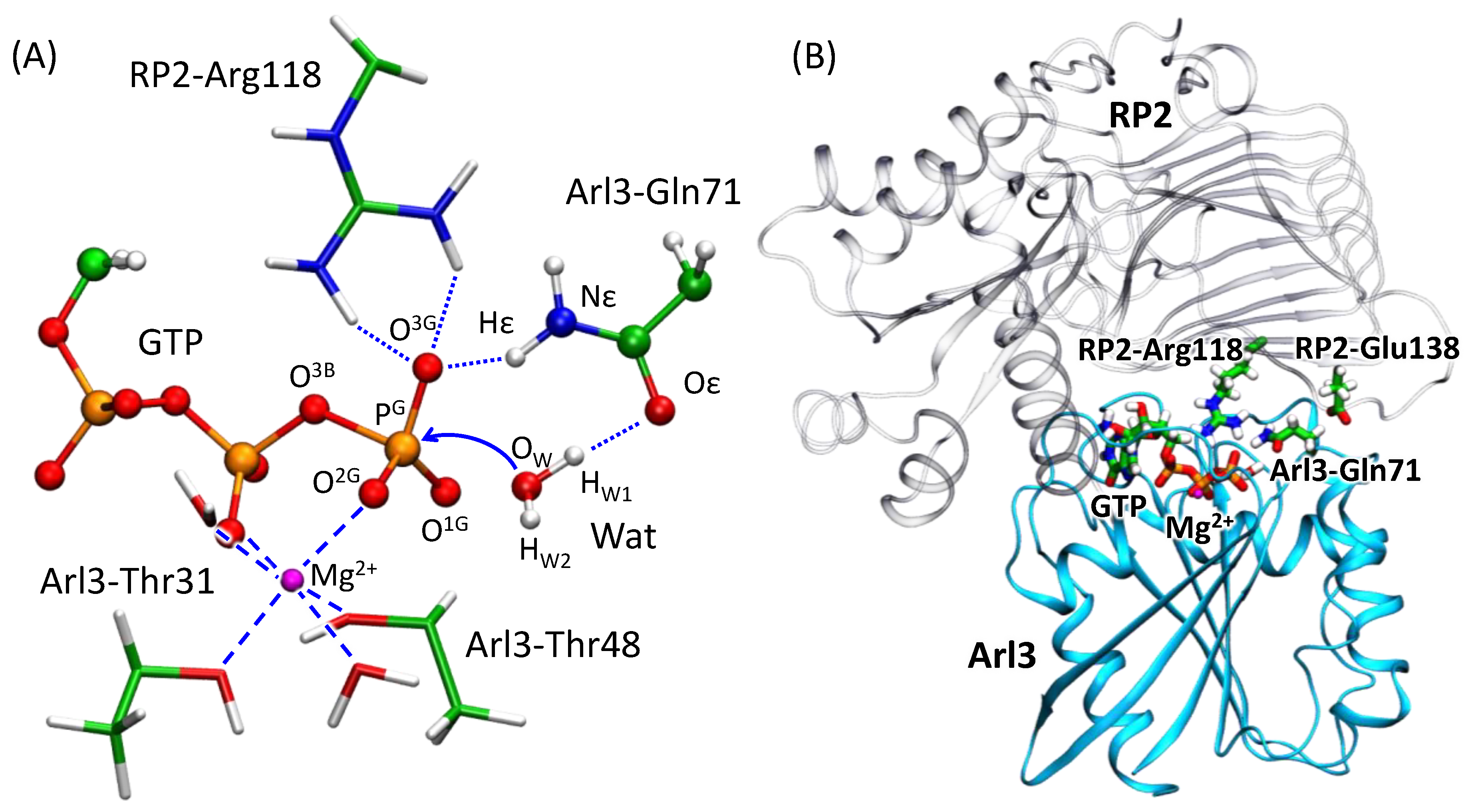
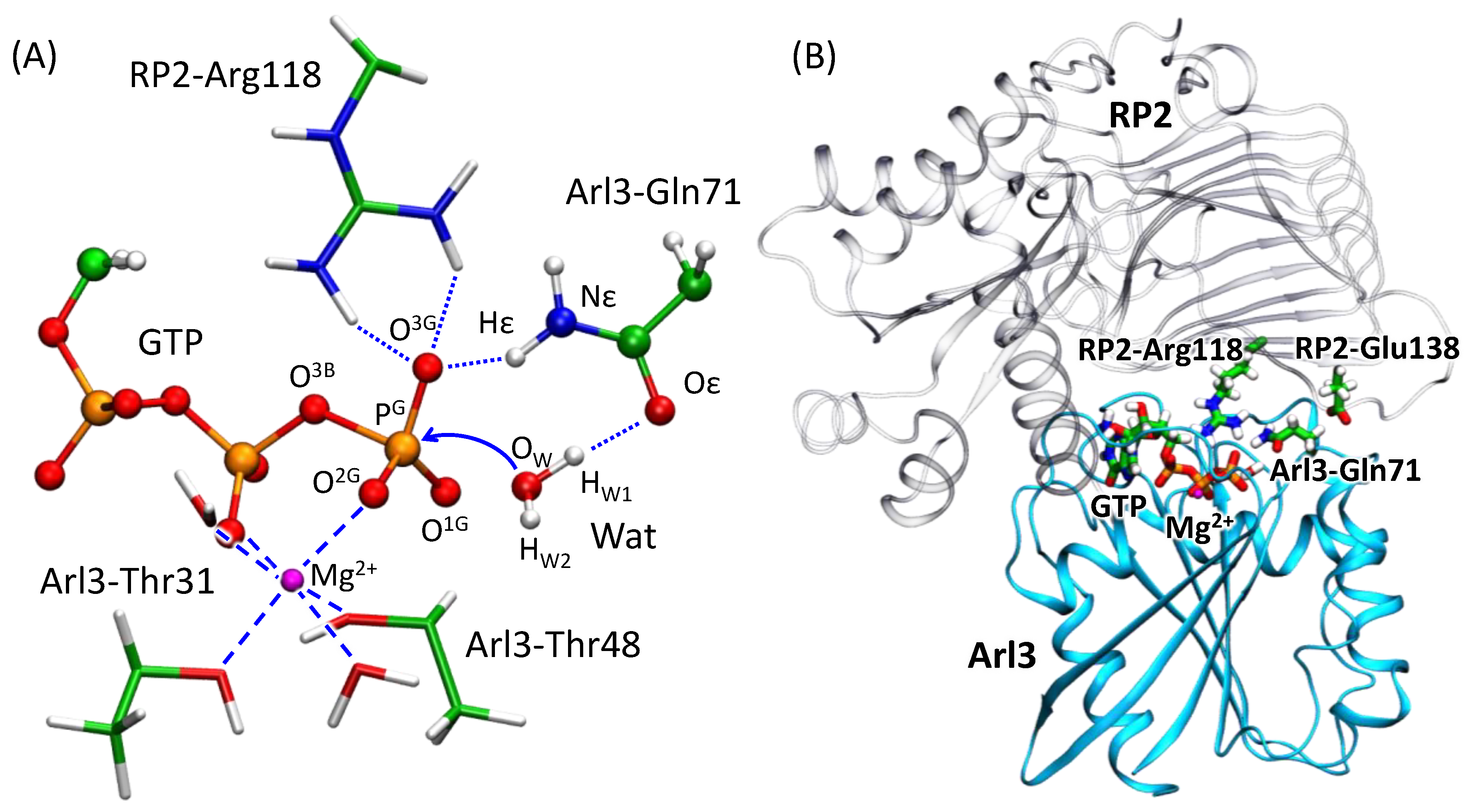
3.1. Dynamics of the Enzyme–Substrate Complex
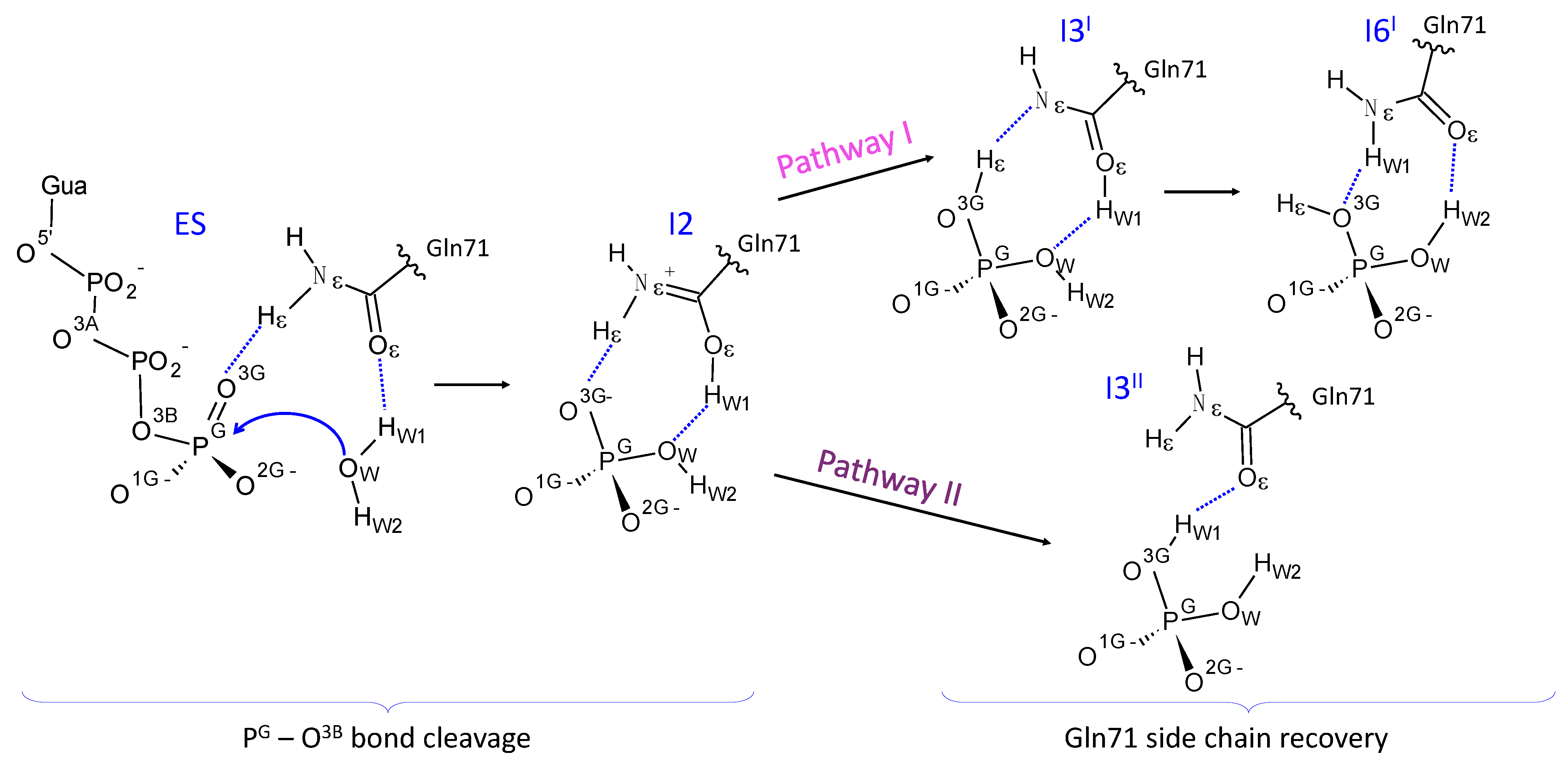
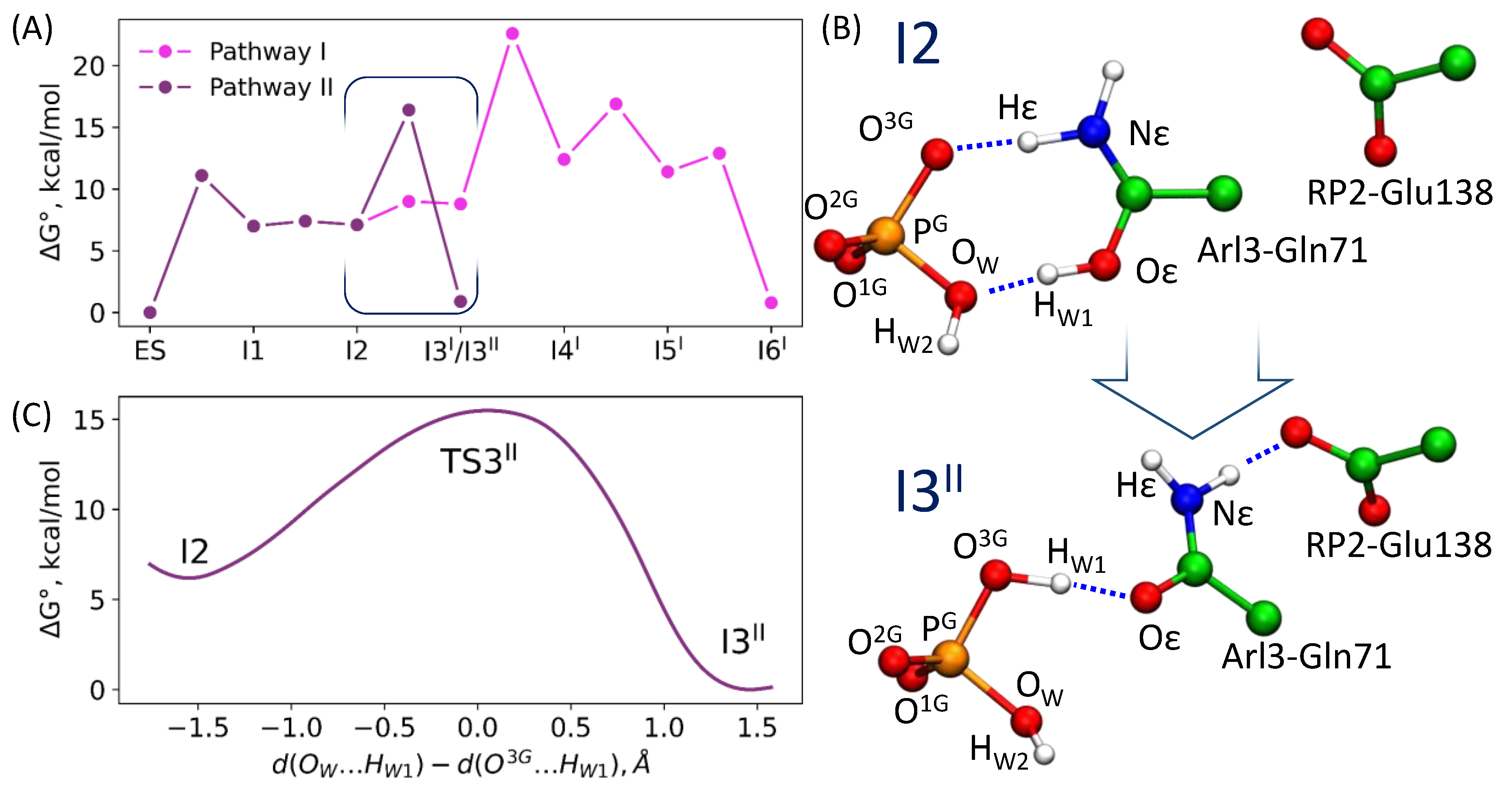
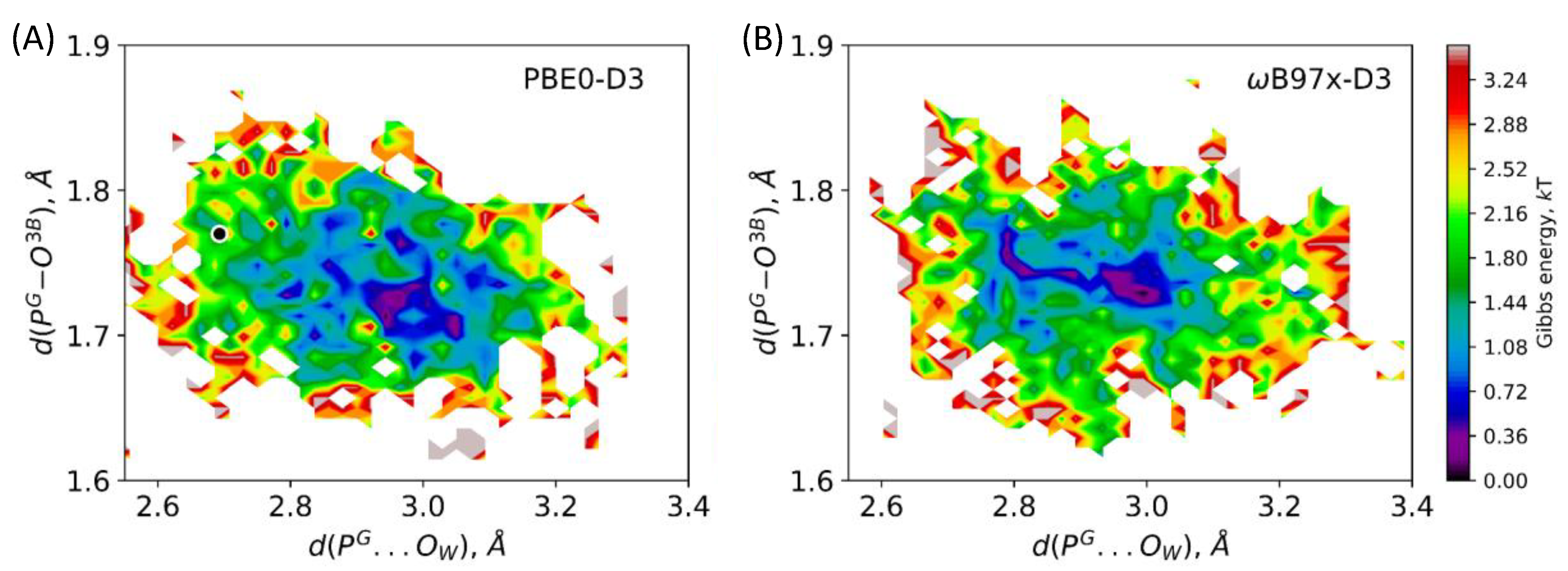
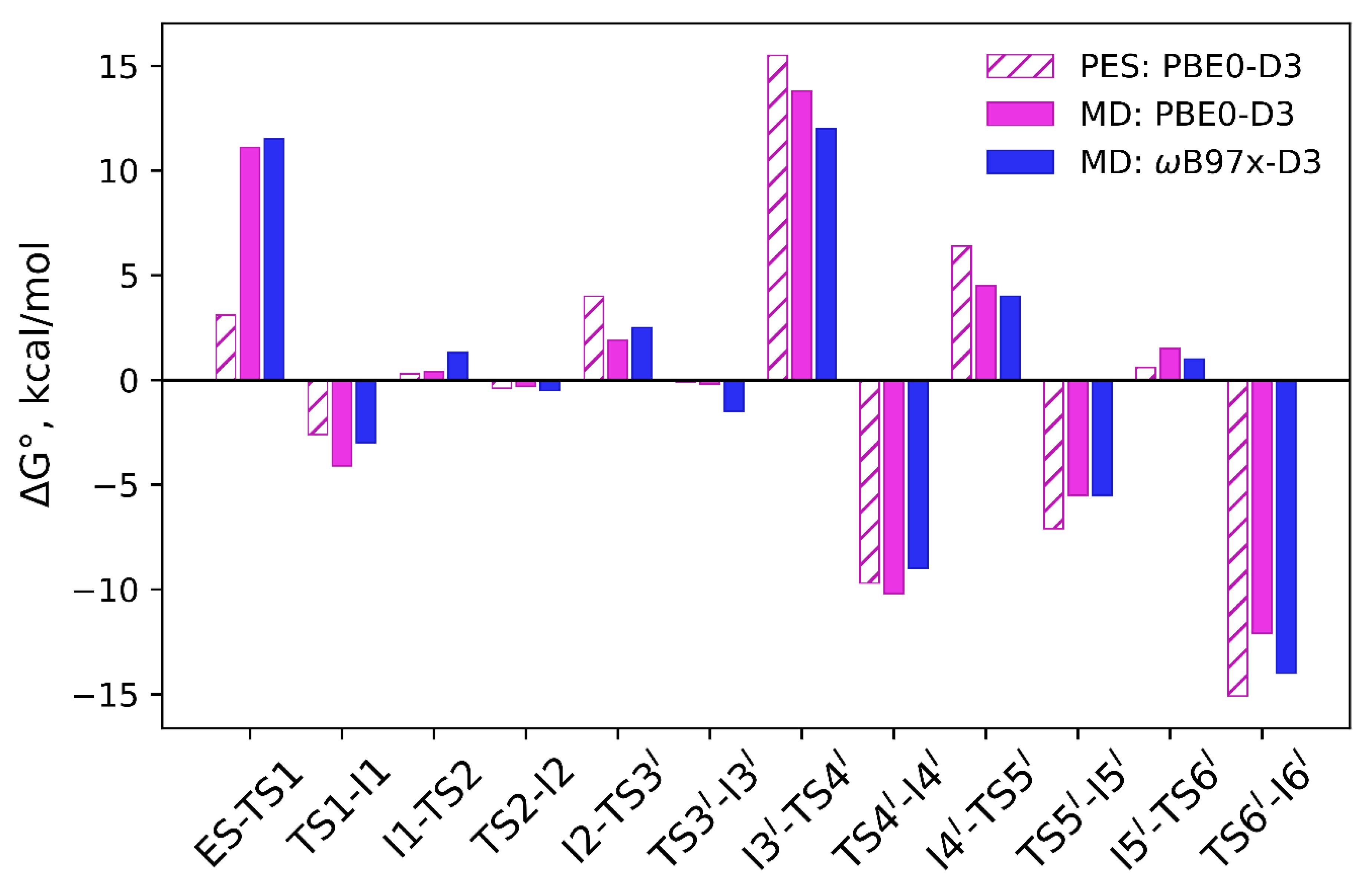
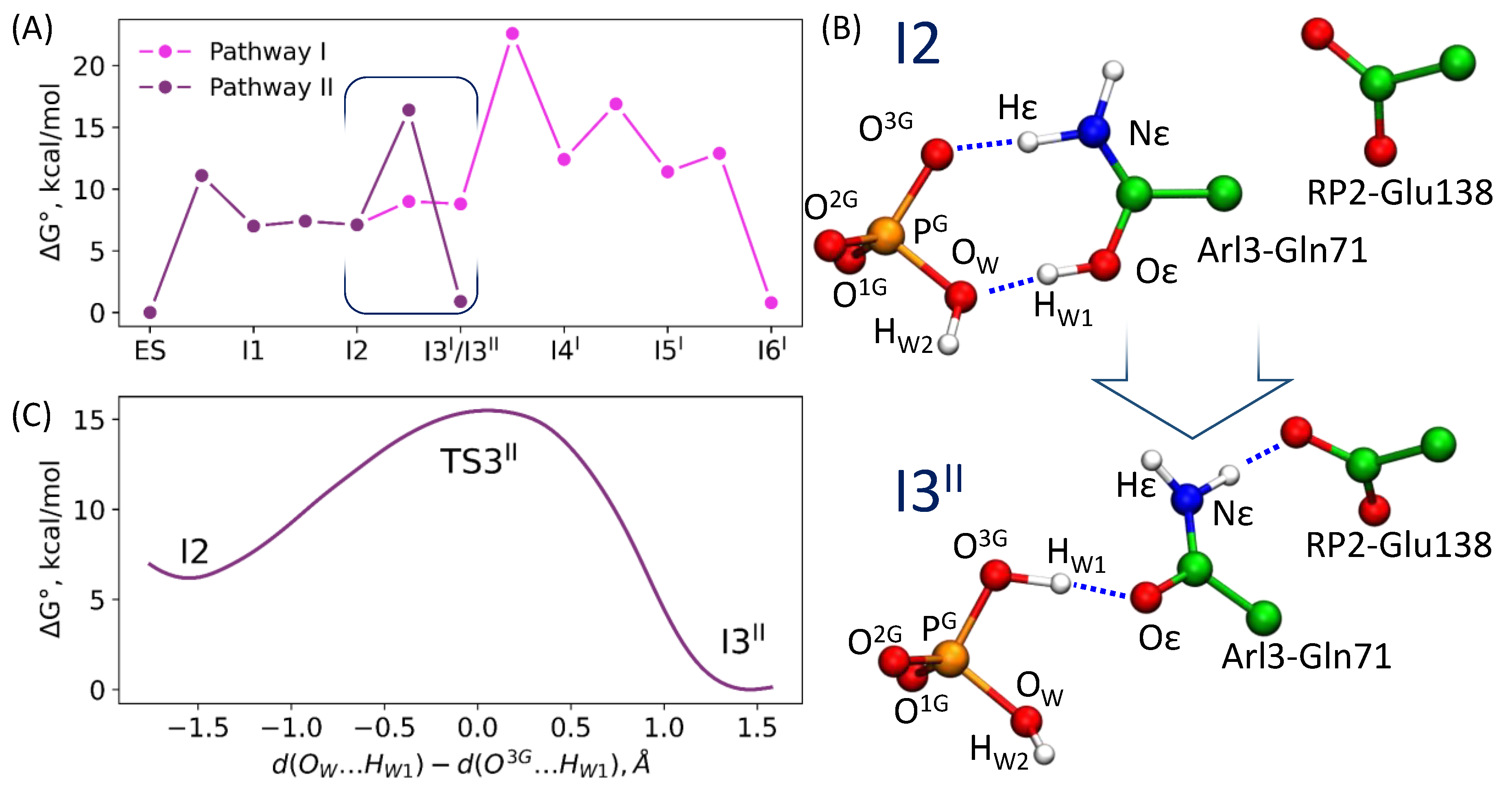
3.2. Comparison of Reaction Energy Profiles for the Pathway Revealed in QM/MM Minimization
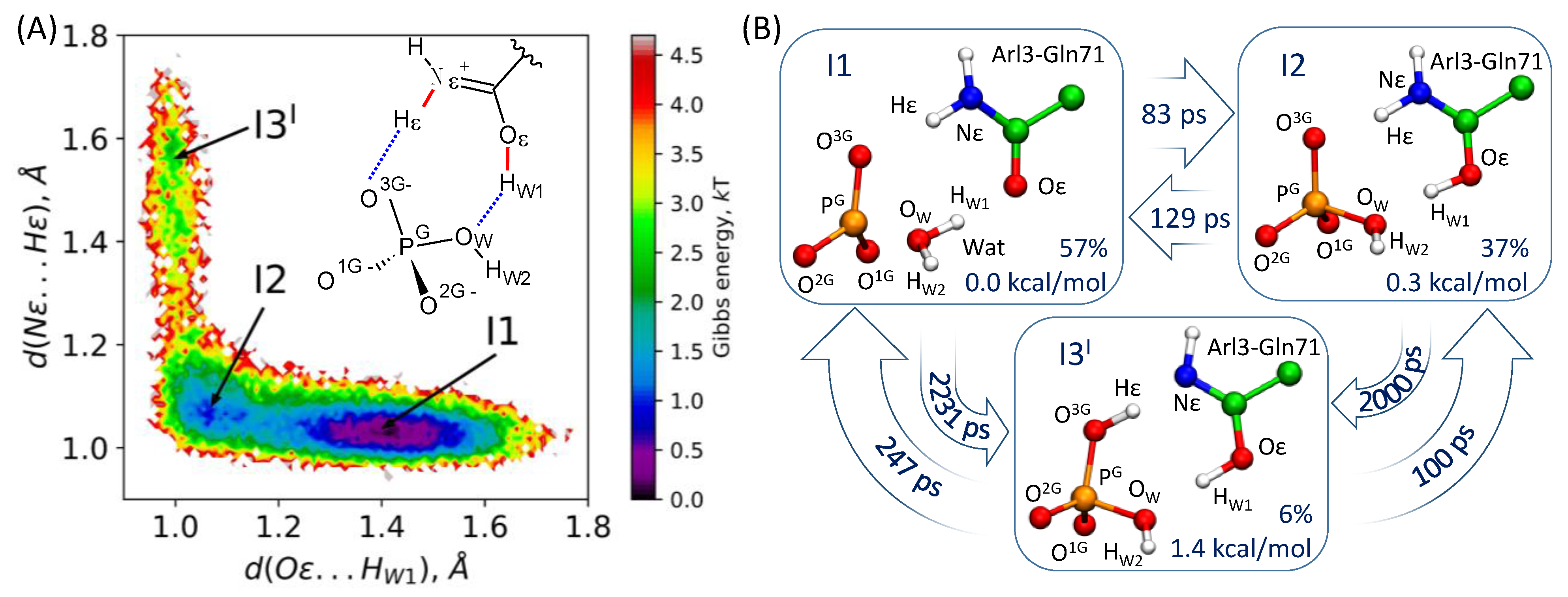
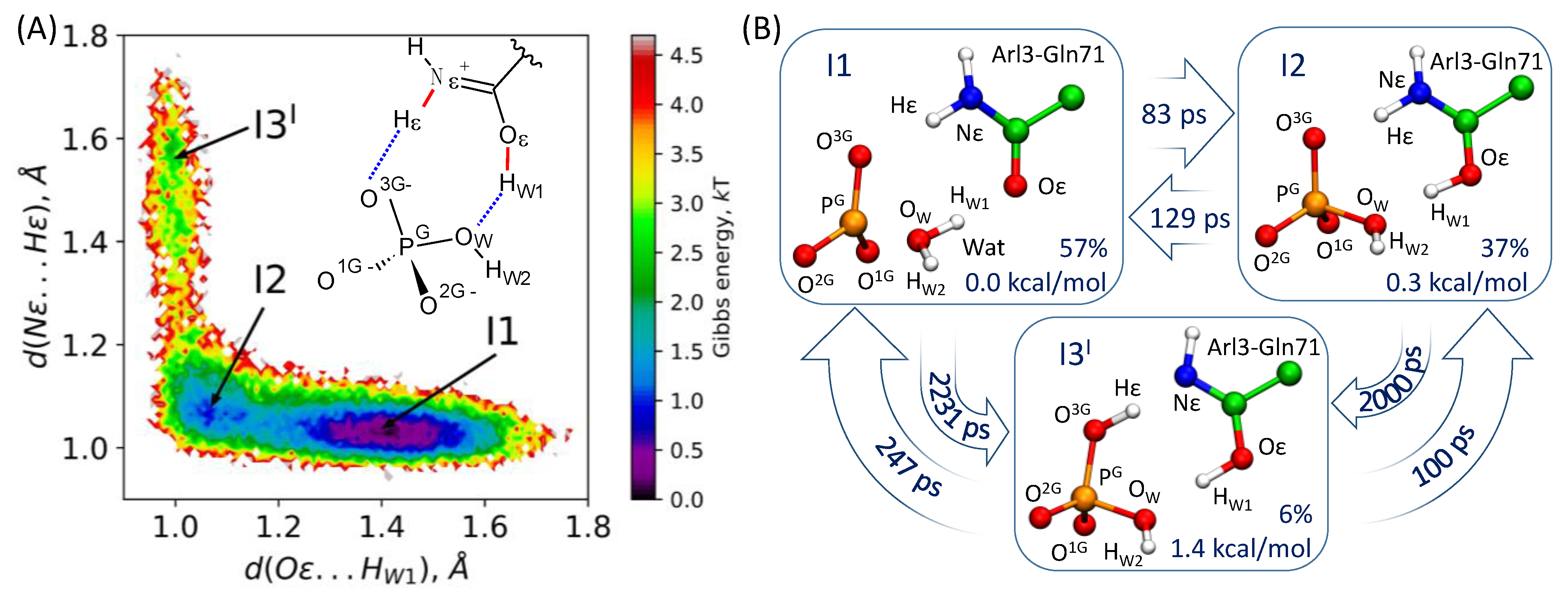
3.3. Markov State Model of the I1-I2-I3I Transitions
3.4. Enzyme Regeneration Pathways
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Senn, H.M.; Thiel, W. QM/MM Methods for Biomolecular Systems. Angew. Chemie Int. Ed. 2009, 48, 1198–1229. [Google Scholar] [CrossRef]
- van der Kamp, M.W.; Mulholland, A.J. Combined Quantum Mechanics/Molecular Mechanics (QM/MM) Methods in Computational Enzymology. Biochemistry 2013, 52, 2708–2728. [Google Scholar] [CrossRef]
- Acevedo, O.; Jorgensen, W.L. Advances in Quantum and Molecular Mechanical (QM/MM) Simulations for Organic and Enzymatic Reactions. Acc. Chem. Res. 2010, 43, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Pal, T.; Xie, L. Biomolecular QM/MM Simulations: What Are Some of the “Burning Issues”? J. Phys. Chem. B 2021, 125, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Barrios Herrera, L.; Chehelamirani, M.; Hostaš, J.; Jalife, S.; Salahub, D.R. Multiscale modeling of enzymes: QM-cluster, QM/MM, and QM/MM/MD: A tutorial review. Int. J. Quantum Chem. 2018, 118, e25558. [Google Scholar] [CrossRef]
