Abstract
The title compound, 1-(4-triethoxysilyl)phenyl)-4,4,4-trifluoro-1,3-butanedione, was synthesized in a three-step sequence starting from 2-(4-bromophenyl)propene. Containing both a trialkoxysilyl and a substituted 1,3-butanedione functional grouping within its structure, this new silane is a viable starting material for the preparation of functionalized sol-gel materials.
Keywords:
Trialkoxysilane; 4; 4; 4-Trifluoro-1-phenyl-1; 3-butanedione (4-TPB); Sol-gel; Molecular imprinting Introduction
The use of molecular imprinting on a silica sol-gel thin film constitutes an important process for the development of chemical sensor materials [1,2,3,4,5,6]. In our present efforts to develop a fluorometric sensing material for organophosphonate nerve agents using a templated sol-gel matrix incorporating the Eu(III) cation [7,8,9], initial studies using the ligand 4,4,4-trifluoro-1-phenyl-1,3-butanedione (1) seemed to indicate that the 4,4,4-trifluoro-1,3-butanedionyl moiety was the superior binding group for use in forming the desired ligand-Eu(III) complex. In view of this observation, it was envisioned that the use of an appropriate trialkoxysilyl monomer incorporating this binding group such as the silane 1-(4-(triethoxysilyl)phenyl)-4,4,4-trifluoro-1,3-butanedione (2) would allow for the covalent integration of the ligand-Eu(III) complex into the sol-gel matrix in the initial stages of the overall molecular-imprinting process. In this study, we wish to report the successful preparation of 2, a new triethoxysilane monomer that we are using in this study.
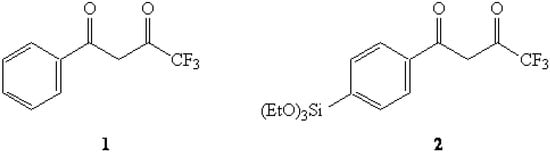
Figure 1.
4,4,4-Trifluoro-1-phenyl-1,3-butanedione (1) and the title compound 2.
Figure 1.
4,4,4-Trifluoro-1-phenyl-1,3-butanedione (1) and the title compound 2.
Results and Discussion
In our attempts to prepare 2, the two main issues of interest were the successful introduction of both the triethoxysilyl and 1,3-diketone functionalities and the use of reaction conditions that would not hydrolyze the acid-labile triethoxysilyl portion of the molecule. While this sensitivity to acid hydrolysis led to some difficulties initially, we were eventually successful in devising an efficient and cost-effective synthesis for this potentially-useful and interesting silane monomer.
In the first of our attempts, the dimethyl ketal of 4-bromoacetophenone was converted to the corresponding Grignard reagent which was then reacted with tetraethoxysilane (TEOS) to afford the expected triethoxysilyl dimethyl ketal 3. It was thought that a selective hydrolysis of the ketal grouping might yield the anticipated ketone 4-(triethoxysilyl)acetophenone (7) which could then be acylated via a Claisen condensation to afford 2. However, numerous difficulties were encountered in our attempts to selectively-hydrolyze the ketal with the triethoxysilyl group also being so affected.
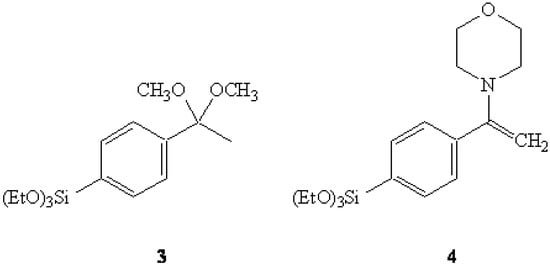
Figure 2.
Ketal 3 and Enamine 4.
Figure 2.
Ketal 3 and Enamine 4.
