Abstract
A method for the preparation of methyl 4-O-methyl-α-D-glucopyranuronate and its single crystal X-ray structure determination are reported. The molecule adopts an almost ideal 4C1 (OC3) conformation.
Introduction
(4-O-Methylglucurono)xylans are important constituents of cell-wall polysaccharides of woods and other plants. These biopolymers are composed mainly of (1→4)-β-linked D-xylopyranoses, some of which are randomly branched at position O-2 with 4-O-methyl-α-D-glucopyranosyluronic acid. In this respect, methyl (benzyl 2,3-di-O-benzyl-4-O-methyl-β-D-glucopyranosid)uronate (1) and methyl 4-O-methyl-α-D-glucopyranuronate (2) represent very useful compounds in the synthesis and structural studies of model aldobiouronic acids (needed in studies related to chemical processing of wood) that reflect these structural features [1,2,3,4]. Regarding the preparation of 2, there are two methods described in the literature. The first is based on the conversion of 4-O-methyl-D-glucuronic acid to the corresponding methyl ester by refluxing in absolute methanol in the presence of Dowex-50 X-8 (H+) resin for 20 h [5]. The product was used in the next reaction step without isolation and neither experimental nor structural description data were given. The second method involves the deacetylation of methyl 1,1,2,3,5-penta-O-acetyl-4-O-methyl-aldehydo-D-glucuronate affording methyl 4-O-methyl-D-glucopyranuronate as an unseparable mixture of α- and β-anomers and, therefore, only uncomplete 1H-NMR data, [α]D and Rf values were given for this mixture [6]. Pure α-anomer was incompletely characterized (1H-NMR, [α]D, Rf) only as a corresponding 1,2,3-tri-O-acetyl derivative [6]. We now report an alternative method for the preparation and isolation of a single anomer – methyl 4-O-methyl-α-D-glucopyranuronate (2) as well as its relevant structural and spectral characteristics, including X-ray crystallography data.
Results and Discussion
According to published results [7], acetolysis of (1) led to the formation of both α- and β-1-O-acetates. On the other hand, acid hydrolysis of benzyl 2,3-di-O-benzyl-4-O-methyl-β-D-gluco-pyranosiduronic acid produced the 1-O-deprotected derivative which, without isolation, was treated with diazomethane to give the corresponding methyl glucopyranuronate as a single α-anomer [7]. It is evident that anomerization must occur during both mentioned reactions. In our hands, conventional debenzylation (hydrogenation, Pd/C) of the known 1 [7] afforded an about 2:1 mixture (by NMR) of α- and β-anomers of methyl 4-O-methyl-D-glucopyranuronate, from which the title α-anomer 2 was isolated by the slow crystallization using acetone as a solvent (Scheme 1).

Scheme 1.
The relevant coupling constant J1,2 = 3.6 Hz in the 1H-NMR as well as the signal at 93.2 ppm in the 13C-NMR spectrum, respectively, are indicative of an α-configuration at the anomeric position in 2. This structural arrangement was unambiguously confirmed by X-ray crystalography. A perspective view and the numbering scheme adopted for the molecule of 2 is depicted in Figure 1. The selected bond lengths and bond angles are listed in Table 1. A list of selected torsion angles is given in Table 2. The hydrogen bond geometry is shown in Table 3. The relevant crystallographic and structure refinement data for glucopyranuronate 2 are given in Table 4. Atomic coordinates and displacement parameters have been deposited with CCDC as supplementary information [8].
Due to absence of such protecting groups which could impose some conformational rigidity on molecule of 2, it is not surprising that the values of the relevant torsion angles O5–C1–C2–C3 = 60.05(13)°, C1–C2–C3–C4 = – 57.05(14)°, C2–C3–C4–C5 = 52.98(16)°, C3–C4–C5–O5 = – 52.65(17)°, C4–C5–O5–C1 = 58.02(16)°, C5–O5–C1–C2 = – 61.29(14)°, as well as puckering parameters [9] Q = 0.576(1) Å, Φ = 94.4(17)°, Θ = 4.9(2)° indicate an almost ideal 4C1 (OC3) conformation for the six-membered (O5–C1–C2–C3–C4–C5) pyranose ring .
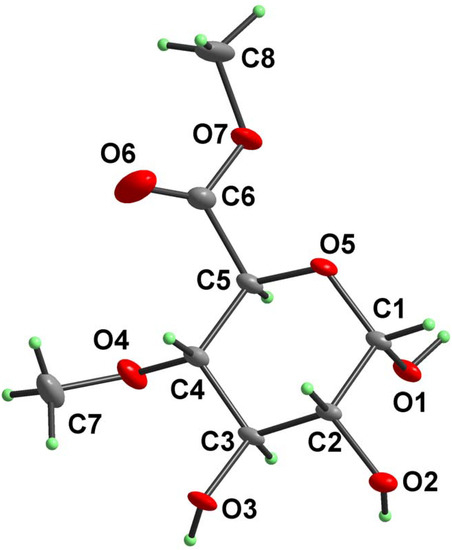
Figure 1.