- Krachtus, D.; Smith, J.; Imhof, P. Quantum Mechanical/Molecular Mechanical Analysis of the Catalytic Mechanism of Phosphoserine Phosphatase. Molecules 2018, 23, 3342. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, P.; Cerqueira, N.M.F.S.A.; Fernandes, P.A.; Romão, M.J.; Ramos, M.J. Catalytic Mechanism of Human Aldehyde Oxidase. ACS Catal. 2020, 10, 9276–9286. [Google Scholar] [CrossRef]
- Sousa, J.P.M.; Neves, R.P.P.; Sousa, S.F.; Ramos, M.J.; Fernandes, P.A. Reaction Mechanism and Determinants for Efficient Catalysis by DszB, a Key Enzyme for Crude Oil Bio-desulfurization. ACS Catal. 2020, 10, 9545–9554. [Google Scholar] [CrossRef]
- Melo, M.C.R.; Bernardi, R.C.; Rudack, T.; Scheurer, M.; Riplinger, C.; Phillips, J.C.; Maia, J.D.C.; Rocha, G.B.; Ribeiro, J.V.; Stone, J.E.; et al. NAMD goes quantum: An integrative suite for hybrid simulations. Nat. Methods 2018, 15, 351–354. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Seritan, S.; Bannwarth, C.; Fales, B.S.; Hohenstein, E.G.; Isborn, C.M.; Kokkila-Schumacher, S.I.L.; Li, X.; Liu, F.; Luehr, N.; Snyder, J.W.; et al. TeraChem: A graphical processing unit-accelerated electronic structure package for large-scale ab initio molecular dynamics. WIREs Comput. Mol. Sci. 2020. [Google Scholar] [CrossRef]
