Abstract
Alzheimer’s disease (AD) is the most predominant age-related neurodegenerative disease, pathologically characterized by the accumulation of aggregates of amyloid beta Aβ1–42 and tau hyperphosphorylation in the brain. It is considered to be the primary cause of cognitive dysfunction. The aggregation of Aβ1–42 leads to neuronal inflammation and apoptosis. Since vitamins are basic dietary nutrients that organisms need for their growth, survival, and other metabolic functions, in this study, the underlying neuroprotective mechanism of nicotinamide (NAM) Vitamin B3 against Aβ1–42 -induced neurotoxicity was investigated in mouse brains. Intracerebroventricular (i.c.v.) Aβ1–42 injection elicited neuronal dysfunctions that led to memory impairment and neurodegeneration in mouse brains. After 24 h after Aβ1–42 injection, the mice were treated with NAM (250 mg/kg intraperitoneally) for 1 week. For biochemical and Western blot studies, the mice were directly sacrificed, while for confocal and “immunohistochemical staining”, mice were perfused transcardially with 4% paraformaldehyde. Our biochemical, immunofluorescence, and immunohistochemical results showed that NAM can ameliorate neuronal inflammation and apoptosis by reducing oxidative stress through lowering malondialdehyde and 2,7-dichlorofluorescein levels in an Aβ1–42-injected mouse brains, where the regulation of p-JNK further regulated inflammatory marker proteins (TNF-α, IL-1β, transcription factor NF-kB) and apoptotic marker proteins (Bax, caspase 3, PARP1). Furthermore, NAM + Aβ treatment for 1 week increased the amount of survival neurons and reduced neuronal cell death in Nissl staining. We also analyzed memory dysfunction via behavioral studies and the analysis showed that NAM could prevent Aβ1–42 -induced memory deficits. Collectively, the results of this study suggest that NAM may be a potential preventive and therapeutic candidate for Aβ1–42 -induced reactive oxygen species (ROS)-mediated neuroinflammation, neurodegeneration, and neurotoxicity in an adult mouse model.
1. Introduction
Alzheimer’s disease (AD), a dementing neurodegenerative disease, is a global health problem that has affected millions of individuals globally, and its prevalence continues to grow [1]. The disease is neuropathologically characterized by the accumulation of insoluble hyperphosphorylated tau proteins (intracellularly, resulting in tangle formation) and amyloid-β (Aβ) peptides (extracellularly, resulting in neuritic-plaque formation) [2]. Amyloid beta (Aβ), with 40–42 amino acid peptides, is produced by the proteolytic processing of amyloid precursor protein (APP), which is considered to be the main cause of AD [3]. Most Aβ peptides that are found in the human brain are Aβ1–40 and Aβ1–42, where Aβ1–42 is considered to be the most neurotoxic species [4]. Aβ1–42 is toxic to neuronal and glial cells [3], although the molecular mechanisms through which Aβ1–42 yields its neurotoxicity are still not yet fully understood. However, studies showed that Aβ1–42 has a vital role in oxidative stress-mediated neurodegeneration [5,6].
Growing experimental evidence has proven that oxidative stress is involved in several neuropathological disorders [5,7]. Oxidative stress induces neurodegenerative diseases, as reactive oxygen species (ROS) generation involves oxidative changes in biomolecules such as lipids, proteins, and nucleic acid, which, in turn, damage cellular function and eventually lead to neurodegeneration in the brain [8]. Increased oxidative stress in neuronal cells disrupts the endogenous antioxidant system and downregulates Nrf2/HO-1 proteins expression levels [9]. The c-Jun N-terminal kinase (JNK) is an important transducing enzyme, reported as a stress-activated kinase (SAPK), and it is activated in response to diverse inciting signals, including oxidative stress, in various neurological illnesses [10,11].
Growing experimental evidence showed that JNK, a multifunctional signaling molecule, is a key regulator of many cellular events, including development, memory formation, and brain tissue repair. However, a great deal of other evidence has also shown that the abnormal activation of JNK is related to several neuropathological diseases [12]. For instance, activated JNK is involved in Aβ1–42 -induced neuroinflammation. Similarly, other studies showed that Aβ1–42 treatment initiates a two- to threefold activation of the JNK/SAPK/c-Jun pathway in numerous neuronal cells, and that Aβ-evoked JNK activation/pathway is involved in neuronal cell death [13]. Likewise, many other supportive studies proved that the abnormal activation of JNK leads to neuroinflammatory responses, synaptic dysfunctions, and cognitive deficits associated with neurodegeneration [14].
Vitamins are important dietary nutrients needed by animals and humans for regular growth and self-maintenance [15]. Nicotinamide (NAM) is the amide form of niacin and an important precursor of nicotinamide adenine dinucleotide (NAD), which is needed for energy metabolism and cellular functions [16]. It is necessary for cells and is widely used by cellular machinery for its metabolic processes in its oxidized form (NAD+) within various organelles, such as the mitochondria [17]. The dietary intake of NAM recommended is approximately 15 mg/day [18]. It has strong neuroprotective effects against several stimuli, such as oxidative stress, free radical generation, stroke, and cerebral ischemia [18]. NAM had inhibitory potential towards neuroinflammation and neuronal apoptosis in a mouse model of traumatic brain injury [17,19]. The neuroprotective effects of NAM were recently reported in a cerebral ischemic rat model [20,21,22].
In the present study, we examined an Aβ1–42 mouse AD model and observed the neuroprotective effects of NAM. Our results showed that the intraperitoneal administration of NAM + Aβ treatment at a dose of 250 mg/kg for 1 week could abrogate Aβ1–42-induced neuroinflammation and neurodegeneration in a mouse model. As such, this study opens the door for new clinical and preclinical studies.
2. Materials and Methods
2.1. Animals
Wild-type 8-week-old male C57BL/12N mice (1. Vehicle group Total = 12 (6 mice for Western blot + 6 mice for Confocal Microscopy); 2. Aβ (Toxic group) Total = 12 (6 mice for Western blot + 6 mice for Confocal Microscopy); 3. Aβ + NAM (Treated group) Total = 12 (6 mice for Western blot + 6 mice for Confocal Microscopy, i.e., n = 36) of 25–30 g weight were obtained from Samtako Bio, Usan, Korea. The mice were managed according to the protocols of the Animal Ethics Committee of the Division of Applied Life Sciences, Gyeongsang National University, Korea (approval ID: 125, animal ethics code: 200331-M0020, 3 June 2020). Animals were adjusted in the university animal house to a 12 h light/dark cycle at 23–25 °C with 60 ± 10% humidity, and food and water were provided in a standard way. Experimental procedures were conducted in accordance with the Animal Ethics Committee (IACUC) of the Division of Applied Life Science, Department of Biology, Gyeongsang National University, Korea (Approval ID: 125). We used male mice in this study because they are resistant to stress, tough ecological conditions, and hormonal alteration [23].
2.2. Drug Treatment Protocol
A stock solution of human Aβ1–42 peptide was prepared with a specific concentration of 1 mg/mL in sterile saline solution, followed by aggregation via incubation at 37 °C for 4 days, where Intracerebroventricular (i.c.v.) administration of the Aβ1–42 peptide or vehicle (0.9% NaCl, 3 µL/5 min/mouse) was stereotaxically performed using a Hamilton microsyringe (0.2 mm anteroposterior (AP), 1 mm mediolateral (ML), and 2.4 mm dorsoventral (DV) to Bregma) in anesthetic state, homogenizing with 0.05 mL/100 g body weight.