Atomic numbering scheme and atomic displacement ellipsoid plot at 30% probability level for compound 2.

Table 1.
Selected bond lengths [Å] and bond angles [°] for compound 2a
| Bond | Distance | Bond angle | Angle |
|---|---|---|---|
| O1–C1 | 1.3922(17) | C1–O5–C5 | 113.76(10) |
a Standard deviations in parentheses.
Table 2.
Selected bond lengths [Å] and bond angles [°] for compound 2a
| Torsion angle | Angle | Torsion angle | Angle |
|---|---|---|---|
| C5–O5–C1–O1 | 58.47(15) | O5–C1–C2–C3 | 60.05(13) |
a Standard deviations in parentheses.
Table 3.
Hydrogen bond geometry in compound 2a
| Notation | X–H…Y | Symmetry code | X–H (Å) | H…Y (Å) | X…Y (Å) | X–H…Y (°) |
|---|---|---|---|---|---|---|
| a | O1–H1…O5i | –x+2, y+1/2, –z+1 | 0.84 | 2.00 | 2.8219(13) | 166.9 |
a Standard deviations in parentheses.
Analysis of the molecular packing in the unit cell revealed six principal hydrogen bonds (Table 3 and Figure 2). The first-level descriptors based on the graph-set theory [10] include chain C1,1(4), formed by hydrogen bonds [a] and [e], chain C1,1(7) (bonds [b]) and chain C1,1(5) formed by bonds [c], [d] and [f]). On the second-level of graph-set theory, the most interesting features are rings R2,2(10) and R2,1(5) formed by hydrogen bonds [c], [d] and [a], [b], respectively. Assignment of the H-bond descriptors, based on the graph-set theory [10] was obtained using the program PLUTO [11]. For convenience, the Xa,d(n) notation has also been adopted in this paper, in which (X) is the pattern descriptor, (a) is number of acceptors, (d) is number of donors and (n) is the number of atoms comprising the pattern.
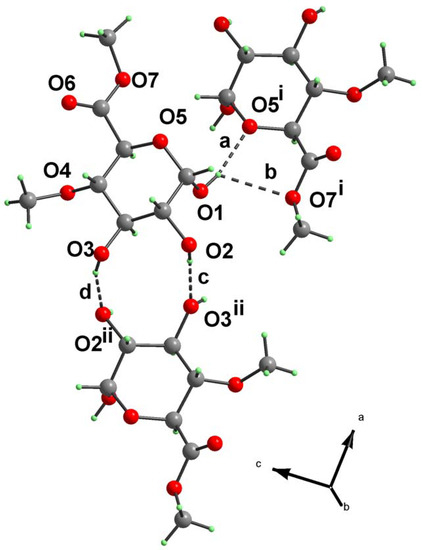
Figure 2.
Hydrogen bonding pattern in compound 2. For notation and symmetry codes see Table 3.
Experimental
General
1H- and 13C-NMR spectra (in D2O, internal standard TSP-d4) were recorded on a Bruker Avance DPX 300 instrument (equipped with gradient-enhanced spectroscopy kit GRASP for generation of Z gradient up to 50 Gauss/cm) operating at working frequencies of 300.13 and 75.46 MHz, respectively. For the assignments of signals, 1D NOESY and C–H heterocorrelated experiments were used. Specific rotations were determined on a Perkin–Elmer 241 polarimeter (25 °C, 10 cm cell). Microanalyses were performed on a Fisons EA 1108 analyser. Melting points were determined with a Boetius PHMK 05 microscope. All reactions were monitored by TLC on Silica Gel 60 plates (E. Merck). Visualisation was effected by spraying the plates with 5% (v/v) solution of H2SO4 in ethanol followed by heating at approx. 200 °C.
X-ray techniques
X-ray quality crystals of the title compound 2 were obtained by slow crystallization from dilute acetone solution. Crystal and experimental data are summarized in Table 4. Preliminary orientation matrix was obtained from the first frames using Siemens SMART software [12]. Final cell parameters were obtained by refinement of 6468 reflections using Siemens SAINT software [12]. The data were empirically corrected for absorption and other effects using the SADABS program [13] based on the method of Blessing [14]. The structure was solved by direct methods and refined by full-matrix least-squares on all F2 data using Bruker SHELXTL [15]. The non-H atoms were refined anisotropically. Hydrogen atoms were constrained to the ideal geometry using an appropriate riding model. Molecular graphics were obtained using the program DIAMOND [16].
Table 4.
Crystallographic and experimental data for compound 2a
| Empirical formula | C8H14O7 |
a Standard deviations in parentheses.
Syntheses
Methyl (benzyl 2,3-di-O-benzyl-4-O-methyl-β-D-glucopyranosid)uronate (1) was prepared from the known benzyl 2,3-di-O-benzyl-β-D-glucopyranoside [17] according to the published five-step synthetic method [7].