- Kästner, J.; Thiel, W. Bridging the gap between thermodynamic integration and umbrella sampling provides a novel analysis method: “Umbrella integration”. J. Chem. Phys. 2005, 123, 144104. [Google Scholar] [CrossRef]
- Kästner, J. Umbrella sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Tsirelson, V.G.; Nemukhin, A.V. Dynamical properties of enzyme–substrate complexes disclose substrate specificity of the SARS-CoV-2 main protease as characterized by the electron density descriptors. Phys. Chem. Chem. Phys. 2020, 22, 19069–19079. [Google Scholar] [CrossRef] [PubMed]
- Krivitskaya, A.V.; Khrenova, M.G. Boronic Acids as Prospective Inhibitors of Metallo-β-Lactamases: Efficient Chemical Reaction in the Enzymatic Active Site Revealed by Molecular Modeling. Molecules 2021, 26, 2026. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Kulakova, A.M.; Nemukhin, A.V. Light-Induced Change of Arginine Conformation Modulates the Rate of Adenosine Triphosphate to Cyclic Adenosine Monophosphate Conversion in the Optogenetic System Containing Photoactivated Adenylyl Cyclase. J. Chem. Inf. Model. 2021, 61. [Google Scholar] [CrossRef]
- Mishra, A.K.; Lambright, D.G. Small GTPases and their GAPs. Biopolymers 2016, 105, 431–448. [Google Scholar] [CrossRef] [Green Version]
- Hanke-Gogokhia, C.; Frederick, J.M.; Zhang, H.; Baehr, W. Binary Function of ARL3-GTP Revealed by Gene Knockouts. In Retinal Degenerative Diseases. Advances in Experimental Medicine and Biology; Ash, J., Anderson, R., LaVail, M., Bowes Rickman, C., Hollyfield, J., Grimm, C., Eds.; Springer: Cham, Switzerland, 2018; Volume 1074, pp. 317–325. [Google Scholar] [CrossRef]
