
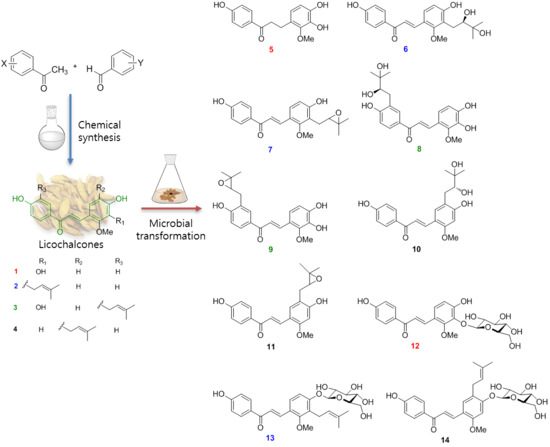
Microbial Transformation of Licochalcones


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
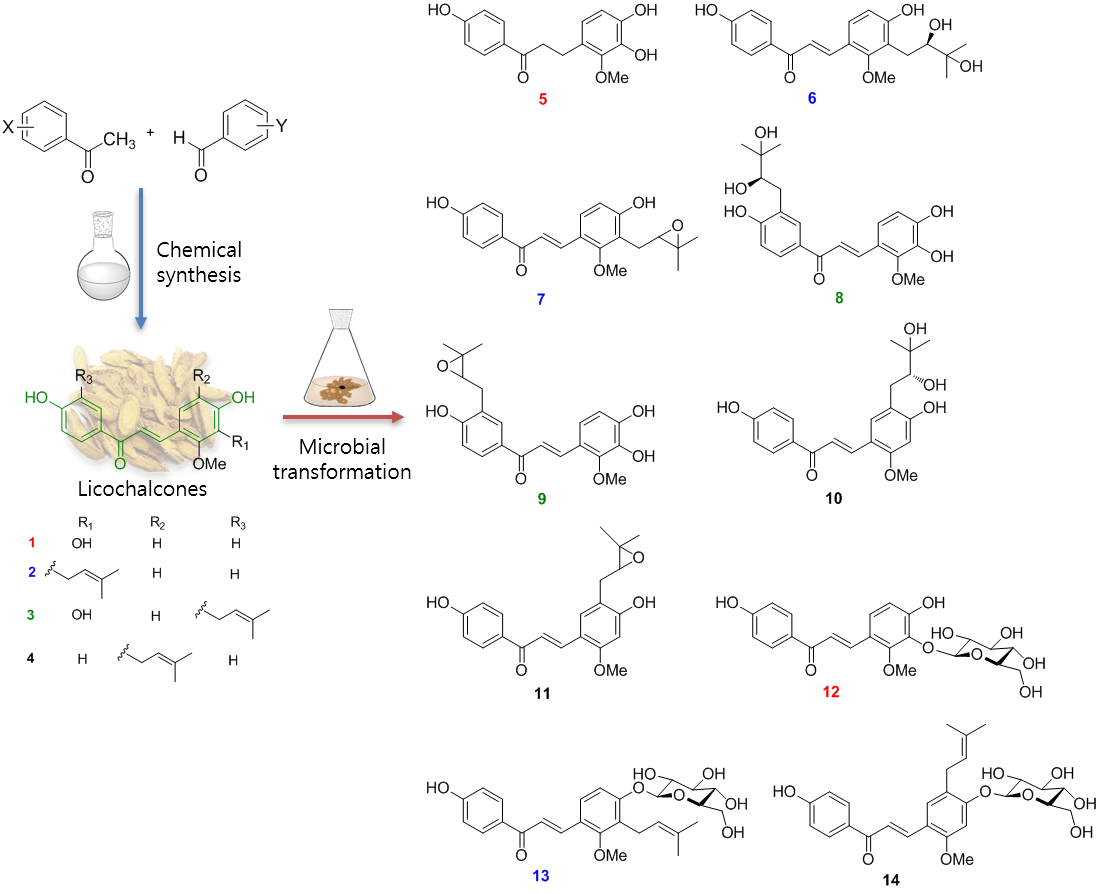
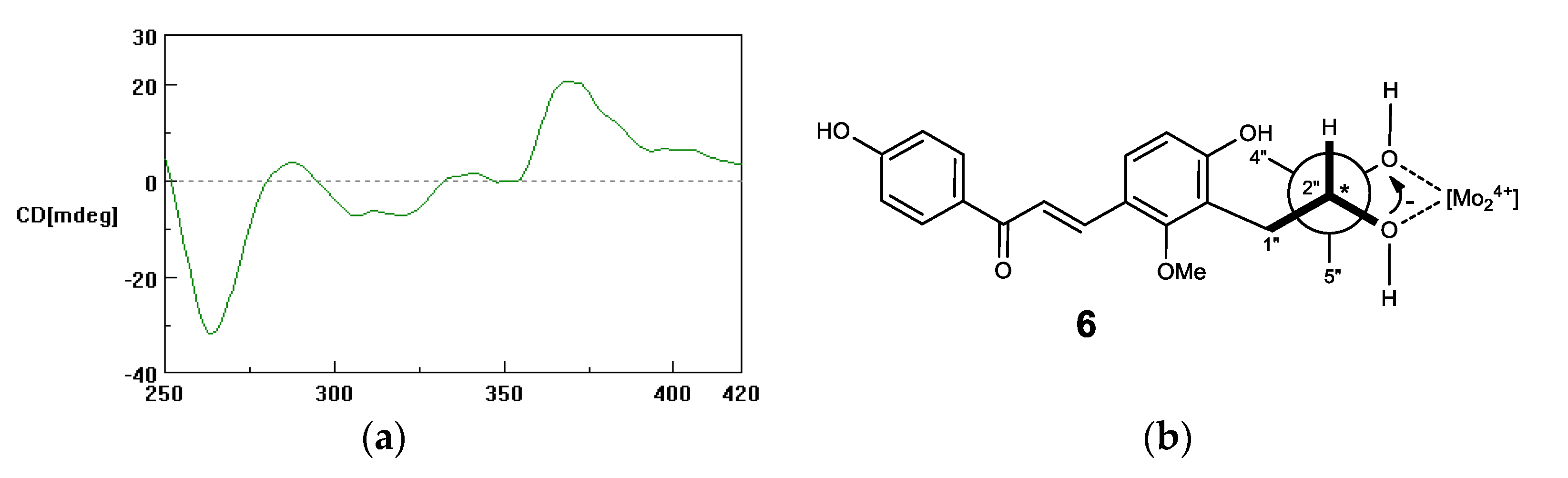
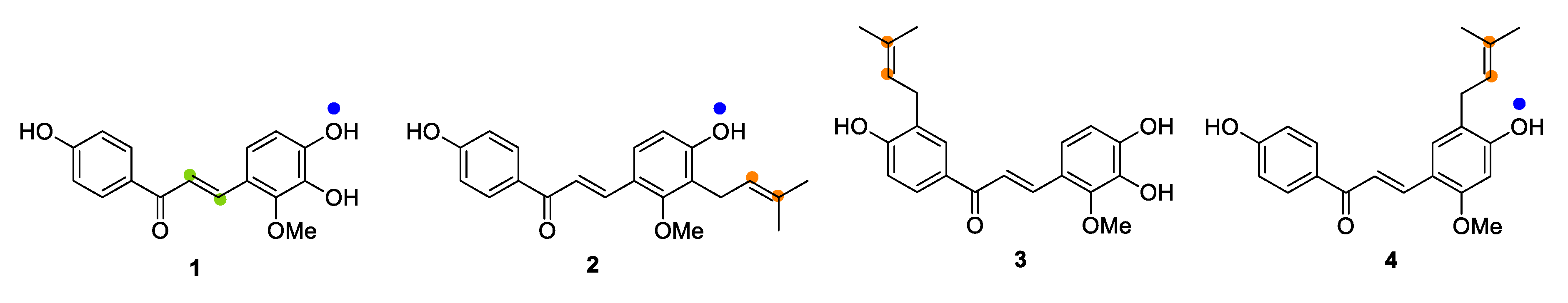
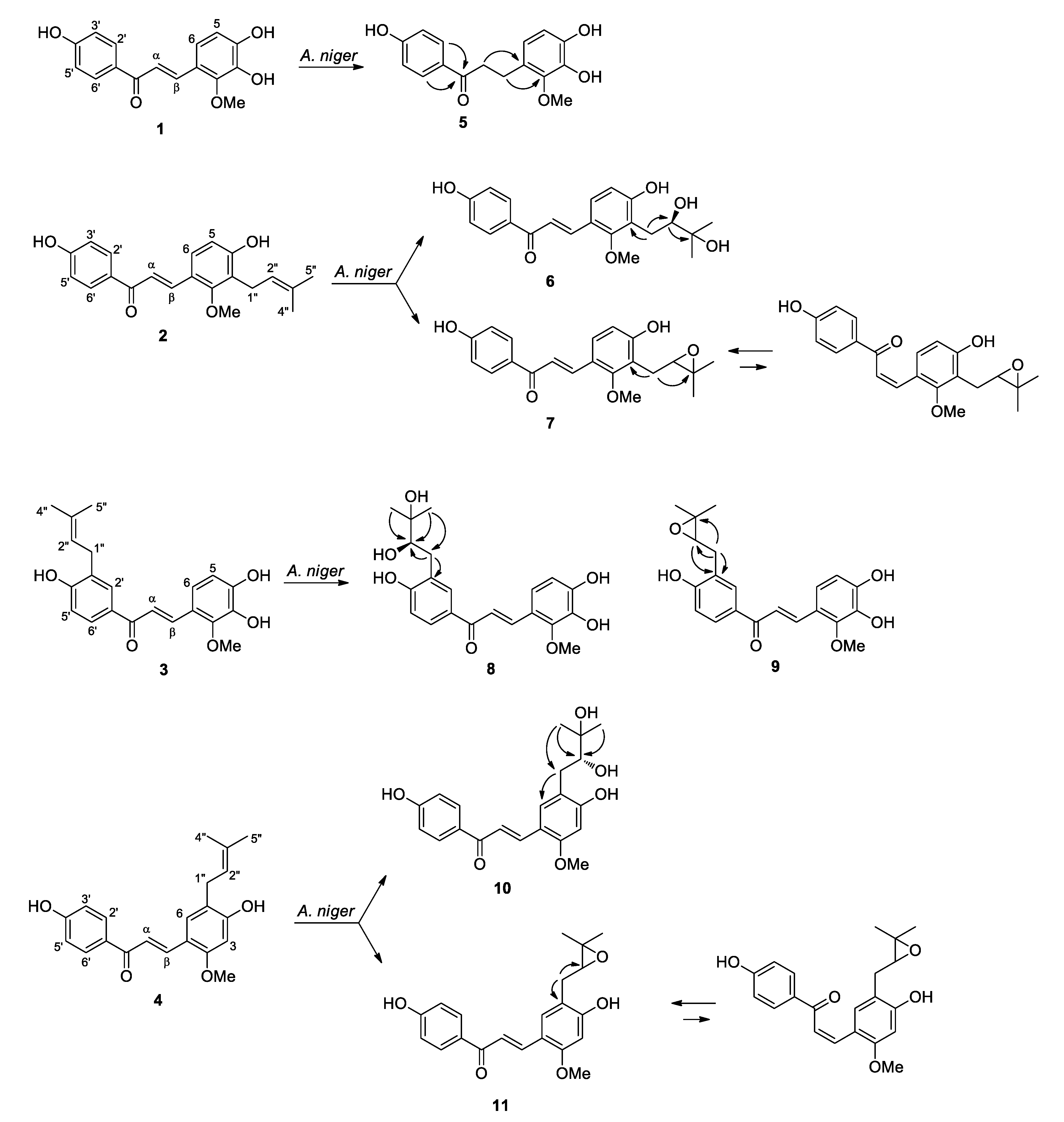
2.1. Microbial Transformation of Licochalcones B (1), C (2), D (3) and H (4) by A. niger
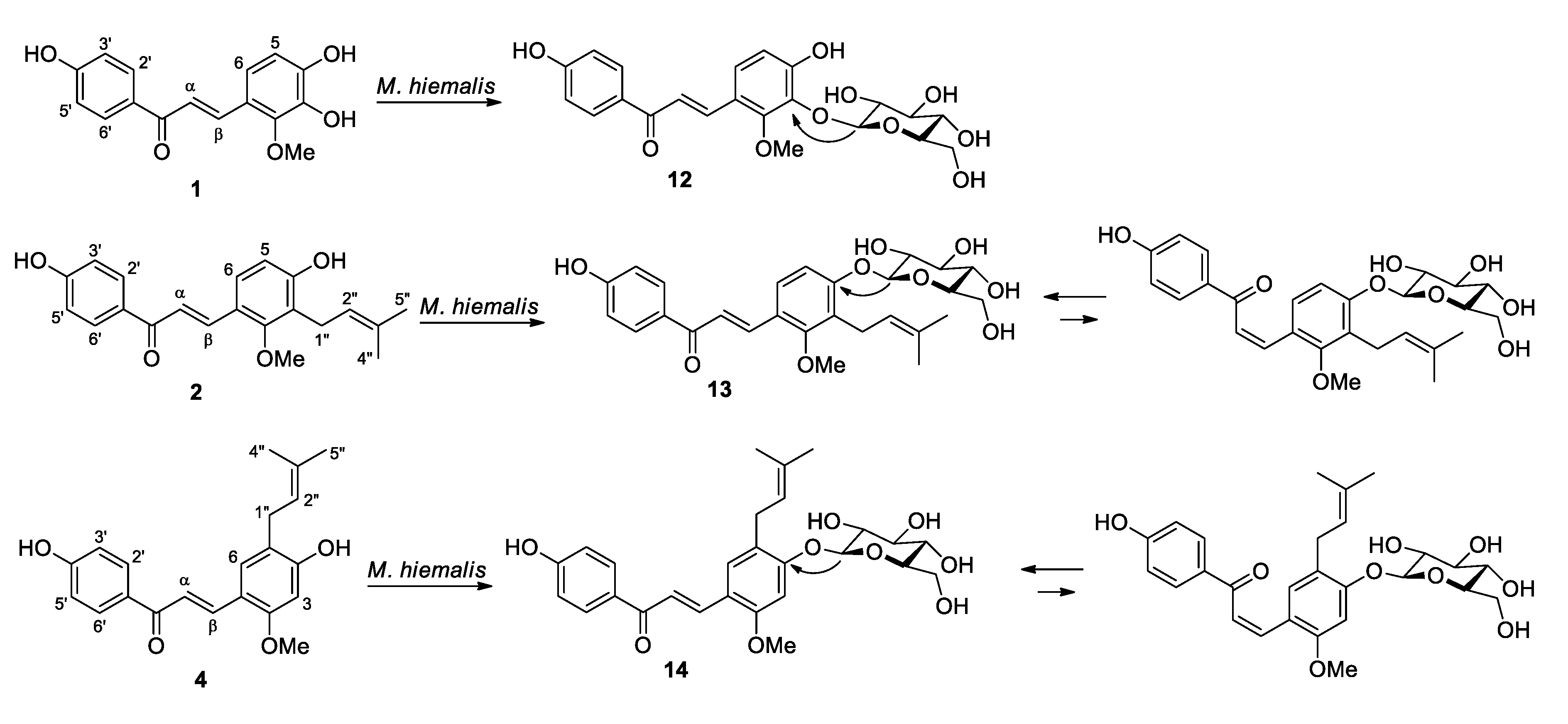
2.2. Microbial Transformation of Licochalcones B (1), C (2), D (3) and H (4) by M. hiemalis
3. Conclusions
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Preparation of Substrates
4.3. Microorganisms and Culture Media
4.4. Procedure for Microbial Transformation
4.5. Extraction and Isolation of Metabolites
4.6. Spectroscopic Data of Metabolites
4.7. Acid Hydrolysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Zhang, H.; Chen, L.; Shan, L.; Fan, G.; Gao, X. Liquorice, a unique “guide drug” of traditional Chinese medicine: a review of its role in drug interactions. J. Ethnopharmacol. 2013, 150, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Asl, M.N.; Hosseinzadeh, H. Review of pharmacological effects of Glycyrrhiza sp. and its bioactive compounds. Phytother. Res. 2008, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Wang, L.Q.; Yuan, B.C.; Liu, Y. The pharmacological activities of licorice. Planta Med. 2015, 81, 1654–1669. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ye, M. Chemical analysis of the Chinese herbal medicine Gan-Cao (licorice). J. Chromatogr. A 2009, 1216, 1954–1969. [Google Scholar] [CrossRef] [PubMed]
