
Synthesis of Human Milk Oligosaccharides: Protein Engineering Strategies for Improved Enzymatic Transglycosylation




Abstract
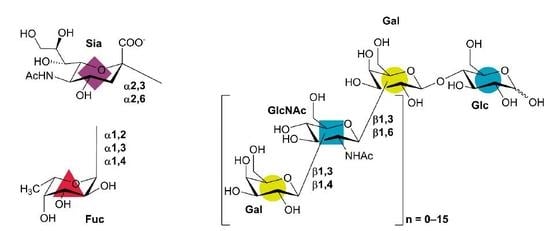
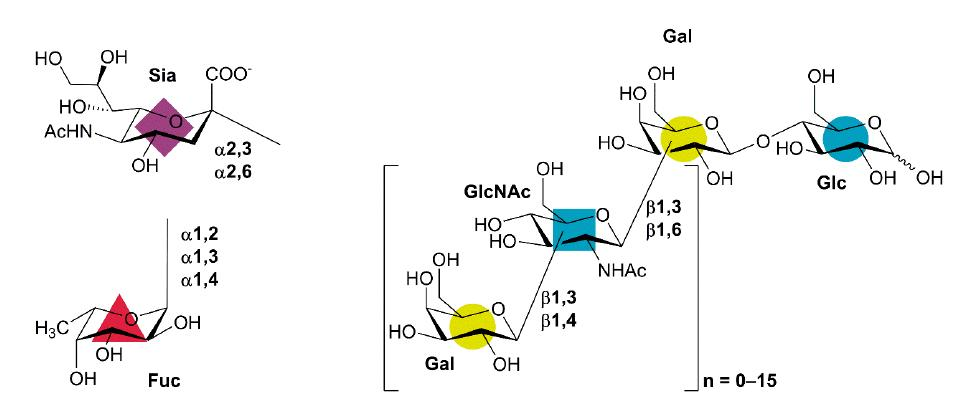
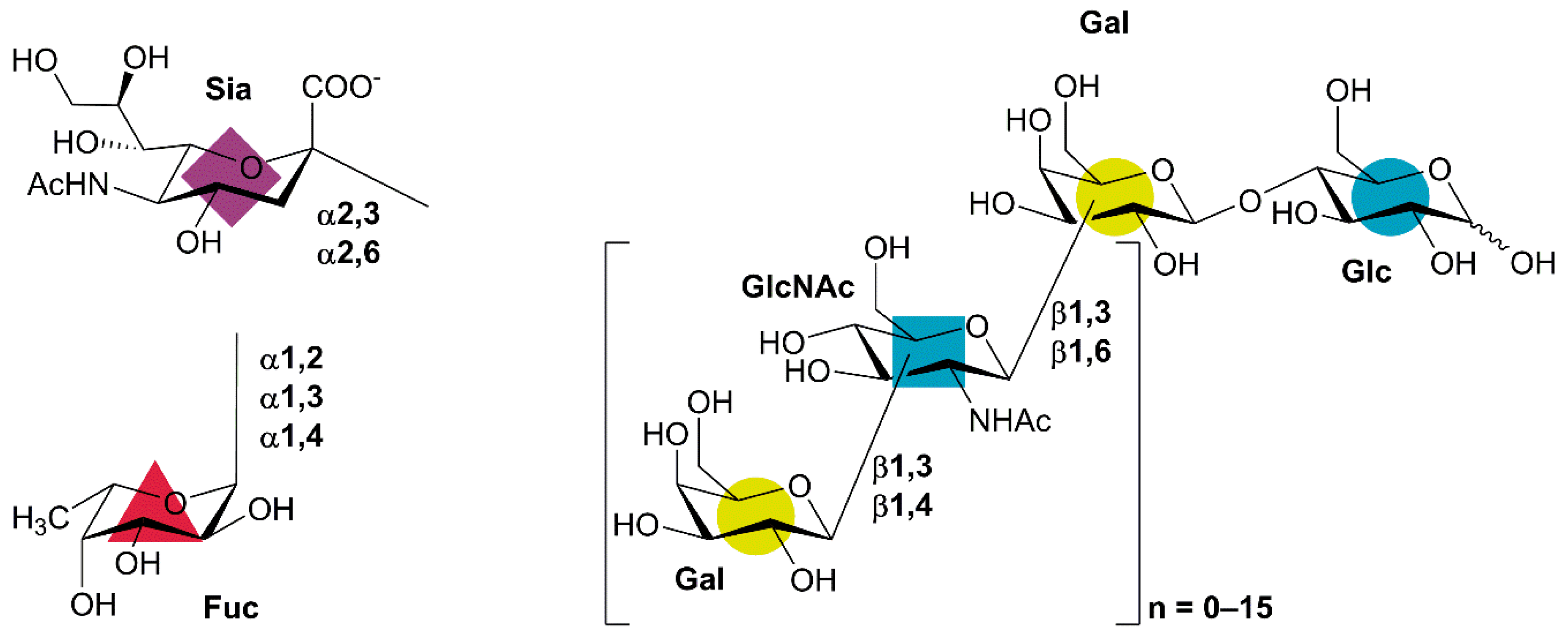
1. Introduction
2. Routes to HMO Production Outside the Mammary Gland
2.1. Microbial Cell Factories
2.2. Chemical Synthesis
2.3. Enzymatic Synthesis in Vitro
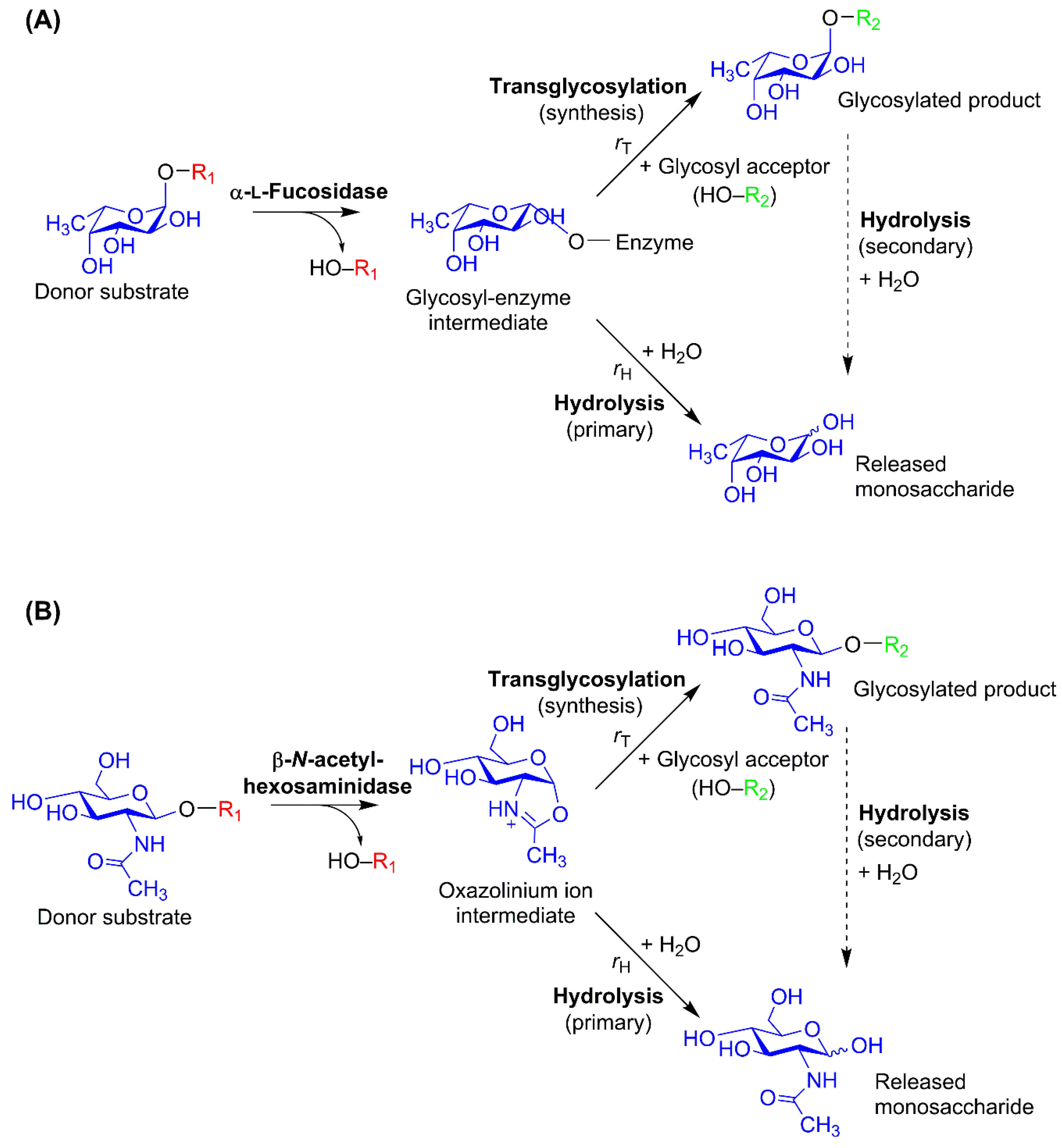
3. Glycosidase-Catalyzed Transglycosylation
4. Improved Transglycosylation through Protein Engineering
4.1. Glycosynthases
4.2. Rational Design
4.2.1. Targeting Conserved Residues in Negative Subsite(s)
4.2.2. Loop Engineering
4.3. Effect of Protein Engineering on Reaction Rates
5. Abundant Natural Substrates from Dairy and Agro-Industrial Side Streams
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bode, L. Human milk oligosaccharides: Every baby needs a sugar mama. Glycobiology 2012, 22, 1147–1162. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.; Cheah, W.Y.; Grinyer, J.; Packer, N. Glycoconjugates in human milk: Protecting infants from disease. Glycobiology 2013, 23, 1425–1438. [Google Scholar] [CrossRef]
- de Moura Bell, J.M.L.N.; Cohen, J.L.; de Aquino, L.F.M.C.; Lee, H.; de Melo Silva, V.L.; Liu, Y.; Domizio, P.; Barile, D. An Integrated Bioprocess to Recover Bovine Milk Oligosaccharides from Colostrum Whey Permeate. J. Food Eng. 2018, 216, 27–35. [Google Scholar] [CrossRef]
- Triantis, V.; Bode, L.; van Neerven, R.J.J. Immunological Effects of Human Milk Oligosaccharides. Front. Pediatr. 2018, 6, 190. [Google Scholar] [CrossRef] [PubMed]
- Elison, E.; Vigsnaes, L.K.; Rindom Krogsgaard, L.; Rasmussen, J.; Sørensen, N.; McConnell, B.; Hennet, T.; Sommer, M.O.A.; Bytzer, P. Oral supplementation of healthy adults with 2′-O-fucosyllactose and lacto-N-neotetraose is well tolerated and shifts the intestinal microbiota. Br. J. Nutr. 2016, 116, 1356–1368. [Google Scholar] [CrossRef] [PubMed]
- Ninonuevo, M.R.; Ward, R.E.; Park, Y.; Clowers, B.H.; Killeen, K.; Lebrilla, C.B.; Grimm, R.; German, J.B.; Yin, H.; Freeman, S.L.; et al. A Strategy for Annotating the Human Milk Glycome. J. Agr. Food Chem. 2006, 54, 7471–7480. [Google Scholar] [CrossRef]
- Urashima, T.; Hirabayashi, J.; Sato, S.; Kobata, A. Human Milk Oligosaccharides as Essential Tools for Basic and Application Studies on Galectins. Trends Glycosci. Glycotechnol. 2018, 30, SE51–SE65. [Google Scholar] [CrossRef]
- Varki, A.; Cummings, R.D.; Aebi, M.; Packer, N.H.; Seeberger, P.H.; Esko, J.D.; Stanley, P.; Hart, G.; Darvill, A.; Kinoshita, T.; et al. Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology 2015, 25, 1323–1324. [Google Scholar] [CrossRef] [PubMed]
- Bych, K.; Mikš, M.H.; Johanson, T.; Hederos, M.J.; Vigsnæs, L.K.; Becker, P. Production of HMOs using microbial hosts—From cell engineering to large scale production. Curr. Opin. Biotechnol. 2018, 56, 130–137. [Google Scholar] [CrossRef]
- Bode, L.; Contractor, N.; Barile, D.; Pohl, N.; Prudden, A.R.; Boons, G.J.; Jin, Y.S.; Jennewein, S. Overcoming the limited availability of human milk oligosaccharides: Challenges and opportunities for research and application. Nutr. Rev. 2016, 74, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Thurl, S.; Munzert, M.; Boehm, G.; Matthews, C.; Stahl, B. Systematic review of the concentrations of oligosaccharides in human milk. Nutr. Rev. 2017, 75, 920–933. [Google Scholar] [CrossRef]
