
Molecular Structure, Thermodynamic and Spectral Characteristics of Metal-Free and Nickel Complex of Tetrakis(1,2,5-thiadiazolo)porphyrazine

 , and
, and
Abstract
1. Introduction
2. Results and Discussion
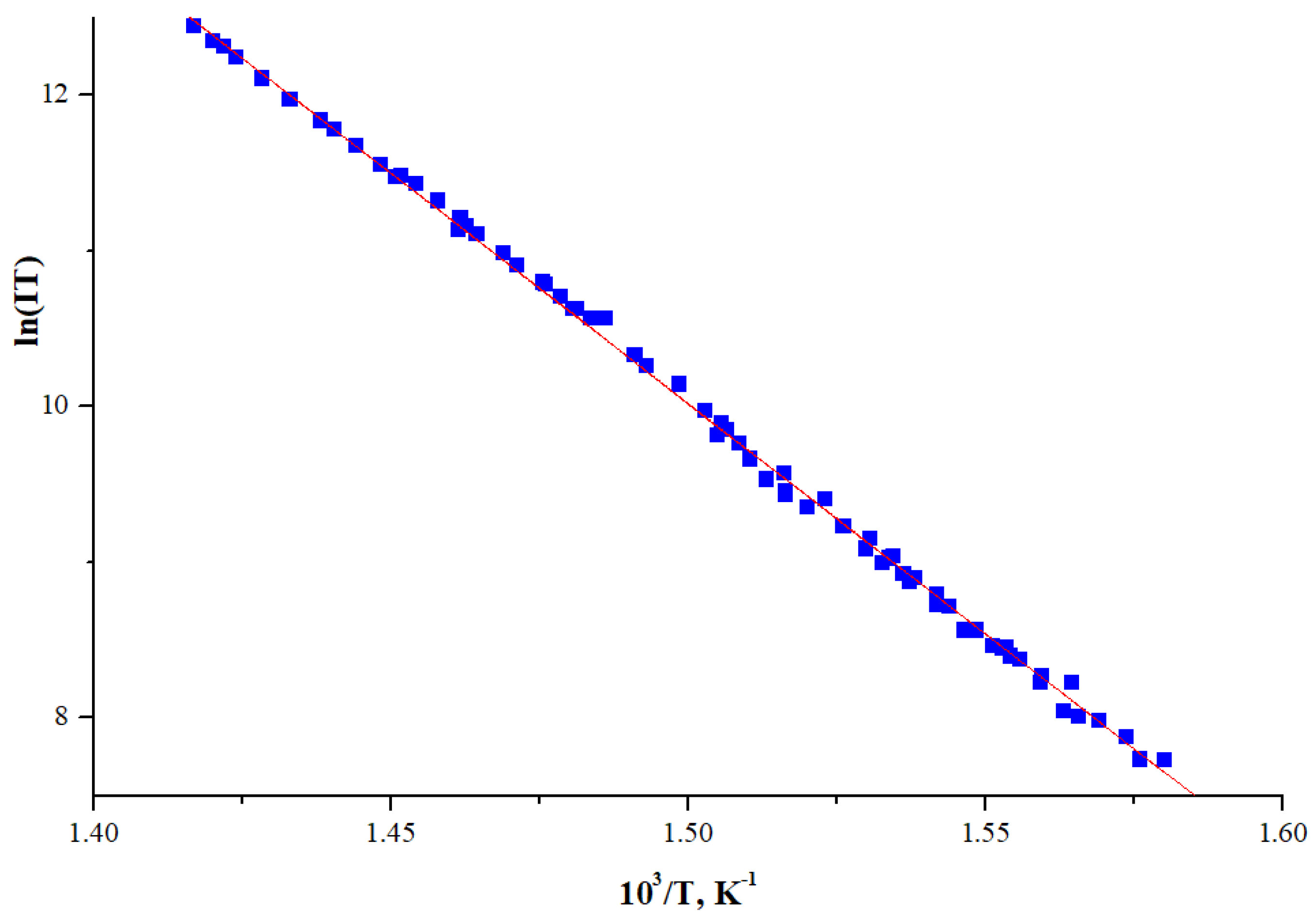
2.1. Sublimation
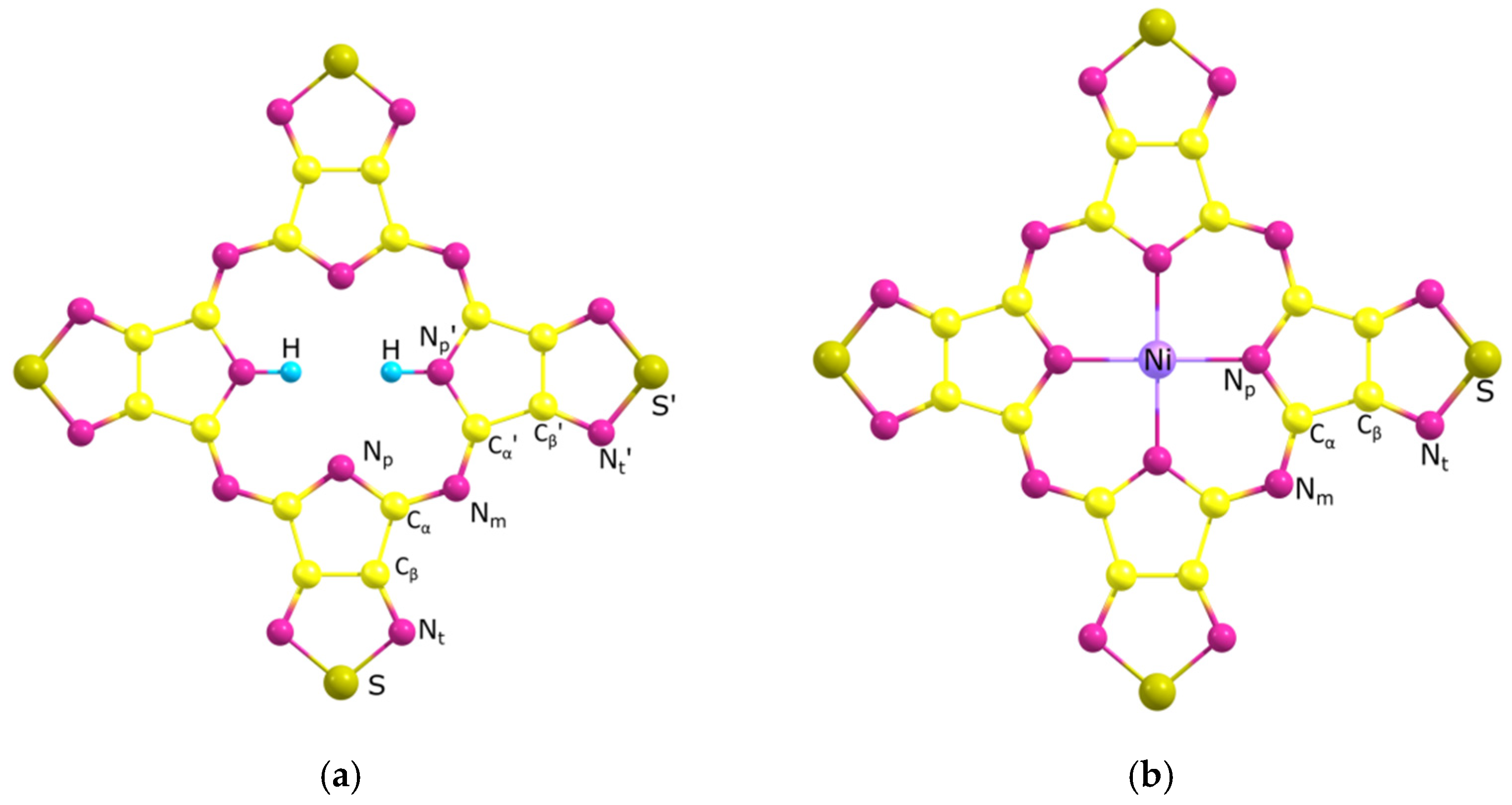
2.2. Molecular Structure
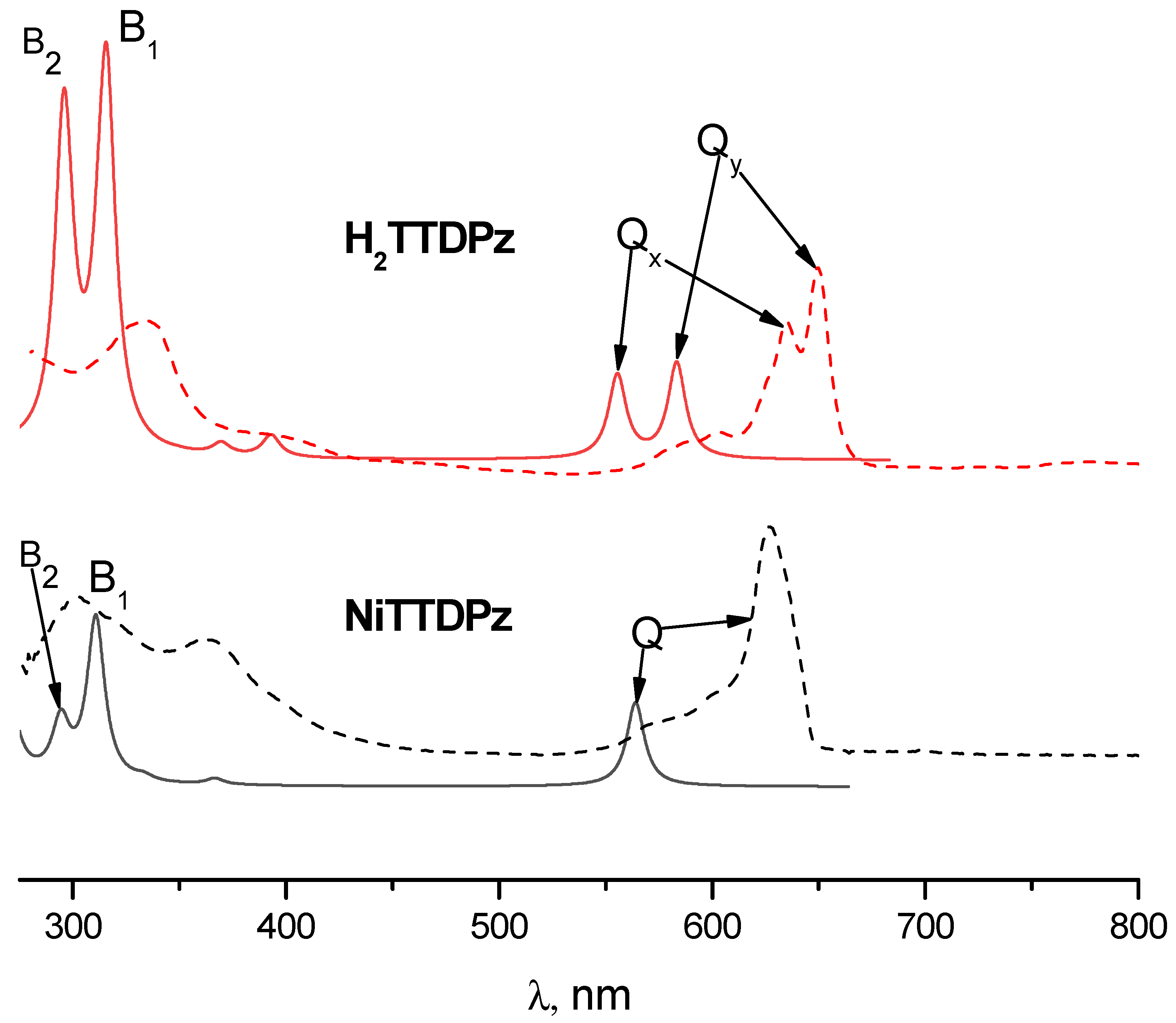
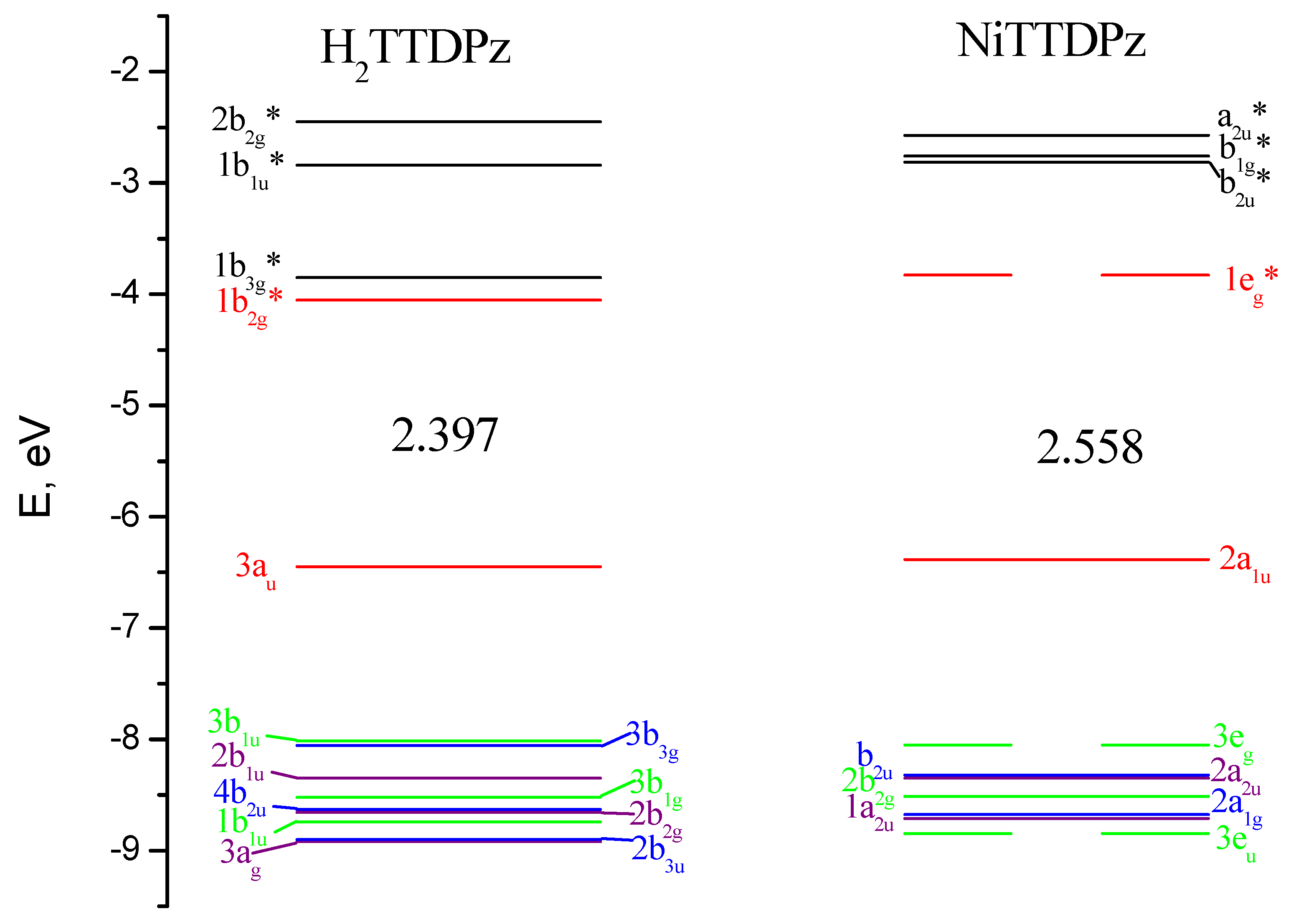
2.3. Electronic Absorption Spectra
2.4. Vibrational Spectra
3. Materials and Methods
3.1. Experimental Details
3.2. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sun, S.-S.; Sariciftci, N.S. Organic Photovoltaics: Mechanisms, Materials, and Devices; CRC Press: Boca Raton, FL, USA, 2017; ISBN 9781420026351. [Google Scholar]
- Kippelen, B.; Brédas, J.L. Organic photovoltaics. Energy Environ. Sci. 2009, 2, 251–261. [Google Scholar] [CrossRef]
- Mazzio, K.A.; Luscombe, C.K. The future of organic photovoltaics. Chem. Soc. Rev. 2015, 44, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Kruk, N.N. Nonlinear optical properties of tetrapyrrole compounds and prospects for their application (a review). J. Appl. Spectrosc. 2008, 75, 461–482. [Google Scholar] [CrossRef]
- De la Torre, G.; Vázquez, P.; Agulló-López, F.; Torres, T. Role of structural factors in the nonlinear optical properties of phthalocyanines and related compounds. Chem. Rev. 2004, 104, 3723–3750. [Google Scholar] [CrossRef]
- Drobizhev, M.; Makarov, N.S.; Rebane, A.; De La Torre, G.; Torres, T. Strong two-photon absorption in Push—Pull phthalocyanines: Role of resonance enhancement and permanent dipole moment change upon excitation. J. Phys. Chem. C 2008, 112, 848–859. [Google Scholar] [CrossRef]
- Senge, M.O.; Fazekas, M.; Notaras, E.G.A.; Blau, W.J.; Zawadzka, M.; Locos, O.B.; Ni Mhuircheartaigh, E.M. Nonlinear Optical Properties of Porphyrins. Adv. Mater. 2007, 19, 2737–2774. [Google Scholar] [CrossRef]
- McEwan, K.; Lewis, K.; Yang, G.-Y.; Chng, L.-L.; Lee, Y.-W.; Lau, W.-P.; Lai, K.-S. Synthesis, Characterization, and Nonlinear Optical Study of Metalloporphyrins. Adv. Funct. Mater. 2003, 13, 863–867. [Google Scholar] [CrossRef]
- Marder, S.R. Organic nonlinear optical materials: Where we have been and where we are going. Chem. Commun. 2006, 2, 131–134. [Google Scholar] [CrossRef]
- Liu, F.; Qin, G.; Li, Z.; Wang, Z.; Peng, M.; Wu, S.; Li, C.; Yang, Y. The design and synthesis of nonlinear optical chromophores containing two short chromophores for an enhanced electro-optic activity †. Cite this Mater. Adv. 2021, 2, 728. [Google Scholar]
- Linstead, R.P.; Whalley, M. Conjugated macrocylces. Part XXII. Tetrazaporphin and its metallic derivatives. J. Chem. Soc. 1952, 4839–4846. [Google Scholar] [CrossRef]
- Donzello, M.P.; Ercolani, C.; Novakova, V.; Zimcik, P.; Stuzhin, P.A. Tetrapyrazinoporphyrazines and their metal derivatives. Part I: Synthesis and basic structural information. Coord. Chem. Rev. 2016, 309, 107–179. [Google Scholar] [CrossRef]
- Novakova, V.; Donzello, M.P.; Ercolani, C.; Zimcik, P.; Stuzhin, P.A. Tetrapyrazinoporphyrazines and their metal derivatives. Part II: Electronic structure, electrochemical, spectral, photophysical and other application related properties. Coord. Chem. Rev. 2018, 361, 1–73. [Google Scholar] [CrossRef]