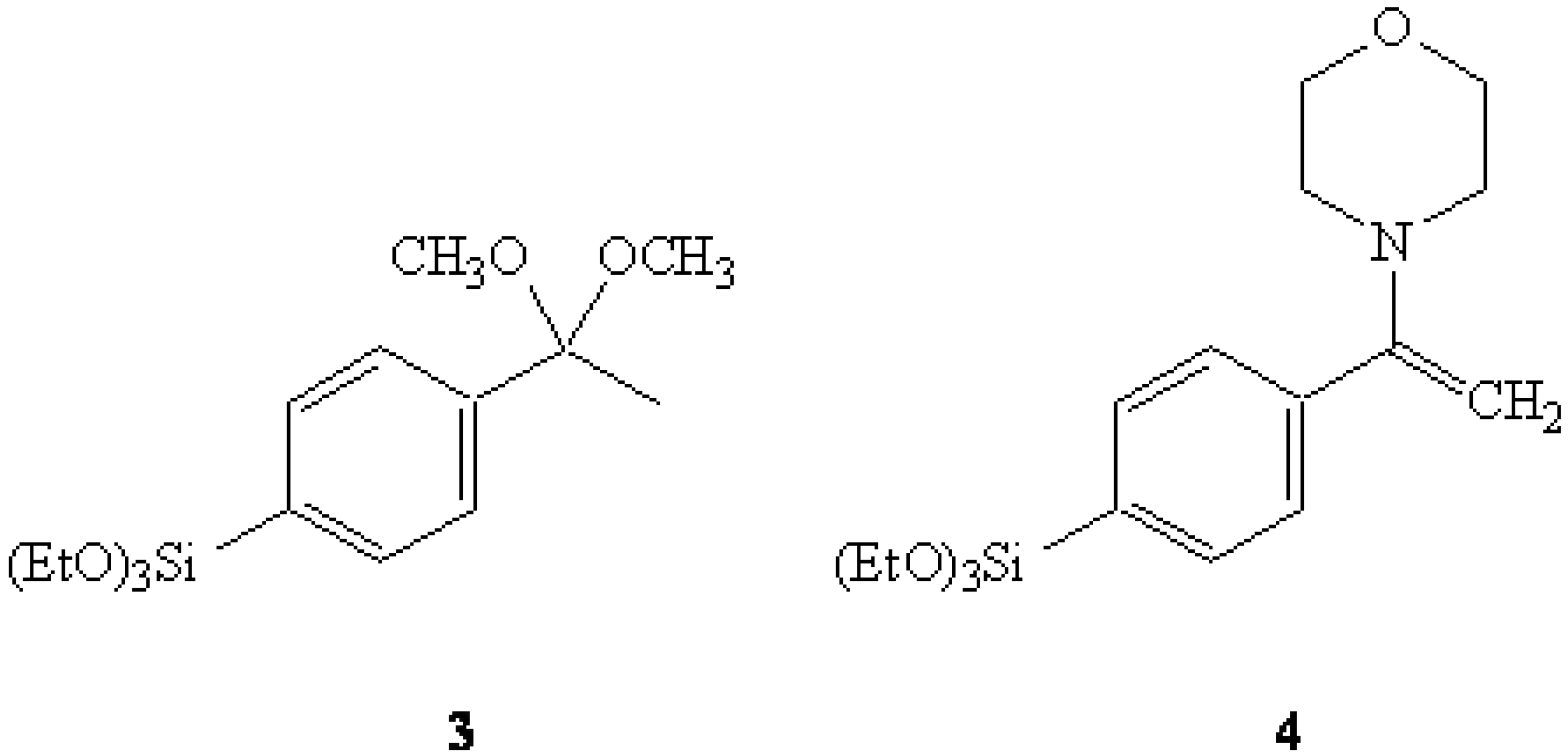
In another related attempt, the morpholine enamine derived from 4-bromoacetophenone was submitted to the same Grignard sequence to yield the anticipated triethoxysilyl enamine 4. It was expected that the acylation of 4 with trifluoroacetic anhydride [10], followed up with the "traditional" Stork work-up, could potentially afford 2. However, the same difficulties with over-hydrolysis were encountered here as well. A successful synthesis of 2 was accomplished by employing a more traditional approach using 2-(4-bromophenyl)propene (5) as the primary starting material [11,12]. The Grignard reagent of 5 was reacted with TEOS to give 2-(4-(triethoxysilyl)phenyl)propene (6) in 81% yield.
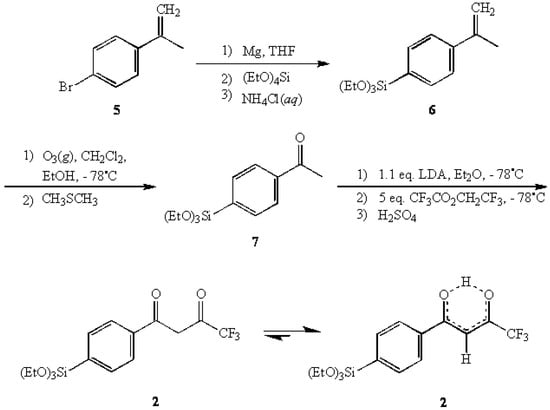
Scheme 1.