- Kunz, C.; Meyer, C.; Collado, M.C.; Geiger, L.; García-Mantrana, I.; Bertua-Ríos, B.; Martínez-Costa, C.; Borsch, C.; Rudloff, S. Influence of Gestational Age, Secretor, and Lewis Blood Group Status on the Oligosaccharide Content of Human Milk. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, G.A.; Baumgärtner, F.; Albermann, C. Production of human milk oligosaccharides by enzymatic and whole-cell microbial biotransformations. J. Biotechnol. 2017, 258, 79–91. [Google Scholar] [CrossRef]
- Baumgärtner, F.; Jurzitza, L.; Conrad, J.; Beifuss, U.; Sprenger, G.A.; Albermann, C. Synthesis of fucosylated lacto-N-tetraose using whole-cell biotransformation. Bioorg. Med. Chem. 2015, 23, 6799–6806. [Google Scholar] [CrossRef]
- Faijes, M.; Castejón-Vilatersana, M.; Val-Cid, C.; Planas, A. Enzymatic and cell factory approaches to the production of human milk oligosaccharides. Biotechnol. Adv. 2019. [Google Scholar] [CrossRef]
- Craft, K.M.; Thomas, H.C.; Townsend, S.D. Interrogation of Human Milk Oligosaccharide Fucosylation Patterns for Antimicrobial and Antibiofilm Trends in Group B Streptococcus. ACS Infect. Dis. 2018, 4, 1755–1765. [Google Scholar] [CrossRef]
- Craft, K.M.; Thomas, H.C.; Townsend, S.D. Sialylated variants of lacto-N-tetraose exhibit antimicrobial activity against Group B Streptococcus. Org. Biomol. Chem. 2018, 17, 1893–1900. [Google Scholar] [CrossRef]
- Gosling, A.; Stevens, G.W.; Barber, A.R.; Kentish, S.E.; Gras, S.L. Recent advances refining galactooligosaccharide production from lactose. Food Chem. 2010, 121, 307–318. [Google Scholar] [CrossRef]
- Torres, D.P.M.; Gonçalves, M.D.P.F.; Teixeira, J.A.; Rodrigues, L.R. Galacto-Oligosaccharides: Production, properties, applications, and significance as prebiotics. Compr. Rev. Food Sci. Food Saf. 2010, 9, 438–454. [Google Scholar] [CrossRef]
- Mano, M.C.R.; Neri-Numa, I.A.; da Silva, J.B.; Paulino, B.N.; Pessoa, M.G.; Pastore, G.M. Oligosaccharide biotechnology: an approach of prebiotic revolution on the industry. Appl. Microbiol. Biotechnol. 2018, 102, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Puccio, G.; Alliet, P.; Cajozzo, C.; Janssens, E.; Corsello, G.; Sprenger, N.; Wernimont, S.; Egli, D.; Gosoniu, L.; Steenhout, P. Effects of infant formula with human milk oligosaccharides on growth and morbidity: A randomized multicenter trial. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 624–631. [Google Scholar] [CrossRef]
- Liu, J.J.; Kwak, S.; Pathanibul, P.; Lee, J.W.; Yu, S.; Yun, E.J.; Lim, H.; Kim, K.H.; Jin, Y.S. Biosynthesis of a Functional Human Milk Oligosaccharide, 2′-Fucosyllactose, and L-Fucose Using Engineered Saccharomyces cerevisiae. ACS Synth. Biol. 2018, 7, 2529–2536. [Google Scholar] [CrossRef]
- Li, L.; Kim, S.A.; Heo, J.E.; Kim, T.J.; Seo, J.H.; Han, N.S. One-pot synthesis of GDP-l-fucose by a four-enzyme cascade expressed in Lactococcus lactis. J. Biotechnol. 2017, 264, 1–7. [Google Scholar] [CrossRef]
- Priem, B.; Gilbert, M.; Wakarchuk, W.W.; Heyraud, A.; Samain, E. A new fermentation process allows large-scale production of human milk oligosaccharides by metabolically engineered bacteria. Glycobiology 2002, 12, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Prudden, A.R.; Liu, L.; Capicciotti, C.J.; Wolfert, M.A.; Wang, S.; Gao, Z.; Meng, L.; Moremen, K.W.; Boons, G.-J. Synthesis of asymmetrical multiantennary human milk oligosaccharides. Proc. Natl. Acad. Sci. 2017, 114, 6954–6959. [Google Scholar] [CrossRef] [PubMed]