- Yoon, G.; Kang, B.Y.; Cheon, S.H. Topoisomerase i inhibition and cytotoxicity of licochalcones A and E from Glycyrrhiza inflata. Arch. Pharmacal Res. 2007, 30, 313–316. [Google Scholar] [CrossRef]
- Kajiyama, K.; Demizu, S.; Hiraga, Y.; Kinoshita, K.; Koyama, K.; Takahashi, K.; Tamura, Y.; Okada, K.; Kinoshita, T. Two prenylated retrochalcones from Glycyrrhiza inflata. Phytochemistry 1992, 31, 3229–3232. [Google Scholar] [CrossRef]
- Haraguchi, H.; Ishikawa, H.; Mizutani, K.; Tamura, Y.; Kinoshita, T. Antioxidative and superoxide scavenging activities of retrochalcones in Glycyrrhiza inflata. Bioorg. Med. Chem. 1998, 6, 339–347. [Google Scholar] [CrossRef]
- Xu, M.; Wu, P.; Shen, F.; Ji, J.; Rakesh, K.P. Chalcone derivatives and their antibacterial activities: Current development. Bioorg. Chem. 2019, 91, 103133. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, C.G.; Yun, S.R.; Kim, J.K.; Jun, J.G. Synthesis of licochalcone analogues with increased anti-inflammatory activity. Bioorg. Med. Chem. Lett. 2014, 24, 181–185. [Google Scholar] [CrossRef]
- Franceschelli, S.; Pesce, M.; Vinciguerra, I.; Ferrone, A.; Riccioni, G.; Antonia, P.; Grilli, A.; Felaco, M.; Speranza, L. Licocalchone-C extracted from Glycyrrhiza glabra inhibits lipopolysaccharide-interferon-γ inflammation by improving antioxidant conditions and regulating inducible nitric oxide synthase expression. Molecules 2011, 16, 5720–5734. [Google Scholar] [CrossRef]