- Schwarz, N.; Hardcastle, A.J.; Cheetham, M.E. Arl3 and RP2 mediated assembly and traffic of membrane associated cilia proteins. Vision Res. 2012, 75, 2–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veltel, S.; Gasper, R.; Eisenacher, E.; Wittinghofer, A. The retinitis pigmentosa 2 gene product is a GTPase-activating protein for Arf-like 3. Nat. Struct. Mol. Biol. 2008, 15, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Kühnel, K.; Veltel, S.; Schlichting, I.; Wittinghofer, A. Crystal Structure of the Human Retinitis Pigmentosa 2 Protein and Its Interaction with Arl3. Structure 2006, 14, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.R.; Stege, P.; Scheffzek, K.; Wittinghofer, A. Confirmation of the arginine-finger hypothesis for the GAP-stimulated GTP-hydrolysis reaction of Ras. Nat. Struct. Biol. 1997, 4, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Kotting, C.; Kallenbach, A.; Suveyzdis, Y.; Wittinghofer, A.; Gerwert, K. The GAP arginine finger movement into the catalytic site of Ras increases the activation entropy. Proc. Natl. Acad. Sci. USA 2008, 105, 6260–6265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, G.N.; Suardíaz, R.; Lopata, A.; Ozohanics, O.; Vékey, K.; Brooks, B.R.; Leveles, I.; Tóth, J.; Vértessy, B.G.; Rosta, E. Structural Characterization of Arginine Fingers: Identification of an Arginine Finger for the Pyrophosphatase dUTPases. J. Am. Chem. Soc. 2016, 138, 15035–15045. [Google Scholar] [CrossRef]