- Donzello, M.P.; Ercolani, C.; Stuzhin, P.A. Novel families of phthalocyanine-like macrocycles-Porphyrazines with annulated strongly electron-withdrawing 1,2,5-thia/selenodiazole rings. Coord. Chem. Rev. 2006, 250, 1530–1561. [Google Scholar] [CrossRef]
- Suzuki, Y.; Fujimori, M.; Yoshikawa, H.; Awaga, K. Packing motifs and magneto-structural correlations in crystal structures of metallo-tetrakis(1,2,5-thiadiazole)porphyrazine series, MTTDPz (M=H 2, Fe, Co, Ni, Cu, Zn). Chem. A Eur. J. 2004, 10, 5158–5164. [Google Scholar] [CrossRef] [PubMed]
- Koifman, O.I.; Ageeva, T.A.; Beletskaya, I.P.; Averin, A.D.; Yakushev, A.A.; Tomilova, L.G.; Dubinina, T.V.; Tsivadze, A.Y.; Gorbunova, Y.G.; Martynov, A.G.; et al. Macroheterocyclic Compounds—A Key Building Block in New Functional Materials and Molecular Devices. Macroheterocycles 2020, 13, 311–467. [Google Scholar] [CrossRef]
- Stuzhin, P.A.; Mikhailov, M.S.; Yurina, E.S.; Bazanov, M.I.; Koifman, O.I.; Pakhomov, G.L.; Travkin, V.V.; Sinelshchikova, A.A. First tellurium-containing phthalocyanine analogues: Strong effect of tellurium on spectral, redox and conductivity properties of porphyrazines with annulated chalcogenodiazole ring(s). Chem. Commun. 2012, 48, 10135–10137. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Kubo, M.; Fujinawa, T.; Suzuki, Y.; Yoshikawa, H.; Awaga, K. Electrochromism and stable n-type doping of highly oriented thin films of tetrakis(thiadiazole)porphyrazine. Angew. Chemie Int. Ed. 2007, 46, 5532–5536. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Miyoshi, Y.; Matsushita, M.M.; Awaga, K. A complementary organic inverter of porphyrazine thin films: Low-voltage operation using ionic liquid gate dielectrics. Chem. Commun. 2011, 47, 5837–5839. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Fujimoto, T.; Yoshikawa, H.; Matsushita, M.M.; Awaga, K.; Yamada, T.; Ito, H. Photoconductivity and FET performance of an n-type porphyrazine semiconductor, tetrakis(thiadiazole)porphyrazine. Org. Electron. 2011, 12, 239–243. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Takahashi, K.; Fujimoto, T.; Yoshikawa, H.; Matsushita, M.M.; Ouchi, Y.; Kepenekian, M.; Robert, V.; Donzello, M.P.; Ercolani, C.; et al. Crystal structure, spin polarization, solid-state electrochemistry, and high n-type carrier mobility of a paramagnetic semiconductor: Vanadyl tetrakis(thiadiazole)porphyrazine. Inorg. Chem. 2012, 51, 456–462. [Google Scholar] [CrossRef]
- Stuzhin, P.; Mikhailov, M.; Travkin, V.; Gudkov, E.; Pakhomov, G. Multilayer Photovoltaic Structures Based on Tetrathiadiazoloporphyrazine/ Subphthalocyanine Heterojunction. Macroheterocycles 2012, 5, 162–165. [Google Scholar] [CrossRef][Green Version]
- Zhabanov, Y.A.; Sliznev, V.V.; Ryzhov, I.V.; Stuzhin, P.A. Peculiarities of electronic structure and chemical bonding in iron and cobalt metal complexes of porphyrazine and tetra(1,2,5-thiadiazole)porphyrazine. J. Porphyr. Phthalocyanines 2020, 24, 1154–1164. [Google Scholar] [CrossRef]