RompunTM (xylazine) and 0.1 mL/100 g body weight ZoletilTM (ketamine). The surgical procedure was stereotaxically arranged in a separate heated room by designing the heating system in such a way as to control body temperature (maintained at 36–37 °C). Using a thermometer, temperature was regularly examined because the anesthesia reduced the animals’ body temperature. Mice were divided after 24 h from Aβ1–42 and vehicle i.c.v. injection into groups: (1) control (C) mice injected i.c.v. with 0.9% saline as a vehicle, (2) mice injected i.c.v. with Aβ1–42 (Aβ1–42 group), and (3) mice injected with Aβ1–42 and NAM (via I.P route) NAM + Aβ1–42 (250 mg/kg for 1 week); dosages of NAM were selected following previously published studies [17]. The NAM-alone group was not examined in the current study, as no ill effects of NAM have previously been reported in the brain [17,23]. Fresh NAM was prepared on daily basis in a normal saline solution followed by the needed volume of injection, and it was injected into the mice for treatment. The treatment schedule is explained in Figure 1.
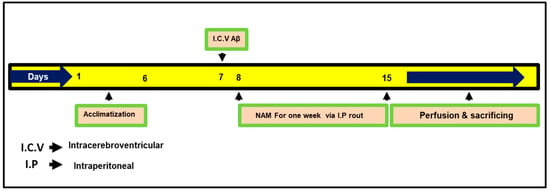
Figure 1.
Presenting study plan for the current research work. Mice were divided into three groups (1) Control (2) Aβ1–42 (3) NAM (nicotinamide) + Aβ1–42.
2.3. Morris Water Maze (MWM) Test
The behavioral pattern of mice was examined by dividing the mice into 3 groups, n = 6 for each. The experimental apparatus was a circular water tank (100 cm in diameter, 40 cm in height) with water (23 ± 1 °C) to a depth of 15.5 cm, and white ink was dissolved to make it opaque. The transparent escape platform (10 cm in diameter, 20 cm in height) was made invisible at 1 cm from the water surface and kept at the midpoint of one quadrant. Every mouse had training once per day for 5 consecutive days using a single hidden platform in one quadrant with three rotating quadrants. For each trial, latency to escape from the water maze (finding the invisible escape platform) was examined after 24 h. On Day 5, for the assessment of memory consolidation, a probe test was performed by removing the invisible platform and freely allowing each mouse to swim for 60 s. The total time that the mice spent in the target quadrant and the number of crossings over the platform site (where the platform was kept during hidden platform training) were measured. Time spent in the target quadrant signified the degree of memory consolidation. Data were documented using video tracking software (SMART, Panlab Harvard Apparatus; Bioscience Company, Holliston, MA, USA).
2.4. Y-Maze Test
The Y maze was composed of black-painted wood, with each arm being 50 cm long, 20 cm high, and 10 cm wide at the bottom and 10 cm wide at the top. The mice were kept at the center of the apparatus one by one and were allowed to move freely through the maze during three 8 min sessions. Visual observation of the series of arm entries was examined. The successive entrance of the mice into the three arms in overlapping triplet sets is called spontaneous alteration. The alteration behavior percentage (%) was measured as (successive triplet sets (entrance into three different arms consecutively)/total number of arm entries-2) × 100. Spontaneous alternation behavior with a higher percentage was thought to enhance cognitive performance.
2.5. Protein Extraction from Mouse Brain
After behavioral studies, the mice were anesthetized and euthanized using a combination of ketamine and xylazine. Brains were directly removed, the cortex and hippocampus were carefully dissected, and the tissue was stored at −80 °C. Both hippocampus and cortical tissue samples were homogenized in PRO-PREPTM protein extraction solution according to the manufacturer’s instructions (iNtRON Biotechnology, Inc., Sungnam, Korea). Centrifugation was performed at 10,000 rpm at 4 °C for 25 min, and supernatants were collected and stored at −80 °C.
2.6. Western Blot Analysis
Western blotting was conducted to measure different protein expressions in the hippocampal and cortical regions. Protein concentration was calculated using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Irvine, CA, USA). Proteins (20–25 μg) were electrophoresed in equal amounts using 4–12% BoltTM Mii Gels (Thermo Fisher Scientific Inc., Waltham, MA, USA) and SDS (sodium dodecyl sulphate (Merck KGaA, Darmstadt, Germany) running buffer 1 × (Novex, Life Technologies). Protein ladders (GangNam-STAIN™, iNtRON Biotechnology Inc., Sungnam, Korea) with a broad range of molecular-weight levels were applied for the detection of proteins’ molecular weights. All membranes were blocked in 5% (w/v) skim milk and further incubated with primary antibodies (1:1000 dilution) overnight at 4 °C to avoid nonspecific bindings. Membranes having primary antibodies were allowed to react with horseradish peroxidase-conjugated (HRP) secondary antibodies. Using ECL (Enhanced chemiluminescent) detection reagents (ATTO Corporation, Tokyo, Japan), proteins were identified to avoid extra bindings, which were further followed by scanning the X-ray films, and the optical densities of the bands were examined with densitometry using Sigma gel software, version 1.0 (SPSS, Chicago, IL, USA).
2.7. ROS Detection Assay
The ROS assay was completed with some alteration on the basis of the oxidation of DCFH-DA (2′,7′-dichlorofluorescin diacetate) to 2’,7’-dichlorodihydrofluorescein. The homogenate of all treated groups was diluted using Lock’s buffer at 1:20 time to obtain a final concentration of 5 mg tissue/mL. The reaction mixture (1 mL) with Locke’s buffer had a pH of 7.4, 0.2 mL homogenate, and 10 mL of DCFH-DA (5 mM). To acquire a fluorescent product DCF (dichlorofluorescin), the mixture was further incubated at room temperature (15 min) and calculated using a spectrofluorometer with excitation at 484 nm and emission at 530 nm. For background fluorescence (conversion of DCFH-DA in the absence of homogenate), a parallel blank was used. Data are shown as per mol DCF formed/min/mg of protein in the tissue homogenate.
2.8. Lipid Peroxidation Determination
For the assessment of oxidative stress, lipid peroxidation (LPO) was carried out. A marker of LPO free malondialdehyde (MDA) was calculated in the tissue homogenate of the hippocampus and cortical parts using a lipid peroxidation (MDA) colorimetric/fluorometric assay kit (BioVision, Milpitas, CA, USA, Cat. #K739-100), followed by the manufacturer’s protocol.
2.9. Tissue Collection and Sample Preparation
Brain samples from the control, Aβ1–42 and NAM + Aβ1–42 treated animals (n = 6 mice/group) were examined. Transcardial perfusion of mice with paraformaldehyde (4% ice-cold) was performed. Brains were kept overnight in 4% paraformaldehyde, transferred to 20% sucrose for 72 h, and frozen in an optimal cutting temperature (OCT) compound (Tissue-Tek O.C.T compound medium, Sakura Finetek USA, Inc., Torrance, CA, USA). Then, 14 μm coronal flat sections were cut using a CM 3050C cryostat (Leica Biosystems, Wetzlar, Germany). These sections were then thaw-mounted on a probe-on plus-charged slide (Thermo Fisher Scientific Inc., Waltham, MA, USA).