- Khrenova, M.G.; Kots, E.D.; Nemukhin, A.V. Reaction Mechanism of Guanosine Triphosphate Hydrolysis by the Vision-Related Protein Complex Arl3–RP2. J. Phys. Chem. B 2016, 120, 3873–3879. [Google Scholar] [CrossRef] [PubMed]
- Grigorenko, B.L.; Nemukhin, A.V.; Shadrina, M.S.; Topol, I.A.; Burt, S.K. Mechanisms of guanosine triphosphate hydrolysis by Ras and Ras-GAP proteins as rationalized by ab initio QM/MM simulations. Proteins Struct. Funct. Bioinforma. 2006, 66, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Khrenova, M.G.; Grigorenko, B.L.; Kolomeisky, A.B.; Nemukhin, A.V. Hydrolysis of guanosine triphosphate (GTP) by the Ras-GAP protein complex: Reaction mechanism and kinetic scheme. J. Phys. Chem. B 2015, 119, 12838–12845. [Google Scholar] [CrossRef] [Green Version]
- Grigorenko, B.L.; Khrenova, M.G.; Nemukhin, A. V Amide-imide tautomerization in the glutamine side chain in enzymatic and photochemical reactions in proteins. Phys. Chem. Chem. Phys. 2018, 20, 23827–23836. [Google Scholar] [CrossRef]
- Grigorenko, B.L.; Kots, E.D.; Nemukhin, A.V. Diversity of mechanisms in Ras–GAP catalysis of guanosine triphosphate hydrolysis revealed by molecular modeling. Org. Biomol. Chem. 2019, 17, 4879–4891. [Google Scholar] [CrossRef]
- Berta, D.; Buigues, P.J.; Badaoui, M.; Rosta, E. Cations in motion: QM/MM studies of the dynamic and electrostatic roles of H+ and Mg2+ ions in enzyme reactions. Curr. Opin. Struct. Biol. 2020, 61, 198–206. [Google Scholar] [CrossRef]