- Arboe Jennum, C.; Hauch Fenger, T.; Bruun, L.M.; Madsen, R. One-pot glycosylations in the synthesis of human milk oligosaccharides. Eur. J. Org. Chem. 2014, 2014, 3232–3241. [Google Scholar] [CrossRef]
- Craft, K.M.; Townsend, S.D. Synthesis of lacto-N-tetraose. Carbohydr. Res. 2017, 440–441, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Agoston, K.; Hederos, M.J.; Bajza, I.; Dekany, G. Kilogram scale chemical synthesis of 2′-fucosyllactose. Carbohydr. Res. 2019, 476, 71–77. [Google Scholar] [CrossRef]
- McKay, M.J.; Nguyen, H.M. Recent Advances in Transition Metal-Catalyzed Glycosylation. ACS Catal. 2012, 2, 1563–1595. [Google Scholar] [CrossRef]
- Xiao, Z.; Guo, Y.; Liu, Y.; Li, L.; Zhang, Q.; Wen, L.; Wang, X.; Kondengaden, S.M.; Wu, Z.; Zhou, J.; et al. Chemoenzymatic Synthesis of a Library of Human Milk Oligosaccharides. J. Org. Chem. 2016, 81, 5851–5865. [Google Scholar] [CrossRef]
- Schmidt, D.; Thiem, J. Chemical synthesis using enzymatically generated building units for construction of the human milk pentasaccharides sialyllacto-N-tetraose and sialyllacto-N-neotetraose epimer. Beilstein J. Org. Chem. 2010, 6, 1–7. [Google Scholar] [CrossRef]
- Yao, W.; Yan, J.; Chen, X.; Wang, F.; Cao, H. Chemoenzymatic synthesis of lacto-N-tetrasaccharide and sialyl lacto-N-tetrasaccharides. Carbohydr. Res. 2015, 401, 5–10. [Google Scholar] [CrossRef]
- Muschiol, J.; Meyer, A.S. A chemo-enzymatic approach for the synthesis of human milk oligosaccharide backbone structures. J. Biosci. 2019, 74, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Schmölzer, K.; Weingarten, M.; Baldenius, K.; Nidetzky, B. Lacto-N-tetraose synthesis by wild-type and glycosynthase variants of the β-N-hexosaminidase from Bifidobacterium bifidum. Org. Biomol. Chem. 2019. [Google Scholar] [CrossRef]
- Wen, L.; Edmunds, G.; Gibbons, C.; Zhang, J.; Gadi, M.R.; Zhu, H.; Fang, J.; Liu, X.; Kong, Y.; Wang, P.G. Toward Automated Enzymatic Synthesis of Oligosaccharides. Chem. Rev. 2018, 118, 8151–8187. [Google Scholar] [CrossRef]
- Nidetzky, B.; Gutmann, A.; Zhong, C. Leloir Glycosyltransferases as Biocatalysts for Chemical Production. ACS Catal. 2018, 8, 6283–6300. [Google Scholar] [CrossRef]
- Choi, Y.H.; Kim, J.H.; Park, J.H.; Lee, N.; Kim, D.H.; Jang, K.S.; Park, I.H.; Kim, B.G. Protein engineering of α2,3/2,6-sialyltransferase to improve the yield and productivity of in vitro sialyllactose synthesis. Glycobiology 2014, 24, 159–169. [Google Scholar] [CrossRef]
- Yu, H.; Yan, X.; Autran, C.A.; Li, Y.; Etzold, S.; Latasiewicz, J.; Robertson, B.M.; Li, J.; Bode, L.; Chen, X. Enzymatic and Chemoenzymatic Syntheses of Disialyl Glycans and Their Necrotizing Enterocolitis Preventing Effects. J. Org. Chem. 2017, 82, 13152–13160. [Google Scholar] [CrossRef]
- Weijers, C.A.G.M.; Franssen, M.C.R.; Visser, G.M. Glycosyltransferase-catalyzed synthesis of bioactive oligosaccharides. Biotechnol. Adv. 2008, 26, 436–456. [Google Scholar] [CrossRef]
- US Food and Drug Administration. 3′-Sialyllactose Sodium Salt. GRAS Notice GRN 000766. Available online: https://www.accessdata.fda.gov/scripts/fdcc/index.cfm?set=GRASNotices&id=766 (accessed on 27 March 2019).