- Kim, J.K.; Shin, E.K.; Park, J.H.; Kim, Y.H.; Park, J.H.Y. Antitumor and antimetastatic effects of licochalcone A in mouse models. J. Mol. Med. 2010, 88, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Li, T.; Xiao, E.; Zhao, H.; Li, Y.; Fu, S.; Gan, L.; Wang, Z.; Zheng, Q.; Wang, Z. Licochalcone B inhibits growth of bladder cancer cells by arresting cell cycle progression and inducing apoptosis. Food Chem. Toxicol. 2014, 65, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Yoon, G.; Kang, I.A.; Oh, H.N.; Chae, J.I.; Shim, J.H. Natural compound licochalcone B induced extrinsic and intrinsic apoptosis in human skin melanoma (A375) and squamous cell carcinoma (A431). Cells. Phytother. Res. 2017, 31, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Nho, S.H.; Yoon, G.; Seo, J.H.; Oh, H.N.; Cho, S.S.; Kim, H.; Choi, H.W.; Shim, J.H.; Chae, J.I. Licochalcone H induces the apoptosis of human oral squamous cell carcinoma cells via regulation of matrin 3. Oncol. Rep. 2019, 41, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.S.; Hsu, Y.K.; Huang, Y.F.; Chen, H.Y.; Hsieh, C.P.; Chen, C.L. Licochalcone A suppresses the proliferation of osteosarcoma cells through autophagy and ATM-Chk2 activation. Molecules 2019, 24, 2435. [Google Scholar] [CrossRef] [PubMed]