The synthesis of 2 form 2-(4-bromopheny)propene (5).
Scheme 1.
The synthesis of 2 form 2-(4-bromopheny)propene (5).
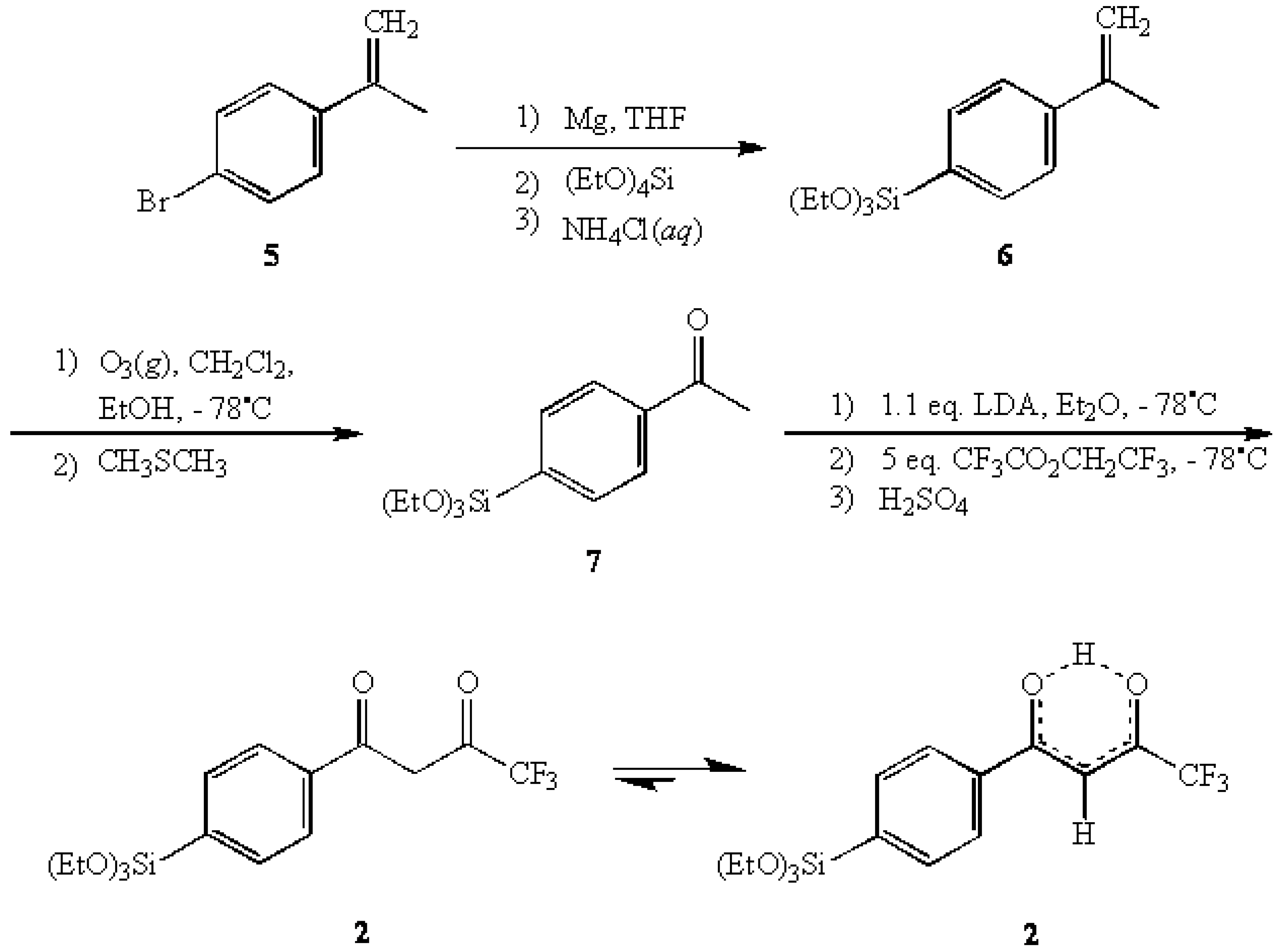
The subsequent ozonolysis of 6 afforded 4-(triethoxysilyl)acetophenone (7) [13,14] in 70% yield which was then reacted with a five-fold excess of 2,2,2-trifluoroethyl trifluoroacetate in an LDA-directed Claisen condensation [15] to provide the title compound 1-(4-(triethoxysilyl)-phenyl)-4,4,4-trifluoro-1,3-butanedione (2) in 73% yield. In characterizing 2, 1H-NMR analysis indicated that the enol tautomer was the predominant form present (>98%) with virtually none of the keto form being detected.