- Woo, J.S.; Kim, B.-G.; Kim, D.H.; Choi, Y.H.; Song, J.-K.; Kang, S.Y.; Seo, W.M.; Yang, J.Y.; Lee, S.M. Method for preparing sialic acid derivative. US Patent US 9637768 B2, 2 May 2017. [Google Scholar]
- Guo, Y.; Jers, C.; Meyer, A.S.; Arnous, A.; Li, H.; Kirpekar, F.; Mikkelsen, J.D. A Pasteurella multocida sialyltransferase displaying dual trans-sialidase activities for production of 3′-sialyl and 6′-sialyl glycans. J. Biotechnol. 2014, 170, 60–67. [Google Scholar] [CrossRef]
- Cheng, J.; Yu, H.; Lau, K.; Huang, S.; Chokhawala, H.A.; Li, Y.; Tiwari, V.K.; Chen, X. Multifunctionality of Campylobacter jejuni sialyltransferase CstII: Characterization of GD3/GT3 oligosaccharide synthase, GD3 oligosaccharide sialidase, and trans-sialidase activities. Glycobiology 2008, 18, 686–697. [Google Scholar] [CrossRef]
- Cheng, J.; Huang, S.; Yu, H.; Lau, K.; Chen, X. Trans-sialidase activity of Photobacterium damsela α2,6-sialyltransferase and its application in the synthesis of sialosides. Glycobiology 2010, 20, 260–268. [Google Scholar] [CrossRef]
- Schmölzer, K.; Ribitsch, D.; Czabany, T.; Luley-goedl, C.; Kokot, D.; Lyskowski, A.; Zitzenbacher, S.; Schwab, H.; Nidetzky, B. Characterization of a multifunctional α2,3-sialyltransferase from Pasteurella dagmatis. Glycobiology 2013, 23, 1293–1304. [Google Scholar] [CrossRef]
- Fischöder, T.; Cajic, S.; Reichl, U.; Rapp, E.; Elling, L. Enzymatic Cascade Synthesis Provides Novel Linear Human Milk Oligosaccharides as Reference Standards for xCGE-LIF Based High-Throughput Analysis. Biotechnol. J. 2018, 14, 1800305. [Google Scholar] [CrossRef]
- Bissaro, B.; Monsan, P.; Fauré, R.; O’Donohue, M.J. Glycosynthesis in a waterworld: new insight into the molecular basis of transglycosylation in retaining glycoside hydrolases. Biochem. J. 2015, 467, 17–35. [Google Scholar] [CrossRef]
- Holck, J.; Larsen, D.M.; Michalak, M.; Li, H.; Kjærulff, L.; Kirpekar, F.; Gotfredsen, C.H.; Forssten, S.; Ouwehand, A.C.; Mikkelsen, J.D.; et al. Enzyme catalyzed production of sialylated human milk oligosaccharides and galactooligosaccharides by Trypanosoma cruzi trans-sialidase. N. Biotechnol. 2014, 31, 156–165. [Google Scholar] [CrossRef]
- Zeuner, B.; Jers, C.; Mikkelsen, J.D.; Meyer, A.S. Methods for improving enzymatic trans-glycosylation for synthesis of human milk oligosaccharide biomimetics. J. Agric. Food Chem. 2014, 62, 9615–9631. [Google Scholar] [CrossRef]
- Lundemo, P.; Karlsson, E.N.; Adlercreutz, P. Eliminating hydrolytic activity without affecting the transglycosylation of a GH1 β-glucosidase. Appl. Microbiol. Biotechnol. 2017, 101, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Ajisaka, K.; Fujimoto, H.; Isomura, M. Regioselective transglycosylation in the synthesis of oligosaccharides: comparison of β-galactosidases and sialidases of various origins. Carbohydr. Res. 1994, 259, 103–115. [Google Scholar] [CrossRef]
- Jers, C.; Michalak, M.; Larsen, D.M.; Kepp, K.P.; Li, H.; Guo, Y.; Kirpekar, F.; Meyer, A.S.; Mikkelsen, J.D. Rational design of a new Trypanosoma rangeli trans-sialidase for efficient sialylation of glycans. PLoS ONE 2014, 9, e83902. [Google Scholar] [CrossRef] [PubMed]
- Zeuner, B.; Muschiol, J.; Holck, J.; Lezyk, M.; Gedde, M.R.; Jers, C.; Mikkelsen, J.D.; Meyer, A.S. Substrate specificity and transfucosylation activity of GH29 α-l-fucosidases for enzymatic production of human milk oligosaccharides. N. Biotechnol. 2018, 41, 34–45. [Google Scholar] [CrossRef]
- Zeuner, B.; Vuillemin, M.; Holck, J.; Muschiol, J.; Meyer, A.S. Loop engineering of an α-1,3/4-l-fucosidase for improved synthesis of human milk oligosaccharides. Enzyme Microb. Technol. 2018, 115, 37–44. [Google Scholar] [CrossRef]
- Saumonneau, A.; Champion, E.; Peltier-Pain, P.; Molnar-Gabor, D.; Hendrickx, J.; Tran, V.; Hederos, M.; Dekany, G.; Tellier, C. Design of an α-l-transfucosidase for the synthesis of fucosylated HMOs. Glycobiology 2016, 26, 261–269. [Google Scholar] [CrossRef][Green Version]
- Zeuner, B.; González-Delgado, I.; Holck, J.; Morales, G.; López-Muñoz, M.-J.; Segura, Y.; Meyer, A.S.; Dalgaard Mikkelsen, J. Characterization and immobilization of engineered sialidases from Trypanosoma rangeli for transsialylation. AIMS Mol. Sci. 2017, 4, 140–163. [Google Scholar] [CrossRef][Green Version]
- Guo, L.; Chen, X.; Xu, L.; Xiao, M.; Lu, L. Enzymatic synthesis of 6′-sialyllactose, a dominant sialylated human milk oligosaccharide, by a novel exo-α-sialidase from Bacteroides fragilis NCTC9343. Appl. Environ. Microbiol. 2018, 84, e00071-18. [Google Scholar] [CrossRef] [PubMed]
- Nyffenegger, C.; Nordvang, R.T.; Zeuner, B.; Łężyk, M.; Difilippo, E.; Logtenberg, M.J.; Schols, H.A.; Meyer, A.S.; Mikkelsen, J.D. Backbone structures in human milk oligosaccharides: trans-glycosylation by metagenomic β-N-acetylhexosaminidases. Appl. Microbiol. Biotechnol. 2015, 99, 7997–8009. [Google Scholar] [CrossRef]
- Jamek, S.B.; Mikkelsen, J.D.; Busk, P.K.; Meyer, A.S.; Holck, J.; Zeuner, B.; Muschiol, J. Loop Protein Engineering for Improved Transglycosylation Activity of a β-N-Acetylhexosaminidase. ChemBioChem 2018, 19, 1858–1865. [Google Scholar] [CrossRef]
- Zeuner, B.; Nyffenegger, C.; Mikkelsen, J.D.; Meyer, A.S. Thermostable β-galactosidases for the synthesis of human milk oligosaccharides. N. Biotechnol. 2016, 33, 355–360. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Murata, T.; Inukai, T.; Suzuki, M.; Yamagashi, M.; Usui, T. Facile enzymatic conversion of lactose into lacto-N-tetraose and lacto-N-neotetraose. Glycoconj. J. 1999, 16, 189–195. [Google Scholar] [CrossRef]
- Davies, G.J.; Wilson, K.S.; Henrissat, B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 1997, 321, 557–559. [Google Scholar] [CrossRef]
- Kasche, V. Mechanism and yields in enzyme catalyzed equilibrium and kinetically controlled synthesis of β-lactam antibiotics, peptides and other condensation products. Enzyme Microb. Technol. 1986, 8, 4–16. [Google Scholar] [CrossRef]
- Koshland, D.E. Stereochemistry and the mechanism of enzymatic reactions. Biol. Rev. 1953, 28, 416–436. [Google Scholar] [CrossRef]
- Vocadlo, D.J.; Withers, S.G. Detailed comparative analysis of the catalytic mechanisms of β-N-acetylglucosaminidases from families 3 and 20 of glycoside hydrolases. Biochemistry 2005, 44, 12809–12818. [Google Scholar] [CrossRef]
- Nordvang, R.T.; Nyffenegger, C.; Holck, J.; Jers, C.; Zeuner, B.; Sundekilde, U.K.; Meyer, A.S.; Mikkelsen, J.D. It all starts with a sandwich: Identification of sialidases with trans-glycosylation activity. PLoS ONE 2016, 11, e0158434. [Google Scholar] [CrossRef]