- Donzello, M.P.; Ercolani, C.; Kadish, K.M.; Ricciardi, G.; Rosa, A.; Stuzhin, P.A. Tetrakis(thiadiazole)porphyrazines. 5. Electrochemical and DFT/TDDFT studies of the free-base macrocycle and its MgII ZnII, and CuII complexes. Inorg. Chem. 2007, 46, 4145–4157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, Z.; Sheng, N.; Jiang, J. Molecular structure, electronic structure and vibrational spectra of metal-free, N,N′-dideuterio, and magnesium tetrakis(thiadiazole)porphyrazines: Density functional calculations. J. Mol. Struct. THEOCHEM 2005, 755, 179–186. [Google Scholar] [CrossRef]
- Zhabanov, Y.A.; Tverdova, N.V.; Giricheva, N.I.; Girichev, G.V.; Stuzhin, P.A. DFT Study of molecular and electronic structure of magnesium (II) tetra(1,2,5-chalcogenadiazolo) porphyrazines, [TXDPzMg] (X = O, S, Se, Te). J. Porphyr. Phthalocyanines 2017, 21, 439–452. [Google Scholar] [CrossRef]
- Otlyotov, A.A.; Ryzhov, I.V.; Kuzmin, I.A.; Zhabanov, Y.A.; Mikhailov, M.S.; Stuzhin, P.A. DFT Study of Molecular and Electronic Structure of Ca(II) and Zn(II) Complexes with Porphyrazine and tetrakis(1,2,5-thiadiazole)porphyrazine. Int. J. Mol. Sci. 2020, 21, 2923. [Google Scholar] [CrossRef]
- Cai, X.; Zhang, Y.; Zhang, X.; Jiang, J. Structures and properties of metal-free and copper tetrakis(thiadiazole) porphyrazine and metal-free tetrakis(selenodiazole) porphyrazine based on density functional theory calculations. J. Mol. Struct. THEOCHEM 2007, 812, 63–70. [Google Scholar] [CrossRef]
- Tverdova, N.V.; Giricheva, N.I.; Savelyev, D.S.; Mikhailov, M.S.; Vogt, N.; Koifman, O.I.; Stuzhin, P.A.; Girichev, G.V. Molecular structure of tetrakis(1,2,5-thiadiazolo)- porphyrazinatozinc(II) in gaseous phase. Macroheterocycles 2017, 10, 27–30. [Google Scholar] [CrossRef][Green Version]
- Strenalyuk, T.; Samdal, S.; Volden, H.V. Molecular structures of chloro(phthalocyaninato)-aluminum(III) and -gallium(III) as determined by gas electron diffraction and quantum chemical calculations: Quantum chemical calculations on fluoro(phthalocyaninato)-aluminum(III) and -gallium(III), chloro(tetrakis(1,2,5-thiadiazole) porphyrazinato)-aluminum(III) and -gallium(III) and comparison with their X-ray structures. J. Phys. Chem. A 2008, 112, 9075–9082. [Google Scholar]
- Tarakanova, E.N.; Hamdoush, M.; Eroshin, A.V.; Ryzhov, I.V.; Zhabanov, Y.A.; Stuzhin, P.A. Tetra(1,2,5-thiadiazolo)porphyrazines. 10. Synthesis, spectral characterization and DFT study of complexes with yttrium(III) and lutetium(III). Polyhedron 2021, 193, 114877. [Google Scholar] [CrossRef]
- Zhabanov, Y.A.; Ryzhov, I.V.; Kuzmin, I.A.; Eroshin, A.V.; Stuzhin, P.A. DFT Study of Molecular and Electronic Structure of Y, La and Lu Complexes with Porphyrazine and Tetrakis(1,2,5-thiadiazole)porphyrazine. Molecules 2020, 26, 113. [Google Scholar] [CrossRef]
- Hochmuth, D.H.; Michel, S.L.J.; White, A.J.P.; Williams, D.J.; Barrett, A.G.M.; Hoffman, B.M. Ci Symmetric and Non-Centrosymmetric Crystalline Complexes of [60]Fullerene with Octakis(dimethylamino)porphyrazinato-Copper(II) and -Nickel(II). Eur. J. Inorg. Chem. 2000, 2000, 593–596. [Google Scholar] [CrossRef]