Conclusions
The route as devised for the preparation of 2 represents an efficient and cost-effective process that is readily amendable to scale-up. Following our successful synthesis of 2, subsequent follow-up studies have indicated that this monomer complexes quite well with the Eu(III) ion and does exhibit fluorometric activity in the presence of our VX-simulant compounds making it a viable candidate for fabricating our desired sol-gel materials. This encouraging observation tends to imply that this monomer may have other potential uses within the realm of molecular imprinting and templated sol-gel production. The results of our current efforts in this project will be the focus of future journal articles.
Experimental
General
All reactions were run under dry N2. Any reactions involving the generation or use of either Grignard reagents or LDA were run in glassware previously flame-dried under vacuum. Anhydrous acetone, TEOS, dimethylsulfide, anhydrous diethyl ether, and 2,2,2-trifluoroethyl trifluoroacetate were obtained from Aldrich and were used as received. LDA was obtained in the form of a commercial solution from Aldrich (1.5 M solution in cyclohexane) and was used as received. Anhydrous THF was prepared by distillation from a solvent still containing Na/benzophenone ketal immediately prior to use. Vacuum distillations were carried out using a Kugelrohr apparatus with all boiling points being reported at 0.20 mmHg. A PCI ozonizer (1 lb/day generation capacity) was used for the ozonolysis reactions with dry O2 being used as the feed gas source. FT-IR spectra were obtained using a Nicolet Avatar 360 spectrophotometer with NaCl plates being used for all thin films. 1H-NMR and 13C-NMR spectra were obtained using a Varian Unity Plus 300 MHz spectrometer and were measured at 300 and 75 MHz respectively, with all chemical shifts reported in ppm using TMS as the internal standard and all determined J values reported in Hz.
2-(4-(Triethoxysilyl)phenyl)propene (6). Into a 250-mL round-bottom three-neck flask were placed magnesium turnings (875 mg, 36.0 mmol) and a magnetic stirring bar and the resulting assembly was flame-dried under vacuum. The flask was then fitted with a reflux condensor and pressure-equalized dropping funnel, was flame-dried under a stream of dry N2 and allowed to cool to RT. The dropping funnel was charged with a solution of 2-(4-bromophenyl)propene (5) (5.91 g, 30.0 mmol) in anhydrous THF (60 mL). Approximately 15 mL of the bromide-THF solution was discharged from the dropping funnel, 1,2-dibromoethane (four drops) was added to the solution in the flask and the resulting mixture was heated to reflux and stirred until the formation of the Grignard reagent had commenced (circa 10 min). The remainder of the bromide-THF solution was then added dropwise with continued heating and stirring over the course of 1 h and the reaction mixture was refluxed with stirring for an additional 90 min. The reaction mixture was allowed to cool to RT, a solution of tetraethoxysilane (6.87 g, 7.35 mL, 33.0 mmol) in anhydrous THF (10 mL) was added rapidly with stirring over the course of 2 min, and the resulting mixture was refluxed with continued stirring for an additional 16 h. The reaction mixture was then allowed to cool to RT, was cooled to 0-5°C by immersion in an icebath, sat. NH4Cl(aq) (15 mL) was added dropwise with vigorous stirring over the course of 5 min and the resulting mixture was stirred for an additional 10 min with continued cooling. The mixture was filtered through Celite® and the collected magnesium salts were washed with three portions of ether (50 mL each). The combined organic extracts were washed with water and saturated aqueous NaCl, dried (MgSO4) and concentrated in vacuo to afford the crude silane product as a thick, golden-yellow oil that was purified by vacuum distillation: 6.78 g (24.2 mmol, 81%); isolated as a clear, colorless thick oil; bp 92-95°C (Kugelrohr, 0.20 mmHg); FT IR (thin film) 1628 (mod.), 1616 (w) cm-1; 1H-NMR (CDCl3) δ 7.69 (2H, d, J = 8.1 Hz), 7.52 (2H, d, J = 8.1 Hz), 5.45 (1H, d, Jgeminal = 1.5 Hz), 5.15 (1H, d, Jgeminal = 1.5 Hz), 3.92 (6H, q, J = 6.3 Hz), 2.19 (3H, s), 1.29 (9H, t, J = 6.3 Hz); 13C- NMR (CDCl3) δ 143.4, 143.3, 135.1, 130.1, 125.2, 113.3, 59.0, 21.9, 18.50.