2.10. Immunofluorescence Analysis
An immunofluorescence study was carried out. Slides with brain tissue samples were washed twice for 10 min in 0.01 M PBS (Phosphate-Buffered Saline) by adding a proteinase K solution. Tissue samples were incubated for 60 min in blocking solution covering 2% normal goat/mouse serum and 0.3% Triton X-100 in PBS. Slides were further incubated overnight in primary antibodies at 4 °C, and further incubated at room temperature for 2 h in secondary antibodies (goat anti-mouse, fluorescein isothiocyanate (FITC)-labeled secondary antibodies (1:50 in PBS) Santa Cruz Biotechnology), after incubating the primary antibodies. The mounting medium was applied to cover the slides with glass coverslips after counterstaining with DAPI (4,6-diamidino-2-phenylindole) (for 10 min). Immunofluorescence was studied using a confocal laser-scanning microscope (Flouview FV 1000MPE, Olympus, Japan); for immunohistological quantitative analysis, ImageJ software was used (v. 1.50).
2.11. Histological Examination
To assess the histological study and degree of neuronal cell death, cresyl violet (Nissl) staining was carried out. Slides containing 14 µm tissue sections were washed twice for 15 min in 0.01 M PBS, and further stained with a 0.5% cresyl violet solution (with a few drops of glacial acetic acid) for about 10–15 min. Sections were then further washed with distilled water and dehydrated in graded ethanol (70%, 95%, and 100%), immersed in xylene, cover-slipped using a mounting medium, and slides were examined with a fluorescent light microscope. An assessment of the results was performed using Image J software.
2.12. Antibodies
All antibodies (primary and secondary) used in the present study are shown in Table 1.

Table 1.
List of antibodies used for Western Blot (WB) and immunofluorescence (IF) analysis.
2.13. Statistical Analysis
To evaluate the scanned Western blots, Sigma gel software (SPSS Inc., Chicago, IL, USA) was used, and Image J software was used to analyze immunohistological findings. One-way analysis of variance (ANOVA) followed by Student’s t-test was used to find out the mean ± SEM, and Graph Pad Prism 6 software San Diego, CA, USA was used to desi gn the graphs. “Asterisk sign (*) indicated significant difference between control and Aβ injection group; hash sign (#) indicated significant difference between Aβ injection group and Aβ + NAM treated group. Significance: (*) # = p ≤ 0.05, (**) ## = p ≤ 0.01.”
3. Results
3.1. Nicotinamide Treatment Attenuated Aβ1–42 -Elevated Oxidative Stress and p-JNK Levels in Mouse Brain
Several studies reported that oxidative stress plays an important role in the pathophysiology of numerous neurodegenerative diseases, including AD [24,25,26]. Similarly, Aβ1–42 -induced oxidative stress was demonstrated to alter the brain’s antioxidant pathways [5]. Therefore, to analyze the effect of NAM on Aβ1–42 -induced oxidative stress, we first performed reactive oxygen species (ROS) and lipid peroxidation (LPO) assays. Our results indicated a significant increase in DCF (ROS levels) and MDA (LPO levels) in Aβ1-–2-injected mouse brains in comparison with saline-injected normal mouse brains (Figure 2A,B). NAM + Aβ1–42-treatment significantly reduced the increased ROS and LPO levels in both regions (cortex and hippocampus) compared with in Aβ1–42-injected mouse brains (Figure 2A,B). Moreover, Aβ peptides were reported to initiate the JNK signaling pathway in Alzheimer’s brains [27]. Therefore, to analyze the effect of NAM on Aβ-induced oxidative stress markers and related p-JNK level, we performed Western blotting and immunofluorescence analysis. Our Western blot results showed a significant effect on the protein expression level of Nrf2/HO, which was reduced, and p-JNK, which was increased in different brain regions in Aβ1–42-injected mouse brains compared to in saline-injected normal mouse brains. However, NAM + Aβ1–42 treatment significantly upregulated the protein expression level of Nrf2/HO-1 and reduced p-JNK expression in comparison with Aβ1–42-injected mouse brains (Figure 2C).
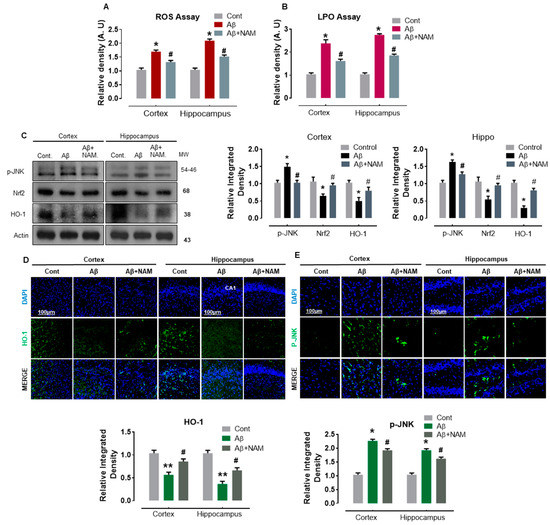
Figure 2.
NAM Ameliorating the JNK activation, oxidative stress, and regulates Nrf2, HO-1 expression in Aβ1–42-induced Mouse Brains. (A,B) representative histograms showing the ROS/LPO assays in vivo. 6 animals were kept per group and each experiment was repeated 3 times, i.e., (n = 6)/(n = 3) (C) Represents Western blot analysis of JNK, HO-1and Nrf2 proteins in mouse cortex and hippocampus, respectively. Scale bar (100 μm). (D,E) the immunofluorescence study was carried out of HO-1 and p-JNK in both cortical and hippocampal regions of mouse model. “Asterisk sign (*) indicated significant difference between control and Aβ injection group; hash sign (#) indicated significant difference between Aβ injection group and Aβ + NAM treated group. Significance: (*) # = p ≤ 0.05, (**) = p ≤ 0.01.”.
Immunofluorescence analysis also confirmed the immunoblotting results for HO-1; HO-1 reactivity was increased together with a significant reduction in p-JNK reactivity in both the indicated regions of NAM + Aβ1–42 treatment mouse brains in comparison with the Aβ1–42-injected mouse brains (Figure 2D,E). These findings indicate that NAM is effective in preventing Aβ1–42-induced oxidative stress-mediated elevated p-JNK expression level in mouse brains.
3.2. Nicotinamide Treatment Abrogates in Aβ1–42-Elevated Astrocyte and Neuroinflammatory Cytokines in Mouse Brains
Early reported studies documented that Aβ-induced glial cell activation with the subsequent release of neuroinflammatory cytokines to initiate inflammatory responses in AD pathophysiology [28,29,30]. Therefore, to analyze the effect of NAM on Aβ1–42-induced astrocyte activation and associated neuroinflammatory markers, we performed Western blotting and immunofluorescence analysis.
Our immunoblot results indicated a significant increase in the protein expression level of GFAP (an astrocyte marker), nuclear factor kappa-light-chain-enhancer of activated B cells (p-NF-KB), and its associated downstream neuroinflammatory mediators, including tumor necrosis factor alpha (TNFα) and interleukin 1 beta (IL-1β) in Aβ1–42-injected mouse brains. NAM + Aβ1–42 treatment significantly reduced astrocyte activation and its associated neuroinflammatory mediators in comparison with in Aβ1–42-injected mouse brains (Figure 3A).
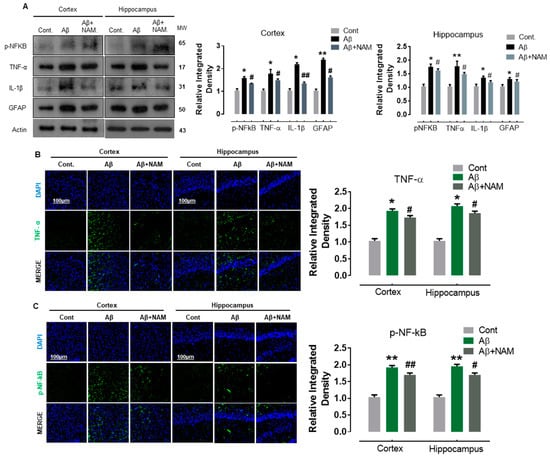
Figure 3.