- Teze, D.; Hendrickx, J.; Czjzek, M.; Ropartz, D.; Sanejouand, Y.-H.; Tran, V.; Tellier, C.; Dion, M. Semi-rational approach for converting a GH1 β-glycosidase into a β-transglycosidase. Protein Eng. Des. Sel. 2014, 27, 13–19. [Google Scholar] [CrossRef]
- Van Rantwijk, F.; Woudenberg-Van Oosterom, M.; Sheldon, R.A. Glycosidase-catalyzed synthesis of alkyl glycosides. J. Mol. Catal. - B Enzym. 1999, 6, 511–532. [Google Scholar] [CrossRef]
- Wilbrink, M.H.; Kate, G.A.; Van, S.S.; Sanders, P.; Sallomons, E.; Johannes, A.; Dijkhuizen, L.; Kamerling, J.P. Galactosyl-lactose sialylation using Trypanosoma cruzi trans-sialidase as the biocatalyst and bovine κ-casein-derived glycomacropeptide asthe donor substrate. Appl. Environ. Microbiol. 2014, 80, 5984–5991. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Jers, C.; Meyer, A.S.; Li, H.; Kirpekar, F.; Mikkelsen, J.D. Modulating the regioselectivity of a Pasteurella multocida sialyltransferase for biocatalytic production of 3′- and 6′-sialyllactose. Enzyme Microb. Technol. 2015, 78, 54–62. [Google Scholar] [CrossRef]
- Choi, K.-W.; Park, K.; Jun, S.-Y.; Park, C.; Park, K.; Cha, J. Modulation of the Regioselectivity of a Thermotoga neapolitana β-glucosidase by site-directed mutagenesis. J. Microbiol. Biotechnol 2008, 18, 901–907. [Google Scholar] [PubMed]
- Bobrov, K.S.; Borisova, A.S.; Eneyskaya, E.V.; Ivanen, D.R.; Shabalin, K.A.; Kulminskaya, A.A.; Rychkov, G.N. Improvement of the Efficiency of Transglycosylation Catalyzed by α-Galactosidase from Thermotoga maritima by Protein Engineering. Biochem. 2013, 78, 1112–1123. [Google Scholar] [CrossRef]
- Talens-Perales, D.; Polaina, J.; Marín-Navarro, J. Structural Dissection of the Active Site of Thermotoga maritima β-Galactosidase Identifies Key Residues for Transglycosylating Activity. J. Agric. Food Chem. 2016, 64, 2917–2924. [Google Scholar] [CrossRef] [PubMed]
- Nyffenegger, C.; Nordvang, R.T.; Jers, C.; Meyer, A.S.; Mikkelsen, J.D. Design of Trypanosoma rangeli sialidase mutants with improved trans-sialidase activity. PLoS ONE 2017, 12, e0171585. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.-Y.; Drone, J.; Hoffmann, L.; Tran, V.; Tellier, C.; Rabiller, C.; Dion, M. Converting a β-glycosidase into a β-transglycosidase by directed evolution. J. Biol. Chem. 2005, 280, 37088–37097. [Google Scholar] [CrossRef]
- Placier, G.; Watzlawick, H.; Rabiller, C.; Mattes, R. Evolved β-galactosidases from Geobacillus stearothermophilus with improved transgalactosylation yield for galacto-oligosaccharide production. Appl. Environ. Microbiol. 2009, 75, 6312–6321. [Google Scholar] [CrossRef]
- Zhang, H.-P.; Dong, Y.-N.; Chen, W.; Zhang, H.; Liu, X.-M.; Xia, Y.; Chen, H.-Q. Enhancement of the hydrolysis activity of β-galactosidase from Geobacillus stearothermophilus by saturation mutagenesis. J. Dairy Sci. 2011, 94, 1176–1184. [Google Scholar]
- Hayes, M.R.; Pietruszka, J. Synthesis of glycosides by glycosynthases. Molecules 2017, 22, 1434. [Google Scholar] [CrossRef]
- Giddens, J.P.; Lomino, J.V.; Amin, M.N.; Wang, L.X. Endo-F3 glycosynthase mutants enable chemoenzymatic synthesis of core-fucosylated triantennary complex type glycopeptides and glycoproteins. J. Biol. Chem. 2016, 291, 9356–9370. [Google Scholar] [CrossRef]
- Mackenzie, L.F.; Wang, Q.; Warren, R.A.J.; Withers, S.G. Glycosynthases: Mutant Glycosidases for Oligosaccharide Synthesis. J. Am. Chem. Soc. 1998, 120, 5583–5584. [Google Scholar] [CrossRef]
- Henze, M.; Schmidtke, S.; Hoffmann, N.; Steffens, H.; Pietruszka, J.; Elling, L. Combination of Glycosyltransferases and a Glycosynthase in Sequential and One-Pot Reactions for the Synthesis of Type 1 and Type 2 N-Acetyllactosamine Oligomers. ChemCatChem 2015, 7, 3131–3139. [Google Scholar] [CrossRef]
- Cobucci-Ponzano, B.; Conte, F.; Bedini, E.; Corsaro, M.M.; Parrilli, M.; Sulzenbacher, G.; Lipski, A.; Dal Piaz, F.; Lepore, L.; Rossi, M.; et al. β-Glycosyl Azides as Substrates for α-Glycosynthases: Preparation of Efficient α-l-Fucosynthases. Chem. Biol. 2009, 16, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Sakurama, H.; Fushinobu, S.; Hidaka, M.; Yoshida, E.; Honda, Y.; Ashida, H.; Kitaoka, M.; Kumagai, H.; Yamamoto, K.; Katayama, T. 1,3-1,4-α-l-Fucosynthase that specifically introduces Lewis a/x antigens into type-1/2 chains. J. Biol. Chem. 2012, 287, 16709–16719. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Gotoh, A.; Katoh, T.; Honda, Y.; Yoshida, E.; Kurihara, S.; Ashida, H.; Kumagai, H.; Yamamoto, K.; Kitaoka, M.; et al. Introduction of H-antigens into oligosaccharides and sugar chains of glycoproteins using highly efficient 1,2-α-l-fucosynthase. Glycobiology 2016, 26, 1235–1247. [Google Scholar] [CrossRef]
- Wada, J.; Honda, Y.; Nagae, M.; Kato, R.; Wakatsuki, S.; Katayama, T.; Taniguchi, H.; Kumagai, H.; Kitaoka, M.; Yamamoto, K. 1,2-α-l-Fucosynthase: A glycosynthase derived from an inverting α-glycosidase with an unusual reaction mechanism. FEBS Lett. 2008, 582, 3739–3743. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, T.; Fukuda, T.; Dozen, S.; Honda, Y.; Kitaoka, M.; Fukamizo, T. A glycosynthase derived from an inverting GH19 chitinase from the moss Bryum coronatum. Biochem. J. 2012, 444, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Martinez, E.A.; Boer, H.; Koivula, A.; Samain, E.; Driguez, H.; Armand, S.; Cottaz, S. Engineering chitinases for the synthesis of chitin oligosaccharides: Catalytic amino acid mutations convert the GH-18 family glycoside hydrolases into transglycosylases. J. Mol. Catal. B Enzym. 2012, 74, 89–96. [Google Scholar] [CrossRef]
- Umekawa, M.; Huang, W.; Li, B.; Fujita, K.; Ashida, H.; Wang, L.-X.; Yamamoto, K. Mutants of Mucor hiemalis Endo-β-N-acetylglucosaminidase Show Enhanced Transglycosylation and Glycosynthase-like Activities. J. Biol. Chem. 2008, 283, 4469–4479. [Google Scholar] [CrossRef]
- Slámová, K.; Kapešová, J.; Kulik, N.; Křen, V. Transglycosylation activity of glycosynthase-type mutants of β-N-acetylhexosaminidase from Talaromyces flavus. In Proceedings of the Carbohydrate Bioengineering Meeting, Toulouse, France, 19–22 May 2019; p. 149. [Google Scholar]
- Osanjo, G.; Dion, M.; Drone, J.; Solleux, C.; Tran, V.; Rabiller, C.; Tellier, C. Directed evolution of the α-l-fucosidase from Thermotoga maritima into an α-l-transfucosidase. Biochemistry 2007, 46, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Teze, D.; Dion, M.; Daligault, F.; Tran, V.; André-Miral, C.; Tellier, C. Alkoxyamino glycoside acceptors for the regioselective synthesis of oligosaccharides using glycosynthases and transglycosidases. Bioorganic Med. Chem. Lett. 2013, 23, 448–451. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490-5. [Google Scholar] [CrossRef]