4-(Triethoxysilyl)acetophenone (7). Into a 200-mL round-bottom three-neck reaction flask equipped with a magnetic stirring bar, a fire-polished bubbler tube, a glass stopper and a gas outlet portal was placed a solution of 6 (3.00 g, 10.7 mmol) dissolved in a 1:4 (v/v) mixture of dry ethanol in dichloromethane (60 mL) and the stirred solution was cooled to -78°C. With continued cooling and stirring, the solution was treated with O3 for 5 min [16]. Following the purgement of excess ozone gas from the solution, dimethyl sulfide (3.5 mL, 3.0 g, 48.3 mmol) was added to the cooled and stirred solution over the course of 2 min and the resulting mixture was allowed to warm to and stir at RT for an additional 12 h. The reaction mixture was concentrated in vacuo and the isolated residue was dissolved into ether (50 mL). The resulting ethereal solution was washed with two portions of water (50 mL each) and saturated aqueous NaCl, dried (MgSO4) and concentrated in vacuo to afford the crude ketone product as a thick, light-yellow oil that was purified by vacuum distillation: 2.12 g (7.51 mmol, 70%); isolated as a clear, colorless thick oil; bp 128-131°C (Kugelrohr, 0.20 mmHg); FT IR (thin film) 1689 cm-1 (vs, C=O); 1H-NMR (CDCl3) δ 7.95 (2H, d, J = 8.1 Hz), 7.79 (2H, d, J = 8.1 Hz), 3.89 (6H, q, J = 7.1 Hz), 2.63 (3H, s), 1.26 (9H, t, J = 7.1 Hz); 13C-NMR (CDCl3) δ 198.7, 138.6, 137.5, 135.3, 127.5, 59.1, 26.9, 18.4.
1-(4-(Triethoxysilyl)phenyl)-4,4,4-trifluoro-1,3-butanedione (2). The general approach described by Zayia [15] was followed with several modifications. A 50-mL round-bottom three-neck reaction flask was flame-dried under vacuum and was equipped with a magnetic stirring bar, three rubber septa and a positive-pressure N2 inlet line. The flask was charged with a solution of 7 (1.00 g, 3.55 mmol) dissolved in anhydrous diethyl ether (15 mL). The flask was cooled to -78°C, and with stirring, a 1.5 M solution of LDA in cyclohexane (2.61 mL, 3.91 mmol of LDA) was added dropwise with a syringe over the course of 2 min and the resulting mixture was stirred at -78°C for an additional 90 min. With continued cooling and stirring, neat 2,2,2-trifluoroethyl trifluoroacetate (2.38 mL, 3.48 g, 17.75 mmol) was quickly added over the course of 1 min with a syringe and the resulting mixture was stirred at -78°C for 4 h. Neat H2SO4 (0.40 mL, 0.732 g, 7.46 mmol) was then added in one portion with vigorous stirring using a microsyringe and the reaction mixture was allowed to warm to RT. The mixture was poured into a 125-mL separatory funnel containing ether (25 mL) and water (25 mL), the organic layer was isolated, and was washed with water, 10% aqueous NaHCO3 and saturated aqueous NaCl, dried (MgSO4) and concentrated in vacuo to afford the title diketone 2 as a thick, clear oil that was used without further purification: 0.98 g (2.59 mmol, 73%); FT IR (thin film) 3520-3280 (mod , O-H), 1605 (vs and broad, enolic form), and 1162 cm-1 (vs and broad, C-O); 1H-NMR (CDCl3) d 14.9 (1H, bs), 7.95 (2H, d, J = 7.8 Hz), 7.80 (2H, d, J = 7.8 Hz), 6.62 (1H, s), 3.87 (6H, q, J = 7.2 Hz), 1.31 (9H, t, J = 7.2 Hz); 13C-NMR (CDCl3) d 185.8, 177.8 (q, 2JCF = 35.9 Hz), 138.9, 135.3, 130.3, 126.5, 117.1 (q, , 1JCF = 284.3 Hz), 92.5, 59.0, 18.2. The product as isolated was found to be of sufficient purity for use in the subsequent sol-gel studies. Attempts to obtain an analytical sample of 2 via vacuum distillation failed due to the labile nature of the product though it was possible to obtain an enriched fraction of >95% purity (bp 142-146°C, Kugelrohr, 0.20 mmHg).
Acknowledgements
This material is based upon work supported by the U.S. Army Research Laboratory and the U.S. Army Research Office under grant number D8AD190110476.