NAM decreases the expression level of proinflammatory cytokines in Aβ1–42-induced Mouse Brains (A) Shows the Western blot results of inflammatory cytokines, i.e., p-NF-kB, TNF-α, GFAP and IL-1β both in cortex and hippocampus. 6 animals were kept per group. (B,C) indicating the confocal results of TNF-α, and p-NF-kB. Experiments were repeated 3 times and the scale bar was kept 100 µm. “Asterisk sign (*) indicated significant difference between control and Aβ injection group; hash sign (#) indicated significant difference between Aβ injection group and Aβ + NAM treated group. Significance: (*) # = p ≤ 0.05, (**) ## = p ≤ 0.01.”.
To assess these immunoblot results further, we performed confocal microscopy. Accordingly, our immunofluorescence results also suggested that p-NF-kB and IL-1β immunoreactivity was significantly increased (cortex and dentate gyrus (DG) region of hippocampus) in Aβ1–42-injected mouse brains in comparison with the saline-injected normal mouse brains. NAM + Aβ1–42 treatment significantly reduced the immunoreactivity of p-NF-kB and IL-1β in both the indicated regions in comparison with Aβ1–42-injected mouse brains (Figure 3B,C).
These results indicate that NAM is effective in abrogating Aβ1–42-induced neuroinflammation in the cortex and hippocampal regions of the mouse brains.
3.3. Nicotinamide Treatment Halts Aβ1–42-Induced Neurodegeneration in Mouse Brains
Previous studies determined that Aβ-induced neurodegeneration involved the initiation of apoptotic pathway in neurodegeneration [31,32,33]. Therefore, to analyze the effect of NAM on Aβ1–42 induced neuro-apoptosis, we performed Western blotting and immunofluorescence analysis. Our immunoblot results showed a significant increase in the protein expression level of BCL2-associated X protein (Bax, a proapoptotic marker), caspase-3, and poly (ADP-ribose) polymerase 1 (PARP-1, a DNA damage marker) accompanied with a significant reduction in B cell lymphoma 2 (Bcl-2, an antiapoptotic marker) in different regions in Aβ1–42-injected mouse brains. NAM+ Aβ1–42 treatment significantly reduced the elevated expression of Bax, caspase-3, and PARP-1, and significantly upregulated Bcl-2 in comparison with Aβ1–42-injected mouse brains (Figure 4A).
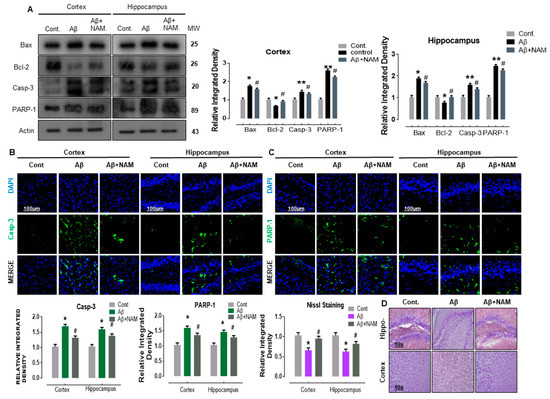
Figure 4.
NAM ameliorates Aꞵ1–42-induced neurodegeneration in an adult mouse brain. (A) Representing the Western blot results of apoptotic markers. (B,C) Indicating the confocal results of neuronal apoptotic proteins. (D) Represents the Nissl Staining results. Experiments were repeated 3 times. Number of animals were keep 6 for Western blot and 6 for confocal microscopy. Magnification 10×. Scale bare 100 µm. Asterisk sign (*) indicated significant difference between control and Aβ injection group; hash sign (#) indicated significant difference between Aβ injection group and Aβ + NAM treated group. Significance: (*) # = p ≤ 0.05, (**) = p ≤ 0.01.
To assess these immunoblot results further, we performed confocal microscopy. Accordingly, our immunofluorescence results also indicated that caspase-3 and PARP-1 immunoreactivity was significantly increased (cortex and hippocampus, DG region) in Aβ1–42-injected mouse brains in comparison with saline-injected normal mouse brains. NAM+ Aβ1–42 treatment significantly reduced the immunoreactivity of caspase-3 and PARP-1 in both the indicated regions in comparison with Aβ1–42-injected mouse brains (Figure 4B,C).
Furthermore, Nissl staining indicated that NAM + Aβ1–42 treatment significantly increased the amount of surviving neurons (cortex and DG region of hippocampus) in comparison with Aβ1–42-injected mouse brains (Figure 4D), suggesting that the extent of neuronal death was significantly reduced. These results indicate that NAM is effective in halting Aꞵ-induced apoptotic neurodegeneration in different regions of mouse brains.
3.4. Nicotinamide Treatment Reversed Aβ1–42-Induced Memory Dysfunction in Mouse Brains
Recently, it was reported that Aβ1–42-induced memory impairment [9]. Therefore, to examine the effects of NAM on learning and memory dysfunction, we performed behavioral analysis (Morris water maze (MWM) and Y-maze tests). In the MWM test, mean latency (time required in seconds (s) to find the hidden platform) was gradually reduced in all experiment groups during the training days except for the Aβ1–42-injected mouse group, which showed longer latency than that of the saline-injected normal mouse group, indicating impaired learning and memory abilities. However, compared with the Aβ1–42-injected mouse group, this effect was significantly reduced by NAM+ Aβ1–42 treatment, as the experiment mice taking less time (in seconds) to reach the hidden platform indicated improved memory performance (Figure 5A). Furthermore, we performed a probe test in which the hidden platform was removed. The time spent in the target quadrant (Figure 5B) and number of platform crossings (Figure 5C) was significantly increased in the NAM+ Aβ1–42 treatment group compared with the Aβ1–42-injected mouse group, showing that NAM reduced Aβ1–42-induced memory impairment.
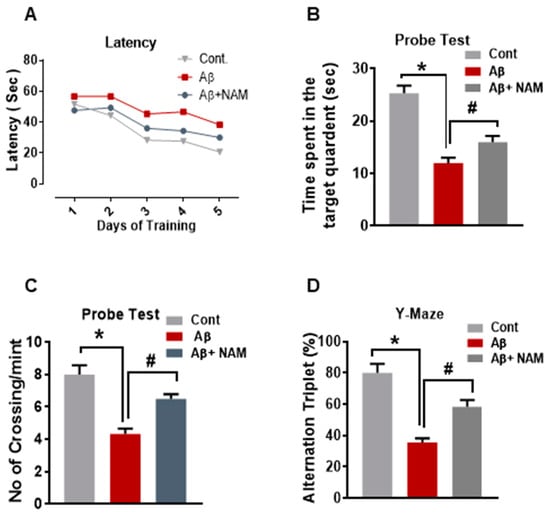
Figure 5.