- Lundemo, P.; Adlercreutz, P.; Karlsson, E.N. Improved Transferase/Hydrolase Ratio through Rational Design of a Family 1 β-Glucosidase from Thermotoga neapolitana. Appl. Environ. Microbiol. 2013, 79, 3400–3405. [Google Scholar] [CrossRef]
- Wu, Y.; Yuan, S.; Chen, S.; Wu, D.; Chen, J.; Wu, J. Enhancing the production of galacto-oligosaccharides by mutagenesis of Sulfolobus solfataricus β-galactosidase. Food Chem. 2013, 138, 1588–1595. [Google Scholar] [CrossRef]
- Teze, D.; Daligault, F.; Ferrières, V.; Sanejouand, Y.H.; Tellier, C. Semi-rational approach for converting a GH36 α-glycosidase into an α-transglycosidase. Glycobiology 2015, 25, 420–427. [Google Scholar] [CrossRef]
- Armand, S.; Andrews, S.R.; Charnock, S.J.; Gilbert, H.J. Influence of the Aglycone Region of the Substrate Binding Cleft of Pseudomonas Xylanase 10A on Catalysis. Biochemistry 2001, 40, 7404–7409. [Google Scholar] [CrossRef]
- Hansson, T.; Kaper, T.; van der Oost, J.; de Vos, W.M.; Adlercreutz, P. Improved oligosaccharide synthesis by protein engineering of β-glucosidase CelB from hyperthermophilic Pyrococcus furiosus. Biotechnol. Bioeng. 2001, 73, 203–210. [Google Scholar] [CrossRef]
- Kelly, R.M.; Leemhuis, H.; Rozeboom, H.J.; van Oosterwijk, N.; Dijkstra, B.W.; Dijkhuizen, L. Elimination of competing hydrolysis and coupling side reactions of a cyclodextrin glucanotransferase by directed evolution. Biochem. J. 2008, 413, 517–525. [Google Scholar] [CrossRef]
- Paris, G.; Ratier, L.; Amaya, M.F.; Nguyen, T.; Alzari, P.M.; Frasch, A.C.C. A sialidase mutant displaying trans-sialidase activity. J. Mol. Biol. 2005, 345, 923–934. [Google Scholar] [CrossRef]
- Tran, V.; Hoffmann, L.; Rabiller, C.; Tellier, C.; Dion, M. Rational design of a GH1 β-glycosidase to prevent self-condensation during the transglycosylation reaction. Protein Eng. Des. Sel. 2010, 23, 43–49. [Google Scholar] [CrossRef]
- Mitchell, F.L.; Miles, S.M.; Neres, J.; Bichenkova, E.V.; Bryce, R.A. Tryptophan as a molecular shovel in the glycosyl transfer activity of Trypanosoma cruzi trans-sialidase. Biophys. J. 2010, 98, 38–40. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pierdominici-Sottile, G.; Palma, J.; Roitberg, A.E. Free-energy computations identify the mutations required to confer trans-sialidase activity into Trypanosoma rangeli sialidase. Proteins Struct. Funct. Bioinforma. 2014, 82, 424–435. [Google Scholar] [CrossRef]
- Teze, D.; Hendrickx, J.; Dion, M.; Tellier, C.; Woods, V.L.; Tran, V.; Sanejouand, Y.-H. Conserved Water Molecules in Family 1 Glycosidases: A DXMS and Molecular Dynamics Study. Biochemistry 2013, 52, 5900–5910. [Google Scholar] [CrossRef]
- Romero-Téllez, S.; Lluch, J.M.; González-Lafont, À.; Masgrau, L. Comparing Hydrolysis and Transglycosylation Reactions Catalyzed by Thermus thermophilus β-Glycosidase. A Combined MD and QM/MM Study. Front. Chem. 2019, 7, 1–19. [Google Scholar] [CrossRef]
- Piens, K.; Fauré, R.; Sundqvist, G.; Baumann, M.J.; Saura-Valls, M.; Teeri, T.T.; Cottaz, S.; Planas, A.; Driguez, H.; Brumer, H. Mechanism-based labeling defines the free energy change for formation of the covalent glycosyl-enzyme intermediate in a xyloglucan endo-transglycosylase. J. Biol. Chem. 2008, 283, 21864–21872. [Google Scholar] [CrossRef]
- Raich, L.; Borodkin, V.; Fang, W.; Castro-López, J.; Van Aalten, D.M.F.; Hurtado-Guerrero, R.; Rovira, C. A Trapped Covalent Intermediate of a Glycoside Hydrolase on the Pathway to Transglycosylation. Insights from Experiments and Quantum Mechanics/Molecular Mechanics Simulations. J. Am. Chem. Soc. 2016, 138, 3325–3332. [Google Scholar] [CrossRef]
- Arab-Jaziri, F.; Bissaro, B.; Tellier, C.; Dion, M.; Fauré, R.; Donohue, M.J.O. Enhancing the chemoenzymatic synthesis of arabinosylated xylo-oligosaccharides by GH51 α-l-arabinofuranosidase. Carbohydr. Res. 2015, 401, 64–72. [Google Scholar] [CrossRef]
- Koné, F.M.T.; Le Béchec, M.; Sine, J.-P.; Dion, M.; Tellier, C. Digital screening methodology for the directed evolution of transglycosidases. Protein Eng. Des. Sel. 2009, 22, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Enam, F.; Mansell, T.J. Linkage-Specific Detection and Metabolism of Human Milk Oligosaccharides in Escherichia coli. Cell Chem. Biol. 2018, 25, 1292–1303. [Google Scholar] [CrossRef]
- Hou, X.E.Z.; Ang, Y.W.; Eehan, E.J.M.; Hen, L.C. Mutation of a Conserved Tryptophan in the Chitin-Binding Cleft of Serratia marcescens Chitinase A Enhances Transglycosylation. Biosci. Biotechnol. Biochem. 2006, 70, 243–251. [Google Scholar]
- Bissaro, B.; Durand, J.; Biarnés, X.; Planas, A.; Monsan, P.; O’Donohue, M.J.; Fauré, R. Molecular Design of Non-Leloir Furanose-Transferring Enzymes from an α-l-Arabinofuranosidase: A Rationale for the Engineering of Evolved Transglycosylases. ACS Catal. 2015, 5, 4598–4611. [Google Scholar] [CrossRef]
- Hassan, N.; Geiger, B.; Gandini, R.; Patel, B.K.C.; Kittl, R.; Haltrich, D.; Nguyen, T.H.; Divne, C.; Tan, T.C. Engineering a thermostable Halothermothrix orenii β-glucosidase for improved galacto-oligosaccharide synthesis. Appl. Microbiol. Biotechnol. 2016, 100, 3533–3543. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Gao, R.; Li, J.; Wang, Q.; Guo, Z.; Yang, J. Engineering T. naphthophila β-glucosidase for enhanced synthesis of galactooligosaccharides by site-directed mutagenesis. Biochem. Eng. J. 2017, 127, 1–8. [Google Scholar]
- Larsbrink, J.; Izumi, A.; Hemsworth, G.R.; Davies, G.J.; Brumer, H. Structural Enzymology of Cellvibrio japonicus Agd31B Protein Reveals α-Transglucosylase Activity in Glycoside Hydrolase Family 31. J. Biol. Chem. 2012, 287, 43288–43299. [Google Scholar] [CrossRef] [PubMed]
- Amaya, M.F.; Watts, A.G.; Damager, I.; Wehenkel, A.; Nguyen, T.; Buschiazzo, A.; Frasch, A.C.; Withers, S.G.; Alzari, P.M. Structural Insights into the Catalytic Mechanism of Trypanosoma cruzi trans-Sialidase. Structure 2004, 12, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Busk, P.K.; Lange, L. Function-Based Classification of Carbohydrate-Active Enzymes by Recognition of Short, Conserved Peptide Motifs. Appl. Environ. Microbiol. 2013, 79, 3380–3391. [Google Scholar] [CrossRef] [PubMed]