- Pia, M.; Gatta, D.; Sara, F.; Mario, F.; Lorenza, S. Biological effects of licochalcones. Mini-Rev. Med. Chem. 2019, 19, 647–656. [Google Scholar]
- Bak, E.J.; Choi, K.C.; Jang, S.; Woo, G.H.; Yoon, H.G.; Na, Y.; Yoo, Y.J.; Lee, Y.; Jeong, Y.; Cha, J.H. Licochalcone F alleviates glucose tolerance and chronic inflammation in diet-induced obese mice through Akt and p38 MAPK. Clin. Nutr. 2016, 35, 414–421. [Google Scholar] [CrossRef]
- Kwak, A.W.; Cho, S.S.; Yoon, G.; Oh, H.N.; Lee, M.H.; Chae, J.I.; Shim, J.H. Licochalcone H synthesized by modifying structure of licochalcone C extracted from Glycyrrhiza inflata induces apoptosis of esophageal squamous cell carcinoma cells. Cell Biochem. Biophys. 2019. [Google Scholar] [CrossRef]
- Zhang, X.; Rakesh, K.P.; Bukhari, S.N.A.; Balakrishna, M.; Manukumar, H.M.; Qin, H.L. Multi-targetable chalcone analogs to treat deadly Alzheimer’s disease: Current view and upcoming advice. Bioorg. Chem. 2018, 80, 86–93. [Google Scholar] [CrossRef]
- Loughlin, W.A. Biotransformations in organic synthesis. Bioresour. Technol. 2000, 74, 49–62. [Google Scholar] [CrossRef]
- Clark, A.M.; Hufford, C.D. Use of microorganisms for the study of drug metabolism: an update. Med. Res. Rev. 1991, 11, 473–501. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gonzalez, M.; Rosazza, J.P.N. Microbial transformations of chalcones: hydroxylation, O-demethylation, and cyclization to flavanones. J. Nat. Prod. 2004, 67, 553–558. [Google Scholar] [CrossRef]