- Eroshin, A.V.; Otlyotov, A.A.; Zhabanov, Y.A.; Veretennikov, V.V.; Islyaikin, M.K. Complexes of Ca(II), Ni(II) and Zn(II) with hemi- and dicarbahemiporphyrazines: Molecular structure and features of metal-ligand bonding. Macroheterocycles 2021. [Google Scholar] [CrossRef]
- Bethe, H. Termaufspaltung in Kristallen. Ann. Phys. 1929, 395, 133–208. [Google Scholar] [CrossRef]
- Van Vleck, J.H. Theory of the variations in paramagnetic anisotropy among different salts of the iron group. Phys. Rev. 1932, 41, 208–215. [Google Scholar] [CrossRef]
- Gouterman, M. Spectra of porphyrins. J. Mol. Spectrosc. 1961, 6, 138–163. [Google Scholar] [CrossRef]
- Gouterman, M.; Wagnière, G.H.; Snyder, L.C. Spectra of porphyrins. Part II. Four orbital model. J. Mol. Spectrosc. 1963, 11, 108–127. [Google Scholar] [CrossRef]
- Weiss, C.; Kobayashi, H.; Gouterman, M. Spectra of porphyrins. Part III. Self-consistent molecular orbital calculations of porphyrin and related ring systems. J. Mol. Spectrosc. 1965, 16, 415–450. [Google Scholar] [CrossRef]
- Stuzhin, P.A.; Ivanova, S.S.; Hamdoush, M.; Kirakosyan, G.A.; Kiselev, A.; Popov, A.; Sliznev, V.; Ercolani, C. Tetrakis(1,2,5-thiadiazolo)porphyrazines. 9. Synthesis and spectral and theoretical studies of the lithium(i) complex and its unusual behaviour in aprotic solvents in the presence of acids. Dalt. Trans. 2019, 48, 14049–14061. [Google Scholar] [CrossRef]
- Zhabanov, Y.A.; Zakharov, A.V.; Giricheva, N.I.; Shlykov, S.A.; Koifman, O.I.; Girichev, G. V To the limit of gas-phase electron diffraction: Molecular structure of magnesium octa(m-trifluoromethylphenyl)porphyrazine. J. Mol. Struct. 2015, 1092, 104–112. [Google Scholar] [CrossRef]
- Bauer, E.M.; Cardarilli, D.; Ercolani, C.; Stuzhin, P.A.; Russo, U. Tetrakis(thiadiazole)porphyrazines. 2. Metal complexes with Mn(II), Fe(II), Co(II), Ni(II), and Zn(II). Inorg. Chem. 1999, 38, 6114–6120. [Google Scholar] [CrossRef]
- Pelipets, O.V. The Thermodynamics of Evaporation of ErCl3, EuBr and EuCl2, and the Molecular Species Structure: High Temperature Mass Spectrometry and Gas-Phase Electron Diffraction (in Russian); Ivanovo State University: Ivanovo, Russia, 2000. [Google Scholar]
- Tyunina, V.V.; Krasnov, A.V.; Badelin, V.G.; Girichev, G.V. Enthalpy of sublimation of hydroxyl-containing amino acids: Knudsen’s effusion mass spectrometric study. J. Chem. Thermodyn. 2016, 98, 62–70. [Google Scholar] [CrossRef]
- Jensen, F. Unifying general and segmented contracted basis sets. segmented polarization consistent basis sets. J. Chem. Theory Comput. 2014, 10, 1074–1085. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019. [Google Scholar] [CrossRef] [PubMed]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis set exchange: A community database for computational sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef]
- Jensen, K.P.; Roos, B.O.; Ryde, U. Performance of density functionals for first row transition metal systems. J. Chem. Phys. 2007, 126, 014103. [Google Scholar] [CrossRef]
- Granovsky, A.A. Firefly version 8. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 15 April 2021).