References and Notes
- Collinson, M. Sol-gel strategies for the preparation of selective materials for chemical analysis. Crit. Rev. Anal. Chem. 1999, 29, 289–311. [Google Scholar] [CrossRef]
- Dickey, F. H. The Preparation of Specific Adsorbents. Proc. Natl. Acad. Sci. USA 1949, 35, 227–229. [Google Scholar] [CrossRef]
- Dickey, F. H. Specific Adsorption. J. Phys. Chem. 1955, 59, 695–707. [Google Scholar] [CrossRef]
- Sasaki, D. Y.; Rush, D. J.; Daitch, C. E.; Alalm, T. M.; Assink, R. A.; Ashely, C. S.; Brinker, J. C.; Shea, K. J. In Molecular and Ionic Recognition with Imprinted Polymers, ACS Symposium Series 703; Bartsch R., A., Maeda, M., Eds.; American Chemical Society: Washington DC, USA, 1998; pp. 314–324. [Google Scholar]
- Makote, R.; Collinson, M. Template Recognition in Inorganic-Organic Hybrid Films Prepared by the Sol-Gel Process. Chem. Mater. 1998, 10, 2440. [Google Scholar] [CrossRef]
- Makote, R.; Collinson, M. Dopamine recognition in templated silicate films. Chem. Comm. 1998, 3, 425. [Google Scholar] [CrossRef]
- Kepley, L. J.; Crooks, R. M.; Ricco, A. J. A. selective SAW-based organophosphonate chemical sensor employing a self-assembled, composite monolayer: a new paradigm for sensor design. Anal. Chem. 1992, 64, 3191–3193. [Google Scholar] [CrossRef]
- Hierlemann, A.; Ricco, A. J.; Bodenhofer, K.; Gopel, W. Effective Use of Molecular Recognition in Gas Sensing: Results from Acoustic Wave and in Situ FT-IR Measurements. Anal. Chem. 1999, 71, 3022–3035. [Google Scholar] [CrossRef]
- Thomas, R. C.; Hierlemann, A.; Staton, A. W.; Hill, M.; Ricco, A. J. Reflectance Infrared Spectroscopy on Operating Surface Acoustic Wave Chemical Sensors during Exposure to Gas-Phase Analytes. Anal. Chem. 1999, 71, 3615–3621. [Google Scholar] [CrossRef]
- Stork, G.; Brizzolara, A.; Landesman, H.; Szmuszkovicz, J.; Terrell, R. The Enamine Alkylation and Acylation of Carbonyl Compounds. J. Am. Chem. Soc. 1963, 85, 207–222. [Google Scholar] [CrossRef]
- Fraenkel, G.; Geckle, J. M. Influence of substituents on NMR and barriers to rotation in tert-benzyllithium compounds. J. Am. Chem. Soc. 1980, 102, 2869–2880. [Google Scholar] [CrossRef]
- Seymour, D.; Wolfstirn, K. B. Preparation and Properties of Trimethylsilylmethanol. J. Am. Chem. Soc. 1948, 70, 1177–1179. [Google Scholar] [CrossRef]
- For an alternate preparation of 7 via a Rh(I)-catalyzed silylation of 4-iodoacetophenone, see: Murata, M.; Ishikura, M.; Nagata, M.; Watanabe, S.; Masuda, Y. Rhodium(I) Catalyzed Silylation of Aryl Halides with Triethoxysilane: Practical Synthetic Route to Aryltriethoxysilanes. Org. Lett. 2002, 4, 1843–1845. [Google Scholar] [CrossRef]
- For an alternate preparation of 7 via a Pd(0)-catalyzed silylation of 4-iodoacetophenone, see: Manoso, A. S.; DeShong, P. Improved Synthesis of Aryltriethoxysilanes via Palladium(0)-Catalyzed Silylation of Aryl Iodides and Bromides with Triethoxysilane. J. Org. Chem. 2001, 66, 7449–7455. [Google Scholar] [CrossRef]
- Zayia, G. H. First General Method for Direct Formylation of Kinetically-Generated Ketone Enolates. Org. Lett. 1999, 1, 989–991. [Google Scholar] [CrossRef]
- Claus, R.; Schreiber, S. Ozonolytic Cleavage of Cyclohexene to Terminally Differentiated Products: Methyl 6-Oxohexonate, 6,6-Dimethoxyhexanal, Methyl 6,6-Dimethoxyhexanoate. Org. Synth. 1990, Collective Volume VII, 168. [Google Scholar]
- Sample Availability: Contact the authors.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).