NAM treatment Improved Memory Deficits in hippocampus and cortex of Aꞵ1–42 injected mice brain.(A) MWM test, where the latency to find the hidden platform was reduced in NAM+ Aβ1–42 treatment group compared to Aβ1–42-injected mouse group. (B) Probe test, the time spent in the target quadrant was increased in NAM+ Aβ1–42 treatment group compared with the Aβ1–42-injected mouse group. (C) Number of platform crossings was increased in NAM+ Aβ1–42 treatment group compared with the Aβ1–42-injected mouse group(D) Y-maze test to evaluate spatial working memory. A higher percentage (%) of spontaneous-alteration behavior was considered to be an indication of increased cognitive performance. Compared to the Aβ1–42-injected mouse group, NAM+ Aβ1–42 treatment showed a significant increase in spontaneous-alteration behavior.8 animals were kept per group. Asterisk sign (*) indicated significant difference between control and Aβ injection group; hash sign (#) indicated significant difference between Aβ injection group and Aβ + NAM treated group. Significance: (*) # = p ≤ 0.05.
After the MWM test, we performed the Y-maze test to evaluate spatial working memory. A higher percentage (%) of spontaneous-alteration behavior was considered to be an indication of increased cognitive performance. The Aβ1–42-injected mouse group exhibited significantly lower percentage (%) of spontaneous alterations than the saline-injected normal mouse group did, indicating impaired working memory. However, compared to the Aβ1–42-injected mouse group, NAM+ Aβ1–42 treatment showed a significant increase in spontaneous-alteration behavior, indicating that NAM attenuated short-term memory deficits in the Aβ1–42-injected mouse group (Figure 5D).
4. Discussion
Oxidative stress [34], (Aβ1–42-induced oxidative stress), has been documented in various (in vitro and in vivo) models of neurological disorders [35,36,37]. Neuroprotective agents [38] and emerging antioxidants as therapeutics still provide hope and are receiving increased attention [39,40]. Accordingly, in this preclinical study, we examined and explored the neuroprotective role of NAM against Aβ1–42-induced neurotoxicity in mouse brains. NAM significantly reversed Aβ1–42-induced oxidative stress, neuroinflammation, neurodegeneration, and memory impairment in Aβ1–42 treated in the mouse AD model (Figure 6).
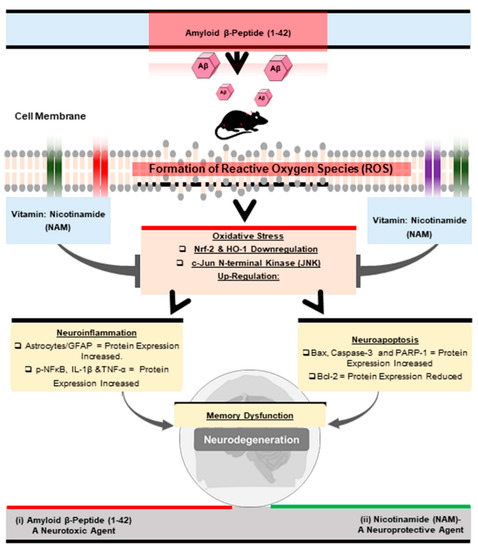
Figure 6.
Shows the graphical abstract of the current study.
Inside our body, oxidative stress (OxS), an abnormal phenomenon [41,42] ROS representing a set of large battery free radical and non-radical species are constantly formed, play an important role in cell signaling to maintain cellular homeostasis [43]. However, increased ROS production is considered to be a major cause in the development of several human diseases [44,45]. More specifically, among all body organs, the brain is the most metabolically active organ, approximately using 20% of total oxygen uptake and is, therefore, more vulnerable to oxidative damage [42]. Free radicals are highly reactive with biomolecules (carbohydrate, proteins, lipids, and DNA) [42,43] that attack brain-cell membranes, cause lipid peroxidation, and damage plasma membrane and cytosolic proteins [46]. Increased oxidative stress biomarkers were detected in AD brains [41,42,47]. Many in vitro and in vivo studies showed that Aβ treatment increased oxidative stress or ROS generation in both neuronal and non-neuronal cells in Aβ-mediated neuronal cell death [41,48]. On the other hand, many studies reported that the administration of different antioxidant vitamins and their combinations or derivatives act as a free radical scavenger, inhibits the oxidation of biomolecules, and thus prevents the development of Aβ1–42-induced ROS formation; therefore, it is effective in decreasing the incidence of disease progression [36,49,50,51,52,53,54]. Additionally, Vitamins C were shown to attenuate Aβ oligomerization and Aβ oligomer-mediated oxidative stress as one of the potential mechanisms in disease prevention [55]. Likewise, in accordance with previous findings, our results also indicated that NAM+ Aβ1–42 treatment significantly reduced Aβ1–42-induced increase in ROS and LPO levels in both the indicated regions of the mouse brain. Furthermore, upon exposure to oxidative stress, cells respond via the antioxidant response element (ARE) pathway. Several genes encoding antioxidant proteins are simultaneously induced [56]. This response is regulated upon the activation of nuclear factor E2-related factor 2 (Nrf2), key transcriptional factor that binds with AREs, activating cytoprotective genes (heme oxygenase-1 (HO-1)) [56,57]. Thus, the function of Nrf2 and its downstream target genes indicates that the Nrf2–ARE pathway is essential in the regulation of the cellular antioxidant defense system [58]. However, studies showed that signaling via Nrf2 is attenuated in both mouse models [59,60]. Recent studies, including ours and those of other research groups, also showed an altered Nrf2/HO-1 signaling pathway in both transgenic (APP/PS1 mouse model of AD) [59,60] and non-transgenic (Aβ-induced AD model) models [60,61]. In an attempt to protect neural cells, many other studies found that neuroprotectants involve the activation of Nrf2/HO-1 signaling pathways as a part of their underlying neuroprotective molecular mechanisms against Aβ-induced cell death [3,62]. On the other hand, Vitamins with antioxidant potentials proved to stimulate the antioxidant (Nrf2/HO-1) pathway, indicating its pivotal role in oxidative stress [63,64]. Likewise, in accordance with previous findings, our results also showed that NAM + Aβ1–42 treatment significantly upregulated Aβ1–42-suppressed Nrf2/HO-1 levels in both the cortex and hippocampus of the mouse brain, indicating the antioxidant effects of NAM. Furthermore, mitogen-activated protein kinases (MAPKs), for example, c-Jun N-terminal kinase (JNK), also known as stress-activated protein kinase (SAPK), is activated in response to a wide range of cellular stress types (genotoxic, proinflammatory cytokines, etc.), including Aβ oligomers [65]. Increasing evidence reported abnormal JNK activation in aged transgenic mouse and human AD brains as one of a potential mechanism in Aβ-induced neurotoxicity [66]. On the other hand, many studies reported that the administration of antioxidant vitamins markedly prevent the activation of MAPK [67]. Likewise, our results also showed that NAM + Aβ1–42 treatment suppressed Aβ1–42-induced elevated p-JNK expression in both the cortex and hippocampus of mouse brains. On the basis of the above results, NAM could be an antioxidant Vitamin that enhances antioxidant response, and limits Aβ1–42-induced oxidative stress and its associated elevated p-JNK protein expression in mouse brains (Figure 2).