- Carvalho, A.T.P.; Szeler, K.; Vavitsas, K.; Åqvist, J.; Kamerlin, S.C.L. Modeling the mechanisms of biological GTP hydrolysis. Arch. Biochem. Biophys. 2015, 582, 80–90. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Denning, E.J.; Priyakumar, U.D.; Nilsson, L.; Mackerell, A.D. Impact of 2′-hydroxyl sampling on the conformational properties of RNA: Update of the CHARMM all-atom additive force field for RNA. J. Comput. Chem. 2011, 32, 1929–1943. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Li, G.-D.; Mao, S.-P.; Chai, J.-D. Long-Range Corrected Hybrid Density Functionals with Improved Dispersion Corrections. J. Chem. Theory Comput. 2013, 9, 263–272. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Prinz, J.-H.; Wu, H.; Sarich, M.; Keller, B.; Senne, M.; Held, M.; Chodera, J.D.; Schütte, C.; Noé, F. Markov models of molecular kinetics: Generation and validation. J. Chem. Phys. 2011, 134, 174105. [Google Scholar] [CrossRef]
- Scherer, M.K.; Trendelkamp-Schroer, B.; Paul, F.; Pérez-Hernández, G.; Hoffmann, M.; Plattner, N.; Wehmeyer, C.; Prinz, J.-H.; Noé, F. PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J. Chem. Theory Comput. 2015, 11, 5525–5542. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Shurki, A.; Warshel, A. Why does the Ras switch “break” by oncogenic mutations? Proteins Struct. Funct. Bioinforma. 2004, 55, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kötting, C.; Gerwert, K. Time-resolved FTIR studies provide activation free energy, activation enthalpy and activation entropy for GTPase reactions. Chem. Phys. 2004, 307, 227–232. [Google Scholar] [CrossRef]