- Wilkens, C.; Busk, P.K.; Pilgaard, B.; Zhang, W.J.; Nielsen, K.L.; Nielsen, P.H. Diversity of microbial carbohydrate-active enzymes in Danish anaerobic digesters fed with wastewater treatment sludge. Biotechnol. Biofuels 2017, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Leyva, A.; Soberón, X.; Sánchez, F.; Argüello, M.; Montero-Morán, G.; Saab-Rincón, G. Protein Design through Systematic Catalytic Loop Exchange in the (β/α)8 Fold. J. Mol. Biol. 2009, 387, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Leyva, A.; Barona-Gómez, F.; Saab-Rincón, G.; Verdel-Aranda, K.; Sánchez, F.; Soberón, X. Exploring the Structure–Function Loop Adaptability of a (β/α)8-Barrel Enzyme through Loop Swapping and Hinge Variability. J. Mol. Biol. 2011, 411, 143–157. [Google Scholar] [CrossRef] [PubMed]
- De Luca, F.; Benvenuti, M.; Carboni, F.; Pozzi, C.; Rossolini, G.M.; Mangani, S.; Docquier, J.-D. Evolution to carbapenem-hydrolyzing activity in noncarbapenemase class D β-lactamase OXA-10 by rational protein design. Proc. Natl. Acad. Sci. 2011, 108, 18424–18429. [Google Scholar] [CrossRef] [PubMed]
- Schiano-di-Cola, C.; Røjel, N.; Jensen, K.; Kari, J.; Sørensen, T.H.; Borch, K.; Westh, P. Systematic deletions in the cellobiohydrolase (CBH) Cel7A from the fungus Trichoderma reesei reveal flexible loops critical for CBH activity. J. Biol. Chem. 2019, 294, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- Borisova, A.S.; Eneyskaya, E.V.; Jana, S.; Badino, S.F.; Kari, J.; Amore, A.; Karlsson, M.; Hansson, H.; Sandgren, M.; Himmel, M.E.; et al. Correlation of structure, function and protein dynamics in GH7 cellobiohydrolases from Trichoderma atroviride, T. reesei and T. harzianum. Biotechnol. Biofuels 2018, 11, 1–22. [Google Scholar] [CrossRef]
- Hoque, M.A.; Zhang, Y.; Chen, L.; Yang, G.; Khatun, M.A.; Chen, H.; Hao, L.; Feng, Y. Stepwise Loop Insertion Strategy for Active Site Remodeling to Generate Novel Enzyme Functions. ACS Chem. Biol. 2017, 12, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Hayes, F.; Hallet, B.; Cao, Y. Insertion mutagenesis as a tool in the modification of protein function. Extended substrate specificity conferred by pentapeptide insertions in the Ω-loop of TEM-1 β-lactamase. J. Biol. Chem. 1997, 272, 28833–28836. [Google Scholar] [CrossRef]
- Guzmán-Rodríguez, F.; Alatorre-Santamaría, S.; Gómez-Ruiz, L.; Rodríguez-Serrano, G.; García-Garibay, M.; Cruz-Guerrero, A. Employment of fucosidases for the synthesis of fucosylated oligosaccharides with biological potential. Biotechnol. Appl. Biochem. 2019, 66, 172–191. [Google Scholar] [CrossRef]
- Thomä-Worringer, C.; Sørensen, J.; López-Fandiño, R. Health effects and technological features of caseinomacropeptide. Int. Dairy J. 2006, 16, 1324–1333. [Google Scholar] [CrossRef]
- Smithers, G.W. Whey and whey proteins-From “gutter-to-gold”. Int. Dairy J. 2008, 18, 695–704. [Google Scholar] [CrossRef]
- Neelima; Sharma, R.; Rajput, Y.S.; Mann, B. Chemical and functional properties of glycomacropeptide (GMP) and its role in the detection of cheese whey adulteration in milk: A review. Dairy Sci. Technol. 2013, 93, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Itoh, T. Variations and Distributions of O-Glycosidically Linked Sugar Chains in Bovine κ-Casein. J. Dairy Sci. 1992, 75, 1768–1774. [Google Scholar] [CrossRef]
- McJarrow, P.; Garman, J.; Harvey, S.; Van Amelsfort, A. Dairy process and product. US Patent WO2003/049547 A2, 19 June 2003. [Google Scholar]
- Luo, J.; Nordvang, R.T.; Morthensen, S.T.; Zeuner, B.; Meyer, A.S.; Mikkelsen, J.D.; Pinelo, M. An integrated membrane system for the biocatalytic production of 3′-sialyllactose from dairy by-products. Bioresour. Technol. 2014, 166, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Morthensen, S.T.; Meyer, A.S.; Pinelo, M. Filtration behavior of casein glycomacropeptide (CGMP) in an enzymatic membrane reactor: Fouling control by membrane selection and threshold flux operation. J. Memb. Sci. 2014, 469, 127–139. [Google Scholar] [CrossRef]
- Nordvang, R.T.; Luo, J.; Zeuner, B.; Prior, R.; Andersen, M.F.; Mikkelsen, J.D.; Meyer, A.S.; Pinelo, M. Separation of 3′-sialyllactose and lactose by nanofiltration: A trade-off between charge repulsion and pore swelling induced by high pH. Sep. Purif. Technol. 2014, 138, 77–83. [Google Scholar] [CrossRef]
- Zeuner, B.; Luo, J.; Nyffenegger, C.; Aumala, V.; Mikkelsen, J.D.; Meyer, A.S. Optimizing the biocatalytic productivity of an engineered sialidase from Trypanosoma rangeli for 3′-sialyllactose production. Enzyme Microb. Technol. 2014, 55, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Sallomons, E.; Wilbrink, M.H.; Sanders, P.; Kamerling, J.P.; Van Vuure, C.A.; Hage, J.A. Methods for providing sialylated oligosaccharides. US Patent 9539270 B2, 10 January 2017. [Google Scholar]
- Pelletier, M.; Barker, W.A.; Hakes, D.J.; Zopf, D.A. Methods for producing sialyloligosaccharides in a dairy source. US Patent 6323008 B1, 27 November 2001. [Google Scholar]
- Lee, S.G.; Shin, D.H.; Kim, B.G. Production of sialyloligosaccharides by trans-sialidase catalyzed reaction using fetuin as a sialic acid donor. Enzyme Microb. Technol. 2002, 31, 742–746. [Google Scholar] [CrossRef]
- Lin, B.X.; Qiao, Y.; Shi, B.; Tao, Y. Polysialic acid biosynthesis and production in Escherichia coli: current state and perspectives. Appl. Microbiol. Biotechnol. 2016, 100, 1–8. [Google Scholar] [CrossRef]
- Bah, C.S.F.; Bekhit, A.E.D.A.; Carne, A.; Mcconnell, M.A. Slaughterhouse blood: An emerging source of bioactive compounds. Compr. Rev. Food Sci. Food Saf. 2013, 12, 314–331. [Google Scholar] [CrossRef]
- Wilbrink, M.H.; Ten Kate, G.A.; Sanders, P.; Gerwig, G.J.; Van Leeuwen, S.S.; Sallomons, E.; Klarenbeek, B.; Hage, J.A.; Van Vuure, C.A.; Dijkhuizen, L.; et al. Enzymatic Decoration of Prebiotic Galacto-oligosaccharides (Vivinal GOS) with Sialic Acid Using Trypanosoma cruzi trans-Sialidase and Two Bovine Sialoglycoconjugates as Donor Substrates. J. Agric. Food Chem. 2015, 63, 5976–5984. [Google Scholar] [CrossRef]
- Dhar, C.; Sasmal, A.; Varki, A. From “Serum Sickness” to “Xenosialitis”: Past, Present, and Future Significance of the Non-human Sialic Acid Neu5Gc. Front. Immunol. 2019, 10, 807. [Google Scholar] [CrossRef]
- Pearce, O.M.T.; Läubli, H. Sialic acids in cancer biology and immunity. Glycobiology 2016, 26, 111–128. [Google Scholar] [CrossRef] [PubMed]
- De Smet, S.; Vossen, E. Meat: The balance between nutrition and health. A review. Meat Sci. 2016, 120, 145–156. [Google Scholar] [CrossRef]
- Harish Prashanth, K.V.; Tharanathan, R.N. Chitin/chitosan: modifications and their unlimited application potential-an overview. Trends Food Sci. Technol. 2007, 18, 117–131. [Google Scholar] [CrossRef]