A growing number of neuropathological studies have shown that neuroinflammation is an early event or feature in the pathophysiology of numerous neurodegeneration [68]. The brain is mainly populated by two broad categories of cells, neurons and neuroglial cells (astrocytes, oligodendrocytes, and ependymal and microglial cells) [69]. Glial cells were characterized as a major brain-derived sources of inflammation, as seen in AD brains [70]. Astrocytes are key elements in maintaining homeostasis and regulators of multiple physiological functions in brain [71]. However, they are responsive to various stimuli, and astrocyte dysfunction may impair neuronal function and thus lead to neurodegeneration [72]. Many studies showed that activated astrocytes are a common neuropathological feature of brain disorders that respond to Aβ metabolically, morphologically, and/or functionally [73]. Previous work from us and other research groups showed that Aβ peptides act as stimuli that significantly contribute to reactive astrocytes (activated for astrocytes [5]), which, in turn, can lead to the production of several potentially toxic inflammatory cytokines or mediators [74]. These reactive astrocytes, with the increased expression of inflammatory cytokines or molecules (IL-1β, TNF-α, COX-2, and iNOS), were well detected [75] in the brains of both AD patients and AD animal models [73]. Additionally, in various central neuroinflammation signaling pathways, Aβ peptides induce the activation of nuclear factor-kappa B (p-NF-κB; a transcription factor), which, in turn, enhances the production and expression of several inflammatory cytokines (IL-6, IL-1β, iNOS, and TNF-α) and causes neuroinflammation, as reported in neuroinflammatory neurodegeneration [76]. Many studies showed that the administration of vitamins or their derivatives inhibited the activation of the NF-κB signaling pathway and its effect on related downstream inflammatory cytokines (IL-6, IL-1β, iNOS, and TNF-α) [77]. Likewise, our results also showed that NAM+ Aβ1–42 treatment significantly inhibits Aβ1–42-activated astrocytosis (GFAP, a specific marker of activated astrocytes) and the activation of transcription factor/p-NF-κB, along with its various related proinflammatory cytokines (TNF-αand IL-1β) in the cortex and hippocampal regions of mouse brains. On the basis of the above results, NAM could be effective in reversing Aβ1–42-activated astrocytosis and the elevated expression of p-NF-κB associated with neuroinflammatory cytokines in mouse brains [5].
In recent years, evidence has indicated that neuronal apoptosis (a form of nerve cell death) and cognitive impairment are central features of many neurodegenerative diseases [78], as seen in human AD brains. ROS serve as physiological modulators of mitochondrial functions, but can damage the mitochondria, which ultimately results in apoptosis [79]. Apoptosis is a fundamental-cell death process that occurs through the activation of specific signal transduction, including the mitochondria, mitochondrial regulatory proteins, and caspase activation. In cellular apoptosis, mitochondria-associated Bcl-2 family proteins are essential modulators that are commonly categorized into two groups: one class promoting cell survival (e.g., Bcl-XL, Bcl-2, and Bcl-w) and the other facilitating cell death (e.g., Bad, Bid, Bax, and Bim). Previous studies suggested that the expression level of antiapoptotic proteins (Bcl-2 and Bcl-XL) was decreased and/or proapoptotic protein (Bax and Bim) levels were increased during Aβ-mediated apoptosis [80]. During Aβ-induced neuroapoptosis, the subsequent disturbance of or decrease in mitochondrial transmembrane potential is also clearly evident, and disrupted mitochondrial membranes may promote the apoptotic process through the leakage of apoptotic-regulated signaling molecules through mitochondria into cytosol [81]. In addition, caspases, particularly the activation of caspase-3 (a cysteine-dependent aspartic protease) by the Aβ peptide, were reported as final effector molecules in apoptotic cell death. For example, in Aβ1–42-induced neuronal apoptosis, many laboratories showed the involvement and activation of different caspases (caspase-9 and caspase-3) in neuronal populations or cell types and in animal models of AD [31]. Others showed that the activated neuronal caspase-3 causes the cleavage of poly (ADP-ribose) polymerase-1 (PARP-1, an essential enzyme important in DNA repair), thereby promoting apoptosis [82]. Indeed, both in in vivo and in vitro studies, and even in postmortem human brain tissue, apoptosis is commonly observed in Aβ-induced toxicity [83]. On the other hand, many studies reported that the administration of different antioxidant vitamins significantly inhibited neuronal apoptosis via the regulation of pro-and antiapoptotic proteins, thereby preventing brain damage [84,85]. Our results also showed that NAM+ Aβ1–42 treatment significantly reversed Aβ1–42-induced increases in Bax, caspase-3, and PARP-1 levels in both the cortex and hippocampal regions of the mouse brain, indicating the antiapoptotic effects of NAM. Additionally, Nissl staining (used for the measurement of neuronal loss) indicated that NAM+ Aβ1–42 treatment significantly reversed Aβ1–42-induced neuronal cell death, as it significantly increased the amount of surviving neurons in the cortex and hippocampal regions of the mouse brain (Figure 4C,D).
With regard to Aβ1–42-induced neuronal apoptosis, our group and others reported that Aβ1–42-induced memory or cognitive dysfunctions in mouse models of AD [31]. On the other hand, the administration of different antioxidant vitamins, either alone or in combination, were well-studied, and proven to improve learning and memory performance in rodent models [84]. Our results also showed that NAM + Aβ1–42 treatment reduced Aβ1–42-induced memory dysfunction, as evidenced from behavioral analysis (MWM and Y-maze tests), thus improving cognition, spatial learning, and memory processing (Figure 5). These results showed that NAM is significantly effective in reversing Aβ1–42-induced apoptosis neurodegeneration and memory impairment in mouse brains.
5. Conclusions
In conclusion, our study provides considerable evidence that NAM can abrogate Aβ1–42 induced oxidative stress, neuroinflammation, and neuronal apoptosis. NAM+ Aꞵ treatment maintains the cellular antioxidant system and regulates Nrf2/HO-1 protein levels. This shows the therapeutic potential of NAM against Aβ1–42-accelerated neurotoxicity, and may also open the door for new therapeutic preclinical research work to be carried out (Figure 6).
Author Contributions
I.U.R. designed, and conducted the experiments, wrote the manuscript, and performed the statistical analysis; R.A., I.K., H.J.L., J.P., R.U. conducted experiments, reviewed, and edited the manuscript. M.J.C. and H.Y.K. helped in resources and validation of data. M.O.K. supplied all of the chemicals reagents, supervised, and approved the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Neurological Disorder Research Program of the National Research Foundation (NRF) funded by the Korean Government (MSIT) (2020M3E5D9080660).
Institutional Review Board Statement
This study was carried out in animals in accordance with approved guidelines (Approval ID: 125) by the animal ethics committee (IACUC) of the Division of Applied Life Science, Gyeongsang National University, Korea.
Informed Consent Statement
Not applicable.
Data Availability Statement
The authors hereby declares that the data presented in this study will be presented upon request from the corresponding author.
Conflicts of Interest
All authors declare no conflict of interest.