- Kim, H.J.; Yim, S.H.; Han, F.; Kang, B.Y.; Choi, H.J.; Jung, D.W.; Williams, D.R.; Gustafson, K.R.; Kennelly, E.J.; Lee, I.S. Biotransformed metabolites of the hop prenylflavanone isoxanthohumol. Molecules 2019, 24, 394. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, Y.; Paudel, S.; Yoon, G.; Cheon, S.H. Concise synthesis of licochalcone C and its regioisomer, licochalcone H. Arch. Pharmacal Res. 2013, 36, 1432–1436. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, H.; Snatzke, G.; Atta-ur-Rahman. The CD in situ complexation method as a tool for determination of absolute configurations of Cottonogenic derivatives. J. Am. Chem. Soc. 1993, 115, 12533–12544. [Google Scholar] [CrossRef]
- Bari, L.D.; Pescitelli, G.; Pratelli, C.; Pini, D.; Salvadori, P. Determination of absolute configuration of acyclic 1,2-diols with Mo2(OAc)4. 1. Snatzke’s method revisited. J. Org. Chem. 2001, 66, 4819–4825. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, L.; Qin, B.; Zhang, X.; Jia, X.; Wang, X.; Jin, D.; You, S. An insight into the curdione biotransformation pathway by Aspergillus niger. Nat. Prod. Res. 2014, 28, 454–460. [Google Scholar] [CrossRef]
- Rozmer, Z.; Perjési, P. Naturally occurring chalcones and their biological activities. Phytochem. Rev. 2016, 15, 87–120. [Google Scholar] [CrossRef]