- Prasad, B., R.; Plotnikov, N.V.; Lameira, J.; Warshel, A. Quantitative exploration of the molecular origin of the activation of GTPase. Proc. Natl. Acad. Sci. USA 2013, 110, 20509–20514. [Google Scholar] [CrossRef] [Green Version]
- Glennon, T.M.; Villà, J.; Warshel, A. How does GAP catalyze the GTPase reaction of Ras?: A computer simulation study. Biochemistry 2000, 39, 9641–9651. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Carloni, P. Enzymatic GTP Hydrolysis: Insights from an ab Initio Molecular Dynamics Study. J. Am. Chem. Soc. 2002, 124, 3763–3768. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Coordinate | Step | Stationary Point | Reaction Coordinates at Stationary Points, Å | |||
|---|---|---|---|---|---|---|
| MD Simulations | PES Scan | |||||
| ωB97x-D3 | PBE0-D3 | PBE0-D3 | ||||
| d(PG…O3B)–d(PG…OW) | 1 | ES | 1.25 | 1.21 | 0.92 | |
| TS1 | −0.19 | −0.21 | 0.02 | |||
| I1 | −1.09 | −1.09 | −0.93 | |||
| d(Oε…H1w)–d(Hε…Nε) | 2, 3I | I1 | 0.45 | 0.33 | 0.30 | |
| TS2 | 0.13 | 0.15 | 0.16 | |||
| I2 | −0.02 | 0.00 | −0.04 | |||
| TS3I | −0.28 | −0.29 | −0.32 | |||
| I3I | −0.60 | −0.49 | −0.43 | |||
| d(O3B…Hε) | 4I | I3I | 3.81 | 3.77 | 3.86 | |
| TS4I | 2.60 | 2.58 | 2.81 | |||
| I4I | 1.40 | 1.42 | 1.41 | |||