References
- Kepp, K.P. Ten challenges of the amyloid hypothesis of Alzheimer’s disease. J. Alzheimer’s Dis. 2016, 55, 447–457. [Google Scholar] [CrossRef]
- Shah, S.A.; Yoon, G.H.; Chung, S.S.; Abid, M.N.; Kim, T.H.; Lee, H.Y.; Kim, M.O. Novel osmotin inhibits SREBP2 via the AdipoR1/AMPK/SIRT1 pathway to improve Alzheimer’s disease neuro-pathological deficits. Mol. Psych. 2017, 22, 407–416. [Google Scholar] [CrossRef]
- Lee, C.; Park, G.H.; Kim, C.Y.; Jang, J.H. [6]-Gingerol attenuates beta-amyloid-induced oxidative cell death via fortifying cellular antioxidant defense system. Food Chem. Toxicol. 2011, 49, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Boyd-Kimball, D. Amyloid beta-peptide (1–42) contributes to the oxidative stress and neurodegeneration found in Alzheimer disease brain. Brain Pathol. 2004, 14, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Amin, F.U.; Shah, S.A.; Kim, M.O. Vanillic acid attenuates Abeta1-42-induced oxidative stress and cognitive impairment in mice. Sci. Rep. 2017, 7, 40753. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Holscher, C. The effect of ageing on neurogenesis and oxidative stress in the APP (swe)/PS1(deltaE9) mouse model of Alzheimer’s disease. Brain Res. 2012, 1449, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Ali, T.; Kim, M.W.; Jo, M.H.; Chung, J.I.; Kim, M.O. Anthocyanins improve hippocampus-dependent memory function and prevent neurodegeneration via JNK/Akt/GSK3beta signaling in LPS-treated adult mice. Mol. Neurobiol. 2019, 56, 671–687. [Google Scholar] [CrossRef]
- Ali, T.; Kim, M.O. Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegen-eration via PI3/Akt/GSk3beta pathway in the mouse hippocampus. J. Pineal Res. 2015, 59, 47–59. [Google Scholar] [CrossRef]
- Borsello, T.; Forloni, G. JNK signalling: A possible target to prevent neurodegeneration. Curr. Pharm. Des. 2007, 13, 1875–1886. [Google Scholar] [CrossRef]
- Ullah, R.; Jo, M.H.; Riaz, M.; Alam, S.I.; Saeed, K.; Ali, W.; Rehman, I.U.; Ikram, M.; Kim, M.O. Glycine, the smallest amino acid, confers neuroprotection against d-galactose-induced neurodegeneration and memory impairment by regulating c-Jun N-terminal kinase in the mouse brain. J. Neuroinflamm. 2020, 17, 303. [Google Scholar] [CrossRef]
- Mehan, S.; Meena, H.; Sharma, D.; Sankhla, R. JNK: A stress-activated protein kinase therapeutic strategies and involvement in Alzheimer’s and various neu-rodegenerative abnormalities. J. Mol. Neurosci. 2011, 43, 376–390. [Google Scholar] [CrossRef]
- Troy, C.M.; Rabacchi, S.A.; Xu, Z.; Maroney, A.C.; Connors, T.J.; Shelanski, M.L.; Greene, L.A. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J. Neurochem. 2001, 77, 157–164. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, M.; Du, Y.; Zhang, W.; Bai, M.; Zhang, Z.; Li, Z.; Miao, J. Inhibition of c-Jun N-terminal kinase activation reverses Alzheimer disease phenotypes in APPswe/PS1dE9 mice. Ann. Neurol. 2015, 77, 637–654. [Google Scholar] [CrossRef]
- Taguchi, K.; Fukusaki, E.; Bamba, T. Simultaneous analysis for water- and fat-soluble Vitamins by a novel single chromatog-raphy technique unifying supercritical fluid chromatography and liquid chromatography. J. Chromatogr. A 2014, 1362, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Hoane, M.R.; Kaplan, S.A.; Ellis, A.L. The effects of nicotinamide on apoptosis and blood–brain barrier breakdown following traumatic brain injury. Brain Res. 2006, 1125, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.I.; Ur Rehman, S.; Kim, M.O. Nicotinamide improves functional recovery via regulation of the rage/Jnk/NF-kappab signaling pathway after brain injury. J. Clin. Med. 2019, 8, 271. [Google Scholar] [CrossRef] [PubMed]
- Rennie, G.; Chen, A.C.; Dhillon, H.; Vardy, J.; Damian, D.L. Nicotinamide and neurocognitive function. Nutr. Neurosci. 2014, 18, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Gharavi, R.; Pitta, M.; Gleichmann, M.; Mattson, M.P. Nicotinamide prevents NAD+ depletion and protects neurons against Excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. NeuroMol. Med. 2009, 11, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Mengistu, G.; Moges, T.; Samuel, A.; Baye, K. Energy and nutrient intake of infants and young children in pastoralist communities of Ethiopia. Nutrients 2017, 41, 1–6. [Google Scholar] [CrossRef]
- Peechakara, B.V.; Gupta, M. Vitamins B3; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Shen, C.C.; Huang, H.M.; Ou, H.C.; Chen, H.L.; Chen, W.C.; Jeng, K.C. Protective effect of nicotinamide on neuronal cells under oxygen and glucose deprivation and hypox-ia/reoxygenation. J. Biomed. Sci. 2004, 11, 472–481. [Google Scholar] [CrossRef]
- Ahmad, R.; Khan, A.; Lee, H.J.; Ur Rehman, I.; Khan, I.; Alam, S.I.; Kim, M.O. Lupeol, a plant-derived triterpenoid, protects mice brains against abeta-induced oxidative stress and neuro-degeneration. Biomedicines 2020, 8, 380. [Google Scholar] [CrossRef] [PubMed]
- Ullah, N.; Lee, H.Y.; Naseer, M.I.; Ullah, I.; Suh, J.W.; Kim, M.O. Nicotinamide inhibits alkylating agent-induced apoptotic neurodegeneration in the developing rat brain. PLoS ONE 2011, 6, e27093. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Perry, G.; Chiba, S.; Atwood, C.S.; Petersen, R.B.; Smith, M.A.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; et al. Oxidative damage is the earliest event in alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Ullah, R.; Khan, M.; Shah, S.A.; Saeed, K.; Kim, M.O. Natural antioxidant anthocyanins—a hidden therapeutic candidate in metabolic disorders with major focus in neurodegeneration. Nutrients 2019, 11, 1195. [Google Scholar] [CrossRef]
- Vukic, V.; Callaghan, D.; Walker, D.; Lue, L.F.; Liu, Q.Y.; Couraud, P.O.; Zhang, W. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alz-heimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol. Dis. 2009, 2009. 34, 95–106. [Google Scholar] [CrossRef]
- Craft, J.M.; Watterson, D.M.; Van Eldik, L.J. Human amyloid beta-induced neuroinflammation is an early event in neuro-degeneration. Glia 2006, 53, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Scuderi, C.; Savani, C.; Steardo, L., Jr.; De Filippis, D.; Cottone, P.; Steardo, L. Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS ex-pression. Br. J. Pharmacol. 2007, 151, 1272–1279. [Google Scholar] [CrossRef]
- Ahmad, A.; Ali, T.; Park, H.Y.; Badshah, H.; Rehman, S.U.; Kim, M.O. Neuroprotective effect of fisetin against amyloid-beta-induced cognitive/Synaptic dysfunction, neuroinflammation, and neurodegeneration in adult mice. Mol. Neurobiol. 2017, 54, 2269–2285. [Google Scholar] [CrossRef]
- Cancino, G.I.; Toledo, E.M.; Leal, N.R.; Hernandez, D.E.; Yévenes, L.F.; Inestrosa, N.C.; Alvarez, A.R. STI571 prevents apoptosis, tau phosphorylation and behavioural impairments induced by Alzheimer’s be-ta-amyloid deposits. Brain 2008, 131, 2425–2442. [Google Scholar] [CrossRef]
- Ali, W.; Ikram, M.; Park, H.Y.; Jo, M.G.; Ullah, R.; Ahmad, S.; Kim, M.O. Oral administration of alpha linoleic acid rescues abeta-induced glia-mediated neuroinflammation and cognitive dysfunction in C57BL/6N mice. Cells 2020, 9, 667. [Google Scholar] [CrossRef]