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Zhurko, G.A.; Zhurko, D.A. ChemCraft Version 1.6 (Build 312); Version 1.6 (Build 312) Ed. Available online: http://www.chemcraftprog.com/index.html (accessed on 28 December 2020).
- Vishnevskiy, Y.V.; Zhabanov, Y.A. New implementation of the first-order perturbation theory for calculation of interatomic vibrational amplitudes and corrections in gas electron diffraction. J. Phys. Conf. Ser. 2015, 633, 12076. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
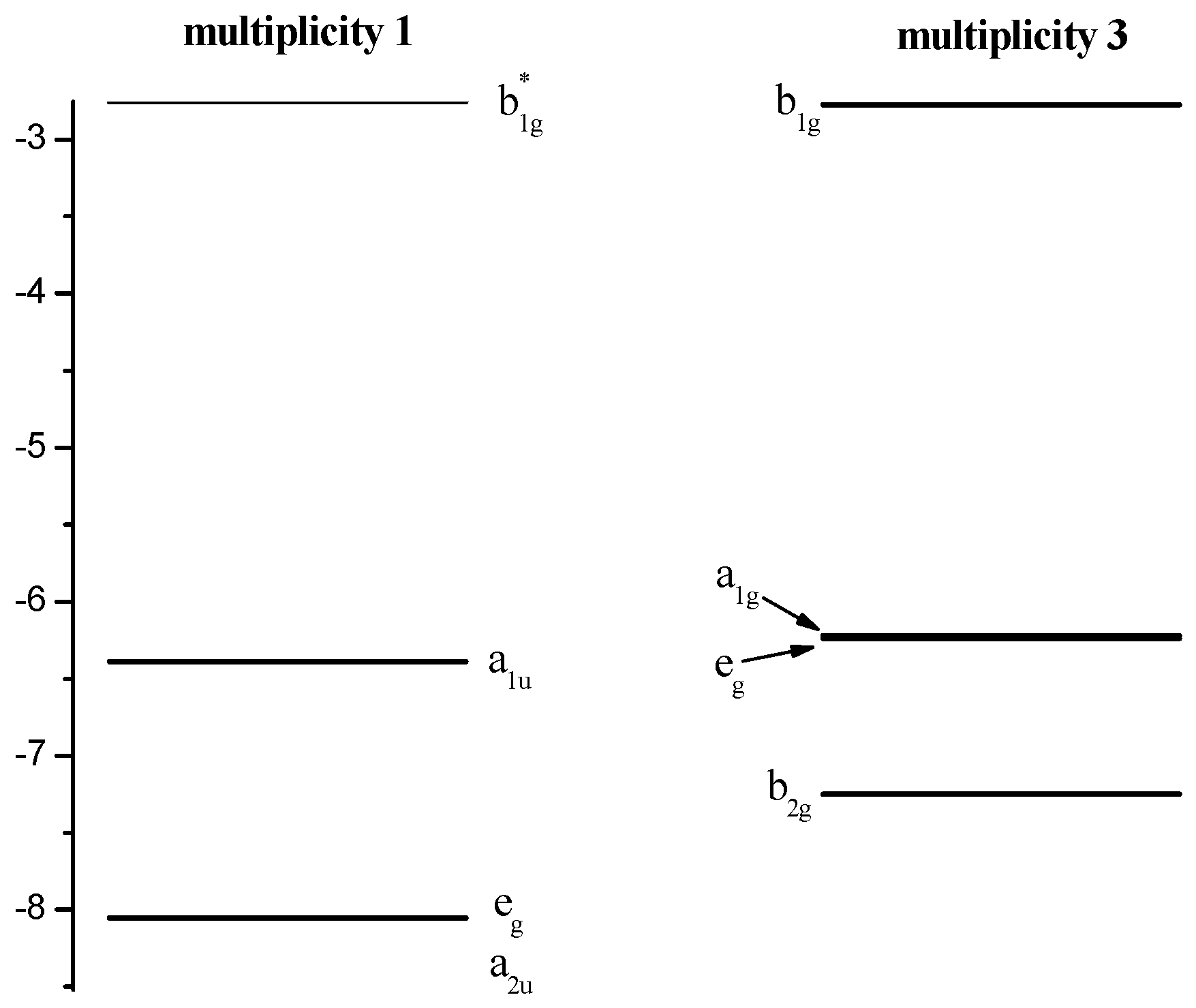{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion | m/z | Intensity, % |
|---|---|---|
| H2TTDPz+ | 546 | 11 |
| C4N4S+ | 136 | 38 |
| C2N2S+ | 84 | 100 |
| S2+ | 64 | 32 |
| C2N2+ | 52 | 25 |
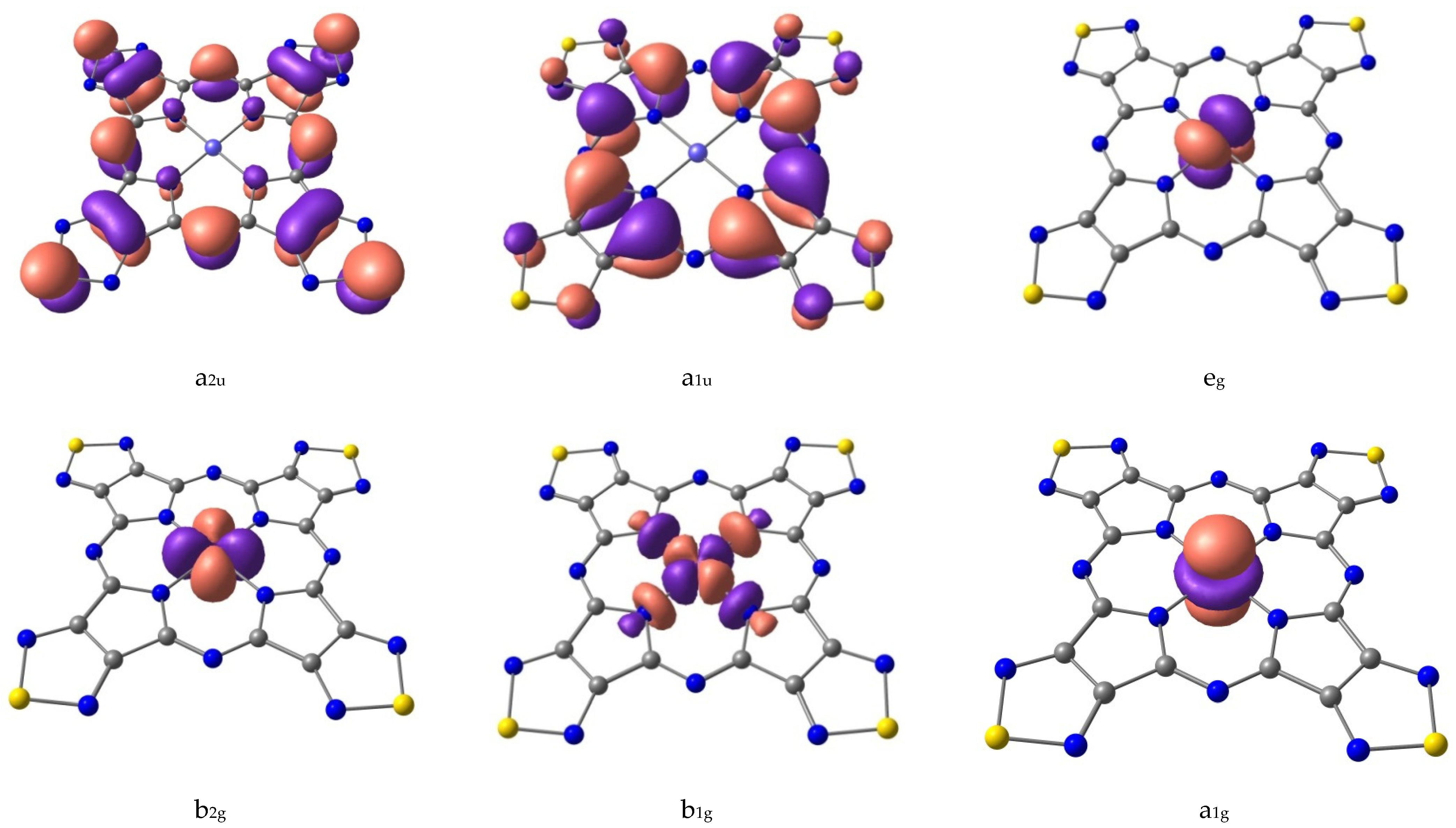
| State | Contributions | ΔE, kJ/mol |
|---|---|---|
| 1A1g | 96/(a2u)2(a1u)2(eg)4(b1g)0/ | 0.0 |
| 1Eg | 99/(a2u)2(a1u)2(eg)3(b1g)1/ | 263.0 |
| 1Eu | 97/(a2u)2(a1u)2(eg)2(b1g)2/ | 578.1 |
| 1A1g | 97/(a2u)2(a1u)2(eg)2(b1g)2/ | 581.5 |
| 1Eu | 96/(a2u)2(a1u)2(eg)2(b1g)2/ | 737.2 |
| 3B1g | 100/(b2g)2(eg)4(a1g)1(b1g)1/ | 82.3 |
| 3Eg | 93/(b2g)2(eg)3(a1g)2(b1g)1/ | 92.6 |
| 3A2g | 30/(b2g)2(eg)2(a1g)2(b1g)2/+70/(b2g)1(eg)4(a1g)2(b1g)1/ | 174.8 |
| 3Eg | 32/(b2g)1(eg)3(a1g)2(b1g)2/+47/(b2g)1(eg)3(a1g)2(b1g)2/+16/(b2g)2(eg)3(a1g)1(b1g)2/ | 331.0 |
| 3B2g | 100/(b2g)1(eg)4(a1g)1(b1g)2/ | 338.5 |
| H2TTDPz PBE0/Pcseg-2 | NiTTDPz (1A1g) PBE0/Pcseg-2 | NiTTDPz (3B1g) PBE0/Pcseg-2 | H2TTDPz [15] X-ray * | NiTTDPz [15] X-ray * | |
|---|---|---|---|---|---|
| Np-M/Np’-M | 2.233/1.011 | 1.927 | 1.976 | 2.281/0.891 | 1.922 |
| Np-Cα/Np’-Cα’ | 1.359/1.375 | 1.374 | 1.364 | 1.371/1.382 | 1.385 |
| Cα-Nm/Cα’-Nm | 1.320/1.301 | 1.303 | 1.313 | 1.331/1.311 | 1.313 |
| Cα-Cβ/Cα’-Cβ’ | 1.459/1.447 | 1.446 | 1.464 | 1.464/1.445 | 1.449 |
| Cβ-Cβ/Cβ’-Cβ’ | 1.408/1.416 | 1.401 | 1.407 | 1.400/1.408 | 1.396 |
| Cβ-Nt/Cβ’-Nt’ | 1.313/1.317 | 1.317 | 1.310 | 1.326/1.329 | 1.326 |
| Nt-S/Nt’-S’ | 1.631/1.622 | 1.626 | 1.634 | 1.646/1.631 | 1.640 |
| (Np…Np)/(Np’…Np’) | 3.958/4.092 | 3.854 | 3.952 | 3.952/4.062 | 3.844 |
| (Np…Np’) | 2.847 | 2.725 | 2.794 | 2.837 | 2.718 |
| ∠ (MNp’Cα) | 122.7 | 125.7 | 124.9 | 122.8 | 125.9 |
| ∠ (NpCαNm)/(Np’Cα’Nm) | 128.2/128.8 | 128.4 | 128.2 | 128.1/129.8 | 128.7 |
| ∠ (CαNmCα’) | 125.0 | 122.0 | 123.8 | 123.5 | 120.7 |
| ∠ (CαNpCα)/(Cα’NpCα’) | 109.3/114.7 | 108.7 | 110.2 | 108.7/113.6 | 108.1 |
| ∠ (NtSNt)/(Nt’S’Nt’) | 100.7/101.3 | 101.1 | 100.4 | 100.5/101.3 | 100.6 |
| State | Composition (%) | λ (nm) | f | Exp λ (nm) |
|---|---|---|---|---|