- Simmler, C.; Lankin, D.C.; Nikolić, D.; van Breemen, R.B.; Pauli, G.F. Isolation and structural characterization of dihydrobenzofuran congeners of licochalcone A. Fitoterapia 2017, 121, 6–15. [Google Scholar] [CrossRef]
- Huang, L.; Nikolic, D.; van Breemen, R.B. Hepatic metabolism of licochalcone A, a potential chemopreventive chalcone from licorice (Glycyrrhiza inflata), determined using liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2017, 409, 6937–6948. [Google Scholar] [CrossRef]
- Liu, J.H.; Yu, B.Y. Biotransformation of bioactive natural products for pharmaceutical lead compounds. Curr. Org. Chem. 2010, 14, 1400–1406. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Z.; Cao, Y.; Paudel, S.; Yoon, G.; Cheon, S.H. Short and efficient synthesis of licochalcone B and D through acid-mediated Claisen-Schmidt condensation. Bull. Korean Chem. Soc. 2013, 34, 3906–3908. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Y.; Han, F.; Lee, I.-S. Microbial Transformation of Licochalcones. Molecules 2020, 25, 60. https://doi.org/10.3390/molecules25010060
Xiao Y, Han F, Lee I-S. Microbial Transformation of Licochalcones. Molecules. 2020; 25(1):60. https://doi.org/10.3390/molecules25010060
Chicago/Turabian StyleXiao, Yina, Fubo Han, and Ik-Soo Lee. 2020. "Microbial Transformation of Licochalcones" Molecules 25, no. 1: 60. https://doi.org/10.3390/molecules25010060
APA StyleXiao, Y., Han, F., & Lee, I.-S. (2020). Microbial Transformation of Licochalcones. Molecules, 25(1), 60. https://doi.org/10.3390/molecules25010060