Methyl 4-O-methyl-α-D-glucopyranuronate (2).
A mixture of methyl (benzyl 2,3-di-O-benzyl-4-O-methyl-β-D-glucopyranosid)uronate (1) (4.0 g, 8.1 mmol) and 10 % Pd on activated charcoal (0.5 g) in acetone–methanol (1:4, v/v, 200 mL) was stirred under a hydrogen atmosphere (normal pressure) at room temperature. After 2 h, when the debenzylation was complete, the product (Rf = 0.49, chloroform–methanol 5:1; ref. [6] gives Rf = 0.38 in the same solvent) was isolated in the usual manner (filtering off the catalyst, evaporation of the solvent on a vacuum evaporator) affording a mixture of α- and β-anomers (in the ratio of about 2:1) of methyl 4-O-methyl-D-glucopyranuronate (1.75 g, 97 %). From this mixture, the pure α-anomer 2 (1.1 g, 61 %) crystallized slowly from acetone at room temperature. Recrystallization from acetone (with seeding) provided the analytical sample of the title compound 2 as colourless crystals, m.p. 130–131 °C; [α]D + 100° (c 1, MeOH), [α]D + 79° (c 1, H2O) {ref. [6] gives [α]D + 47.5° (c 0.6, 1:1 H2O–EtOH) for a mixture of α- and β-anomers}; 1H-NMR: δ 5.25 (d, 1 H, J1,2 = 3.6 Hz, H-1), 4.39 (d, 1 H, J4,5 = 9.6 Hz, H-5), 3.84 (s, 3 H, COOCH3), 3.81 (t, 1 H, J2,3 = J3,4 = 9.6 Hz, H-3), 3.59 (dd, 1 H, J1,2 = 3.6 Hz, J2,3 = 9.6 Hz, H-2), 3.48 (s, 3 H, OCH3), 3.35 (t, 1 H, J3,4 = J4,5 = 9.6 Hz, H-4); 13C-NMR: δ 172.9 (COOCH3), 93.2 (C-1), 82.2 (C-4), 72.8 (C-3), 71.9 (C-2), 70.0 (C-5), 60.8 (OCH3), 54.2 (COOCH3). Anal. Calcd for C8H14O7 (222.19): C, 43.24; H, 6.35. Found: C, 43.09; H, 6.40.
Acknowledgements
The authors thank K. Paule, Dipl. Ing. J. Tonka, and A. Karovičová (Institute of Chemistry) for microanalyses, optical rotation, and NMR spectra measurements. Financial support of this work by the Scientific Grant Agency (VEGA, Slovak Academy of Sciences, Grant Nos. 2/3077/24 and 2/3162/24) is gratefully appreciated.
References
- Timell, T.E. Adv. Carbohydr. Chem. 1964, 19, 247.
- Timell, T.E. Can. J. Chem. 1959, 37, 827.
- Kováč, P.; Petráková, E.; Kočiš, P. Carbohydr. Res. 1981, 93, 144.
- Hirsch, J.; Koóš, M.; Kováč, P. Carbohydr. Res. 1998, 310, 145.
- Das, N.N.; Das, S.C.; Dutt, A.S.; Roy, A. Carbohydr. Res. 1981, 94, 73.
- Kanie, O.; Takeda, T.; Ogihara, Y. Carbohydr. Res. 1989, 190, 53.
- Kováč, P.; Palovčík, R. Chem. Zvesti 1978, 32, 501.
- CCDC 246572 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (Fax: +44-1223-336033; e-mail: deposit@ccdc.cam.ac.uk or www: http://www.ccdc.cam.ac.uk).
- Cremer, D.; Pople, J. A. J. Am. Chem. Soc. 1975, 97, 1354.
- Bernstein, J.; Davis, R. E.; Shimoni, L.; Chang, N.-L. Angew. Chem., Int. Ed. Engl. 1995, 34, 1555.
- Motherwell, W. D. S.; Shields, G. P.; Allen, F. H. Acta Crystallogr., Sect. B 1999, 55, 1044.
- Siemens AXS. In SMART & SAINT; Madison, WI, USA, 1995.
- Sheldrick, G. M. Program SADABS; University of Göttingen: Germany, 2001. [Google Scholar]
- Blessing, R. H. Acta Crystallogr., Sect. A. 1995, 51, 33.
- Bruker AXS Inc. SHELXTL Version 6.10; Madison, WI, USA, 2001. [Google Scholar]
- Brandenburg, K. DIAMOND: Visual Crystal Structure Information System, Version 2.1e; Crystal Impact GbR: Bonn, Germany, 2001. [Google Scholar]
- Klemer, A. Chem. Ber. 1959, 92, 218.
- Sample Availability: Samples of methyl 4-O-methyl-α-D-glucopyranuronate may be obtained from the authors.
© 2005 by MDPI (http://www.mdpi.org) Reproduction is permitted for noncommercial purposes.