| 11B2u | 583 | 0.32 | 650 | |
| 11B3u | 556 | 0.28 | 635 | |
| 21B3u | 393 | 0.07 | ||
| 41B3u | 316 | 1.21 | 335 | |
| 41B2u | 312 | 0.18 | ||
| 51B3u | 298 | 0.15 | ||
| 51B2u | 296 | 1.03 | ||
| 81B2u | 242 | 0.57 | ||
| 81B3u | 237 | 0.31 | ||
| 91B3u | 234 | 0.11 | ||
| 111B3u | 215 | 0.08 |
| State | Composition (%) | λ (nm) | f | Exp λ (nm) |
|---|---|---|---|---|
| 11Eu | (93) | 564 | 0.28 | 627 |
| 21Eu | (94) | 367 | 0.02 | |
| 31Eu | (7) (86) | 333 | 0.02 | |
| 41Eu | (8) (78) (8) | 311 | 0.55 | 362 |
| 51Eu | (76) (8) | 294 | 0.19 | |
| 61Eu | (97) | 269 | 0.31 | |
| 71Eu | (6) (89) | 257 | 0.26 | |
| 81Eu | (5) (88) | 244 | 0.07 | |
| 91Eu | (83) (7) (7) | 240 | 0.34 | |
| 101Eu | (16) (69) | 231 | 0.05 | |
| 131Eu | (13) (11) (48) (10) (5) (10) | 217 | 0.07 |
| Frequency, cm−1 | Irel, % | Symmetry | Assignment * | Exp, cm−1 |
|---|---|---|---|---|
| H2TTDPz | ||||
| 535 (ω34) | 12 | B3u | OPB (Nt-Cα-Cβ-Cβ) (23), θ (Cβ-Nt-S-Nt) (36) | 518 |
| 607 (ω44) | 14 | B2u | r(Cα-Cβ) (17), r(Nt-S) (11), φ(Cβ-Nt-S) (18), φ(Nt-S-Nt) (19) | 587 |
| 686 (ω44) | 29 | B1u | φ(Np-Cα-Cβ) (7), φ(Cα-Np’-Cα) (6), φ(Nm-Cα-Cβ) (10), φ(Cα-Cβ-Cβ) (8), φ(Cβ-Cβ-Nt) (8), φ(Cβ-Nt-S) (6) | 666 |
| 687 (ω45) | 22 | B2u | φ(Cα-Np-Cα) (6), φ(Np-Cα-Cβ) (10), φ(Np’-Cα-Cβ) (8), φ(Nm-Cα-Cβ) (11), φ(Cα-Cβ-Cβ) (10), φ(Cβ-Cβ-Nt) (8), φ(Cβ-Nt-S) (6) | 666 |
| 785 (ω54) | 10 | B2u | r(Nt-S) (5), φ(Np-Cα-Nm) (5), φ(Np’-Cα-Nm) (13), φ(Cα-Np’-H) (10),φ(Cα-Nm-Cα) (12), φ(Nm-Cα-Cβ) (11), | 753 |
| 817 (ω59) | 24 | B3u | OPB (H-Cα-Cα-Np’) (24), OPB (Cβ-Np-Nm-Cα) (11),θ (Cα-H-Np-Cα-Nm-Cβ) (24), θ (Cα-Nm-Cα-Np-Cβ) (8), θ (Cα-Nm-Cα-Np’-Cβ) (8) | 817 |
| 1083 (ω75) | 100 | B1u | r(Np-Cα) (13), r(Nm-Cα) (12), r(Cα-Cβ) (8), φ(Np-Cα-Cβ) (8), φ(Nm-Cα-Cβ) (8), φ(Cα-Cβ-Cβ) (8), φ(Cβ-Cβ-Nt) (6) | 1019 |
| 1171 (ω78) | 87 | B2u | r(Np’-Cα) (9), r(Nm-Cα) (6), r(Cα-Cβ) (7), φ(Cα-Np’-H) (14), φ(Np’-Cα-Nm) (5), φ(Nm-Cα-Cβ) (8), φ(Cα-Nm-Cα) (6), φ(Cβ-Cβ-Nt) (7), φ(Cβ-Nt-S) (6) | 1133 |
| 1264 (ω81) | 42 | B2u | r(Np-Cα) (5), r(Np’-Cα) (6), φ(Cα-Np-Cα) (6), φ(Np-Cα-Nm) (8), φ(Cα-Np’-H) (26) | 1217 |
| 1310 (ω85) | 56 | B1u | r(Np’-Cα) (5), r(Cα-Cβ) (7), r(Cβ-Cβ) (5), φ(Np’-Cα-Nm) (7), φ(Np-Cα-Nm) (7), φ(Cα-Cβ-Nt) (9) | 1263 |
| 1348 (ω86) | 19 | B2u | r(Np-Cα) (9), φ(Cα-Np-Cα) (5), φ(Np-Cα-Nm) (11), φ(Cα-Np’-H) (28), φ(Nm-Cα-Cβ) (9), φ(Cα-Nm-Cα) (7) | 1288 |
| 1381 (ω88) | 14 | B2u | r(Cα-Cβ) (7), r(Cβ-Cβ) (7), φ(Cα-Np’-H) (15), φ(Cα-Cβ-Nt) (9), φ(Cβ-Cβ-Nt) (7), φ(Cβ-Nt-S) (6) | 1340 |
| 1577 (ω98) | 9 | B1u | r(Np-Cα) (5), r(Nm-Cα) (53), r(Cα-Cβ) (9), r(Cβ-Nt) (12) | 1506 |