| d(H2w…Nε) | 5I | I4I | 4.10 | 3.98 | 3.80 | |
| TS5I | 2.86 | 2.79 | 2.31 | |||
| I5I | 1.69 | 1.55 | 1.54 | |||
| d(H1w…Ow) | 6I | I5I | 2.40 | 2.54 | 1.73 | |
| TS6I | 1.88 | 1.94 | 1.63 | |||
| I6I | 0.99 | 1.00 | 1.02 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khrenova, M.G.; Bulavko, E.S.; Mulashkin, F.D.; Nemukhin, A.V. Mechanism of Guanosine Triphosphate Hydrolysis by the Visual Proteins Arl3-RP2: Free Energy Reaction Profiles Computed with Ab Initio Type QM/MM Potentials. Molecules 2021, 26, 3998. https://doi.org/10.3390/molecules26133998
Khrenova MG, Bulavko ES, Mulashkin FD, Nemukhin AV. Mechanism of Guanosine Triphosphate Hydrolysis by the Visual Proteins Arl3-RP2: Free Energy Reaction Profiles Computed with Ab Initio Type QM/MM Potentials. Molecules. 2021; 26(13):3998. https://doi.org/10.3390/molecules26133998
Chicago/Turabian StyleKhrenova, Maria G., Egor S. Bulavko, Fedor D. Mulashkin, and Alexander V. Nemukhin. 2021. "Mechanism of Guanosine Triphosphate Hydrolysis by the Visual Proteins Arl3-RP2: Free Energy Reaction Profiles Computed with Ab Initio Type QM/MM Potentials" Molecules 26, no. 13: 3998. https://doi.org/10.3390/molecules26133998
APA StyleKhrenova, M. G., Bulavko, E. S., Mulashkin, F. D., & Nemukhin, A. V. (2021). Mechanism of Guanosine Triphosphate Hydrolysis by the Visual Proteins Arl3-RP2: Free Energy Reaction Profiles Computed with Ab Initio Type QM/MM Potentials. Molecules, 26(13), 3998. https://doi.org/10.3390/molecules26133998