- Synowiecki, J.; Al-Khateeb, N.A. Production, Properties, and Some New Applications of Chitin and Its Derivatives. Crit. Rev. Food Sci. Nutr. 2003, 43, 145–171. [Google Scholar] [CrossRef]
- Arbia, W.; Arbia, L.; Adour, L.; Amrana, A. Chitin Extraction from Crustacean Shells by Biological Methods—A review. Food Technol. Biotechnol. 2013, 51, 12–25. [Google Scholar]
- Jamek, S.B.; Nyffenegger, C.; Muschiol, J.; Holck, J.; Meyer, A.S.; Mikkelsen, J.D. Characterization of two novel bacterial type A exo-chitobiose hydrolases having C-terminal 5/12-type carbohydrate-binding modules. Appl. Microbiol. Biotechnol. 2017, 101, 4533–4546. [Google Scholar] [CrossRef]
- Vaaje-Kolstad, G.; Horn, S.J.; Sørlie, M.; Eijsink, V.G.H. The chitinolytic machinery of Serratia marcescens—A model system for enzymatic degradation of recalcitrant polysaccharides. FEBS J. 2013, 280, 3028–3049. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Thi, N.; Doucet, N. Combining chitinase C and N-acetylhexosaminidase from Streptomyces coelicolor A3(2) provides an efficient way to synthesize N-acetylglucosamine from crystalline chitin. J. Biotechnol. 2016, 220, 25–32. [Google Scholar] [CrossRef]
- Mekasha, S.; Toupalová, H.; Linggadjaja, E.; Tolani, H.A.; Anděra, L.; Arntzen, M.; Vaaje-Kolstad, G.; Eijsink, V.G.H.; Agger, J.W. A novel analytical method for d-glucosamine quantification and its application in the analysis of chitosan degradation by a minimal enzyme cocktail. Carbohydr. Res. 2016, 433, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Warmerdam, A.; Paudel, E.; Jia, W.; Boom, R.M.; Janssen, A.E.M. Characterization of β-Galactosidase Isoforms from Bacillus circulans and Their Contribution to GOS Production. Appl. Biochem. Biotechnol. 2013, 170, 340–358. [Google Scholar] [CrossRef]
- Hotchkiss, A.T.; Nuñez, A.; Strahan, G.D.; Chau, H.K.; White, A.K.; Marais, J.P.J.; Hom, K.; Vakkalanka, M.S.; Di, R.; Yam, K.L.; et al. Cranberry Xyloglucan Structure and Inhibition of Escherichia coli Adhesion to Epithelial Cells. J. Agric. Food Chem. 2015, 63, 5622–5633. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Noro, O.; Azuma, Y. Analysis of the Oligosaccharides of Xyloglucan in Peanut Hulls (Studies on Production of Fucose-containing Xyloglucan Oligosaccharide Part II). Nippon Shokuhin Kagaku Kogaku Kaishi 2000, 47, 560–563. [Google Scholar] [CrossRef]
- Arumugam, N.; Biely, P.; Puchart, V.; Singh, S.; Pillai, S. Structure of peanut shell xylan and its conversion to oligosaccharides. Process Biochem. 2018, 72, 124–129. [Google Scholar] [CrossRef]
- Cao, H.T.T.; Mikkelsen, M.D.; Lezyk, M.J.; Bui, L.M.; Tran, V.T.T.; Silchenko, A.S.; Kusaykin, M.I.; Pham, T.D.; Truong, B.H.; Holck, J.; et al. Novel Enzyme Actions for Sulphated Galactofucan Depolymerisation and a New Engineering Strategy for Molecular Stabilisation of Fucoidan Degrading Enzymes. Mar. Drugs 2018, 16, 422. [Google Scholar] [CrossRef] [PubMed]
- Berteau, O.; McCort, I.; Goasdoué, N.; Tissot, B.; Daniel, R. Characterization of a new α-l-fucosidase isolated from the marine mollusk Pecten maximus that catalyzes the hydrolysis of α-l-fucose from algal fucoidan (Ascophyllum nodosum). Glycobiology 2002, 12, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Perrella, N.N.; Withers, S.G.; Lopes, A.R. Identity and role of the non-conserved acid/base catalytic residue in the GH29 fucosidase from the spider Nephilingis cruentata. Glycobiology 2018, 28, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Zhang, H.; Chen, X.; Lu, L.; Xu, L.; Xiao, M. Cloning, characterization, and production of three α-l-fucosidases from Clostridium perfringens ATCC 13124. J. Basic Microbiol. 2016, 56, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.M.; Park, Y.C.; Seo, J.H. Production of 3-Fucosyllactose in Engineered Escherichia coli with α-1,3-Fucosyltransferase from Helicobacter pylori. Biotechnol. J. 2019, 1800498, 1–7. [Google Scholar] [CrossRef]
- Champion, E.; McConnell, B.; Dekany, G. Ternary mixtures of 6′SL, LNnT and LST c. US Patent US2018/0161350 A1, 14 June 2018. [Google Scholar]
- Vogel, A.; Schmiedel, R.; Champion, E.; Dekany, G. Mutated sialidases. US Patent US2018/0163185 A1, 14 June 2018. [Google Scholar]
- Champion, E.; Vogel, A.; Bartsch, S.; Dekany, G. Mutated fucosidase. US Patent WO2016/063261 A1, 28 April 2016. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Enzyme | Donor | Acceptor | HMO product | A:D | Yield | Ref. |
|---|---|---|---|---|---|---|
| Arthrobacter ureafaciens sialidase | CGMP | Lac | n.d. | ~45 | 5% | [130] |
| Bifidobacterium infantis sialidase | CGMP | Lac | n.d. | ~45 | 1% | [130] |
| Trypanosoma rangeli sialidase (engineered) | CGMP | Lac | 3′-SL | 44 | 31% | [52] |
| Trypanosoma rangeli sialidase (engineered, membrane reactor) | CGMP | Lac | 3′-SL | ≤25 | 37% | [134] |
| Pasteurella multocida sialidase (sialyltransferase) | CGMP | Lac | 6′-SL, 3′-SL | 11 | 53% | [42,70] |
| Haemophilus parasuis sialidase | CGMP | Lac | 3′-SL (and isomer) | 39 | 19% (28%) | [66] |
| Trypanosoma cruzi transsialidase | CGMP | Lac | 3′-SL | 5 | 32% | [48] |
| Trypanosoma cruzi transsialidase | Fetuin | Lac | 3′-SL | 3 | 76% | [137] |
| Bacteroides fragilis sialidase | Hydrolyzed colominic acid | Lac | 6′-SL | ~15 | 22% | [57] |
| Fusarium graminearum fucosidase | Citrus peel xyloglucan | Lac | 2′-FL | 50 | 14% | [53] |
| β-N-acetylhexosaminidases (engineered, metagenomic) | Chitobiose | Lac | LNT2 (and isomers) | 5 | 5% (30%) | [59] |
| Bacillus circulans β-galactosidases | Lac | LNT2 | LNnT | 1 | 19% | [61] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeuner, B.; Teze, D.; Muschiol, J.; Meyer, A.S. Synthesis of Human Milk Oligosaccharides: Protein Engineering Strategies for Improved Enzymatic Transglycosylation. Molecules 2019, 24, 2033. https://doi.org/10.3390/molecules24112033
Zeuner B, Teze D, Muschiol J, Meyer AS. Synthesis of Human Milk Oligosaccharides: Protein Engineering Strategies for Improved Enzymatic Transglycosylation. Molecules. 2019; 24(11):2033. https://doi.org/10.3390/molecules24112033
Chicago/Turabian StyleZeuner, Birgitte, David Teze, Jan Muschiol, and Anne S. Meyer. 2019. "Synthesis of Human Milk Oligosaccharides: Protein Engineering Strategies for Improved Enzymatic Transglycosylation" Molecules 24, no. 11: 2033. https://doi.org/10.3390/molecules24112033
APA StyleZeuner, B., Teze, D., Muschiol, J., & Meyer, A. S. (2019). Synthesis of Human Milk Oligosaccharides: Protein Engineering Strategies for Improved Enzymatic Transglycosylation. Molecules, 24(11), 2033. https://doi.org/10.3390/molecules24112033