| 1611 (ω103) | 12 | B1u | r(Nm-Cα) (5), r(Cα-Cβ) (15), r(Cβ-Nt) (13), φ(Cα-Cβ-Cβ) (14), (Cβ-Cβ-Nt) (14) | 1532 |
| 1637 (ω104) | 7 | B2u | r(Nm-Cα) (34), r(Cα-Cβ) (23), r(Cβ-Nt) (16), φ(Cα-Np’-H) (6) | 1566 |
| 3554 (ω107) | 45 | B1u | r(Np’-H) (69), r(Np’-Cα) (5), φ(Cα-Np’-Cα) (6), φ(Cα-Np’-H) (6), φ(Cα-Np’-Nm) (6), φ(Np’-Cα-Cβ) (6) | 3291 |
| NiTTDPz | ||||
| 533 (ω35) | 7 | A2u | θ (Nm-Cα-Cβ-Nt) (29), θ (Cβ-Nt-S-Nt) (22), θ(Cα-Nm-Cα-Np-Cβ) (32) | 511 |
| 711 (ω46–ω47) | 33 | Eu | r(Np-Ni) (10), r(Nm-Cα) (12), r(Cα-Cβ) (5), φ(Cα-Np-Cα) (7), φ(Np-Cα-Cβ) (14), φ(Nm-Cα-Cβ) (11), φ(Cα-Cβ-Cβ) (6), φ(Cβ-Cβ-Nt) (6), φ(Cβ-Nt-S) (7) | 689 |
| 788 (ω55) | 3 | A2u | θ(Cα-Nm-Cα-Np-Cβ) (43), θ (Nm-Cα-Cβ-Nt) (45) | 741 |
| 818 (ω59–ω60) | 2 | Eu | r(Nt-S) (44), φ(Cα-Nm-Cα) (7), φ(Np-Cα-Nm) (8), φ(Np-Cα-Cβ) (8), φ(Nt-S-Nt) (5) | 763 |
| 843 (ω62–ω63) | 11 | Eu | r(Nt-S) (75), φ(Cβ-Cβ-Nt) (5), φ(Cβ-Nt-S) (10) | 827 |
| 921 (ω71–ω72) | 8 | Eu | r(Np-Ni) (5), r(Cβ-Nt) (5), r(Nt-S) (12), φ(Cα-Nm-Cα) (12), φ(Np-Cα-Nm) (8), φ(Nm-Cα-Cβ) (11), φ(Cα-Cβ-Nt) (8), φ(Cβ-Cβ-Nt) (6), φ(Cβ-Nt-S) (9) | 895 |
| 1161 (ω77–ω78) | 100 | Eu | r(Np-Cα) (41), r(Nm-Cα) (14), r(Cα-Cβ) (14), r(Cβ-Nt) (6) | 1108/1109 |
| 1325(ω84–ω85) | 83 | Eu | r(Np-Cα) (25), r(Np-Ni) (5), r(Cα-Cβ) (5), r(Cβ-Cβ) (6), r(Cβ-Nt) (17), φ(Cα-Np-Cα) (6), φ(Np-Cα-Nm) (9), φ(Np-Cα-Cβ) (6) | 1269 |
| 1396 (ω86–ω87) | 11 | Eu | r(Np-Cα) (12), r(Nm-Cα) (15), r(Cα-Cβ) (27), r(Cβ-Cβ) (20) | 1347 |
| 1629 (ω102–ω103) | 20 | Eu | r(Nm-Cα) (33), r(Cα-Cβ) (25), r(Cβ-Nt) (20) | 1552 |
| Frequency, cm−1 | Irel, % | Symmetry | Assignment * | Exp, cm−1 |
|---|---|---|---|---|
| H2TTDPz | ||||
| 223 (ω15) | 5 | Ag | r (Nm-Cα) (14), r (Cα-Cβ) (25), φ (Cα-Nm-Cα) (23) | 223 |
| 518 (ω27) | 5 | B3g | r (Nm-Cα) (10), r(Cβ-Nt) (7), r (Nt-S) (9), φ (Np-Cα-Cβ) (9), φ (Cα-Cβ-Cβ) (24), φ (Cα-Cβ-Nt) (13) | 576/504 |
| 714 (ω47) | 10 | Ag | r (Np-Cα) (12), r (Nm-Cα) (12), φ (Cα-Nm-Cα) (31) | 697 |
| 733 (ω49) | 10 | Ag | r (Nm-Cα) (11), φ (Cα-Np-Cα) (23), φ (Np-Cα-Cβ) (24), φ (Nm-Cα-Cβ) (14) | 711 |
| 855 (ω67) | 6 | Ag | r (Cα-Cβ) (8), r(Cβ-Nt) (9), r(Nt-S) (17), φ(Cβ-Nt-S) (21), φ(Nt-S-Nt) (9) | 823 |
| 1239 (ω80) | 33 | B3g | r (Np-Cα) (49), r (Cα-Cβ) (18) | 1177/1158 |
| 1293 (ω83) | 3 | Ag | r (Np-Cα) (19), r(Cβ-Cβ) (18), r(Cβ-Nt) (11), φ (Cα-Np-Cα) (10), (Np-Cα-Nm) (10) | 1254/1237 |
| 1400 (ω90) | 34 | Ag | r (Cα-Cβ) (22), r(Cβ-Cβ) (42) | 1336 |
| 1495 (ω93) | 21 | Ag | r (Cα-Cβ) (19), r(Cβ-Nt) (60) | 1429/1443 |
| 1595 (ω99) | 12 | Ag | r (Nm-Cα) (44), r (Cα-Cβ) (30), r(Cβ-Nt) (10) | 1532 |
| 1657 (ω106) | 100 | Ag | r (Nm-Cα) (85) | 1555 |
| NiTTDPz | ||||
| 253 (ω17) | 7 | A1g | r (Np-Ni) (30), r (Nm-Cα) (7), r (Cα-Cβ) (26), φ (Cα-Nm-Cα) (12) | |
| 735 (ω50) | 10 | A1g | r (Np-Cα) (7), r (Nm-Cα) (6), r (Nt-S) (9), φ (Cα-Nm-Cα) (20), φ (Np-Cα-Nt) (8), φ(Cβ-Nt-S) (17), φ(Nt-S-Nt) (20) | 709 |
| 755 (ω51) | 11 | B1g | r (Np-Ni) (13), r (Nm-Cα) (9), r (Cα-Cβ) (6), (Cα-Np-Cα) (20), φ (Np-Cα-Cβ) (9), φ (Nm-Cα-Cβ) (20) | 800 |
| 844 (ω65) | 13 | A1g | r (Nt-S) (76), φ(Cβ-Nt-S) (10),), φ(Cβ-Cβ-Nt) (5) | 865 |
| 1068 (ω74) | 4 | A2g | r (Np-Cα) (18), r (Nm-Cα) (34), r(Cβ-Nt) (6), φ (Cα -Nm-Cα) (6), φ(Cβ-Cβ-Nt) (6), φ(Cβ-Nt-S) (7), φ(Np-Ni-Np) (5) | 1024 |
| 1242 (ω81) | 27 | B2g | r (Np-Cα) (41), r (Cα-Cβ) (26), r (Cβ-Nt) (10) | 1182 |
| 1413 (ω88) | 16 | A1g | r (Np-Cα) (19), r (Nm-Cα) (13), r (Cα-Cβ) (10), r(Cβ-Cβ) (28) | 1266 |
| 1435 (ω89) | 48 | B1g | r (Cα-Cβ) (29), r(Cβ-Cβ) (40), φ(Cα-Cβ-Nt) (9), φ(Cβ-Nt-S) (7) | 1248 |
| 1501 (ω92) | 15 | B1g | r (Cα-Cβ) (17), r(Cβ-Cβ) (9), r (Cβ-Nt) (63) | 1353 |
| 1536 (ω94) | 7 | B2g | r (Np-Cα) (11), r (Nm-Cα) (41), r(Cβ-Nt) (21), φ(Np-Cα-Cβ) (8) | 1362 |
| 1609 (ω100) | 1 | A2g | r (Nm-Cα) (71), r (Cα-Cβ) (16), r(Cβ-Nt) (5) | 1534 |
| 1634 (ω104) | 10 | A1g | r (Nm-Cα) (43), r (Cα-Cβ) (30), r(Cβ-Nt) (6) | |
| 1681 (ω105) | 100 | B1g | r (Nm-Cα) (84) | 1572 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhabanov, Y.A.; Eroshin, A.V.; Ryzhov, I.V.; Kuzmin, I.A.; Finogenov, D.N.; Stuzhin, P.A. Molecular Structure, Thermodynamic and Spectral Characteristics of Metal-Free and Nickel Complex of Tetrakis(1,2,5-thiadiazolo)porphyrazine. Molecules 2021, 26, 2945. https://doi.org/10.3390/molecules26102945
Zhabanov YA, Eroshin AV, Ryzhov IV, Kuzmin IA, Finogenov DN, Stuzhin PA. Molecular Structure, Thermodynamic and Spectral Characteristics of Metal-Free and Nickel Complex of Tetrakis(1,2,5-thiadiazolo)porphyrazine. Molecules. 2021; 26(10):2945. https://doi.org/10.3390/molecules26102945
Chicago/Turabian StyleZhabanov, Yuriy A., Alexey V. Eroshin, Igor V. Ryzhov, Ilya A. Kuzmin, Daniil N. Finogenov, and Pavel A. Stuzhin. 2021. "Molecular Structure, Thermodynamic and Spectral Characteristics of Metal-Free and Nickel Complex of Tetrakis(1,2,5-thiadiazolo)porphyrazine" Molecules 26, no. 10: 2945. https://doi.org/10.3390/molecules26102945
APA StyleZhabanov, Y. A., Eroshin, A. V., Ryzhov, I. V., Kuzmin, I. A., Finogenov, D. N., & Stuzhin, P. A. (2021). Molecular Structure, Thermodynamic and Spectral Characteristics of Metal-Free and Nickel Complex of Tetrakis(1,2,5-thiadiazolo)porphyrazine. Molecules, 26(10), 2945. https://doi.org/10.3390/molecules26102945