- Ali, T.; Yoon, G.H.; Shah, S.A.; Lee, H.Y.; Kim, M.O. Osmotin attenuates amyloid beta-induced memory impairment, tau phosphorylation and neurodegeneration in the mouse hippocampus. Sci. Rep. 2015, 5, srep11708. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative stress in neurodegenerative diseases: From molecular mechanisms to clinical applications. Oxidative Med. Cell. Longev. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, Y.; Kawamoto, T.; Bennett, B.M. Role of the lipid peroxidation product, 4-hydroxynonenal, in the development of nitrate tolerance. Chem. Res. Toxicol. 2014, 27, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Yatin, S.M.; Varadarajan, S.; Link, C.D.; Butterfield, D.A. In vitro and in vivo oxidative stress associated with Alzheimer’s amyloid beta-peptide (1–42). Neurobiol. Aging 1999, 20, 325–330. [Google Scholar] [PubMed]
- Praticò, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M.-Y. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of alzheimer amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [CrossRef] [PubMed]
- Dunkel, P.; Chai, C.L.; Sperlágh, B.; Huleatt, P.B.; Mátyus, P. Clinical utility of neuroprotective agents in neurodegenerative diseases: Current status of drug development for Alzheimer’s, Parkinson’s and Huntington’s diseases, and amyotrophic lateral sclerosis. Expert Opin. Investig. Drugs 2012, 21, 1267–1308. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.N.; Firuzi, O.; Gama, M.J.; Van Horssen, J.; Saso, L. Oxidative stress and antioxidants in neurological diseases: Is there still hope? Curr. Drug Targets 2017, 18, 705–718. [Google Scholar] [CrossRef]
- Floyd, R.A. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc. Soc. Exp. Boil. Med. 1999, 222, 236–245. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 2007, 43, 658–677. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Stone, T.W.; Smith, R.A. Oxidative stress in neurodegeneration and available means of protection. Front. Biosci. 2008, 13, 3288–3311. [Google Scholar] [CrossRef]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, R. Free radicals, oxidative stress and antioxidant vitamins. Comptes Rendus des Seances de la Soc. Boil. Fil. 1993, 187, 277–285. [Google Scholar]
- Yatin, S.M.; Aksenov, M.; Butterfield, D.A. The antioxidant Vitamin E modulates amyloid beta-peptide-induced creatine kinase activity inhibition and increased protein oxidation: Implications for the free radical hypothesis of Alzheimer’s disease. Neurochem. Res. 1999, 24, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Boil. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.-Q.; Park, B.-C.; Lee, J.-S.; Choi, H.-D.; Lee, Y.-S.; Yang, J.-H.; Kim, J.-A. Mycelial extract of cordyceps ophioglossoides prevents neuronal cell death and ameliorates. BETA-Amyloid peptide-induced memory deficits in rats. Biol. Pharm. Bull. 2004, 27, 1126–1129. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-N.; Ho, Y.-J.; Lai, C.-C.; Chiu, C.-T.; Wang, J.-Y. 1,25-Dihydroxyvitamin D3 attenuates endotoxin-induced production of inflammatory mediators by inhibiting MAPK activation in primary cortical neuron-glia cultures. J. Neuroinflamm. 2015, 12, 1–12. [Google Scholar] [CrossRef]
- Mocchegiani, E.; Costarelli, L.; Giacconi, R.; Malavolta, M.; Basso, A.; Piacenza, F.; Ostan, R.; Cevenini, E.; Gonos, E.S.; Franceschi, C.; et al. Vitamin E–gene interactions in aging and inflammatory age-related diseases: Implications for treatment. A systematic review. Ageing Res. Rev. 2014, 14, 81–101. [Google Scholar] [CrossRef]
- Cheeseman, K.H.; Slater, T.H. An introduction to free radical biochemistry. Br. Med. Bull. 1993, 49, 481–493. [Google Scholar] [CrossRef]
- Koppal, T.; Subramaniam, R.; Drake, J.; Prasad, M.R.; Dhillon, H.; Butterfield, D.A. Vitamins E protects against Alzheimer’s amyloid peptide (25–35)-induced changes in neocortical synapto-somal membrane lipid structure and composition. Brain Res. 1998, 786, 270–273. [Google Scholar] [CrossRef]
- Morris, M.C.; Beckett, L.A.; Scherr, P.A.; Hebert, L.E.; Bennett, D.A.; Field, T.S.; Evans, D.A. Vitamin E and vitamin C supplement use and risk of incident alzheimer disease. Alzheimer Dis. Assoc. Disord. 1998, 12, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Guarino, P.D. Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM-AD VA co-operative randomized trial. JAMA 2014, 311, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Murata, N.; Ozawa, Y.; Kinoshita, N.; Irie, K.; Shirasawa, T.; Shimizu, T. Vitamin C restores behavioral deficits and amyloid-beta oligomerization without affecting plaque for-mation in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 26, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fields, J.; Zhao, C.; Langer, J.; Thimmulappa, R.K.; Kensler, T.W.; Yamamoto, M.; Biswal, S.; Doré, S. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic. Biol. Med. 2007, 43, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Johnson, J.A. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J. Biochem. Mol. Biol. 2004, 37, 139–143. [Google Scholar] [CrossRef]
- Kanninen, K.; Malm, T.M.; Jyrkkänen, H.-K.; Goldsteins, G.; Keksa-Goldsteine, V.; Tanila, H.; Yamamoto, M.; Ylä-Herttuala, S.; Levonen, A.-L.; Koistinaho, J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell. Neurosci. 2008, 39, 302–313. [Google Scholar] [CrossRef]
- Wang, Y.; Miao, Y.; Mir, A.Z.; Cheng, L.; Wang, L.; Zhao, L.; Cui, Q.; Zhao, W.; Wang, H. Inhibition of beta-amyloid-induced neurotoxicity by pinocembrin through Nrf2/HO-1 pathway in SH-SY5Y cells. J. Neurol. Sci. 2016, 368, 223–230. [Google Scholar] [CrossRef]
- Saeed, K.; Shah, S.A.; Ullah, R.; Alam, S.I.; Park, J.S.; Saleem, S.; Kim, M.O. Quinovic acid impedes cholesterol dyshomeostasis, oxidative stress, and neurodegeneration in an amy-loid-beta-induced mouse model. Oxid. Med. Cell Longev. 2020, 2020, 9523758. [Google Scholar] [CrossRef]
- Wruck, C.J.; Götz, M.E.; Herdegen, T.; Varoga, D.; Brandenburg, L.O.; Pufe, T. Kavalactones protect neural cells against amyloid beta peptide-induced neurotoxicity via extracellular signal-regulated kinase 1/2-dependent nuclear factor erythroid 2-related factor 2 activation. Mol. Pharmacol. 2008, 73, 1785–1795. [Google Scholar] [CrossRef]
- El-Din, S.S.; Rashed, L.; Medhat, E.; Aboulhoda, B.E.; Badawy, A.D.; ShamsEldeen, A.M.; Abdelgwad, M. Active form of vitamin D analogue mitigates neurodegenerative changes in Alzheimer’s disease in rats by targeting Keap1/Nrf2 and MAPK-38p/ERK signaling pathways. Steroids 2020, 156, 108586. [Google Scholar] [CrossRef]
- Nakai, K.; Fujii, H.; Kono, K.; Goto, S.; Kitazawa, R.; Kitazawa, S.; Hirata, M.; Shinohara, M.; Fukagawa, M.; Nishi, S. Vitamin D activates the NRF2-KEAP1 antioxidant pathway and ameliorates nephropathy in diabetic rats. Am. J. Hypertens. 2014, 27, 586–595. [Google Scholar] [CrossRef]
- Jia, L.; Liu, J.; Song, Z.; Pan, X.; Chen, L.; Cui, X.; Wang, M. Berberine suppresses amyloid-beta-induced inflammatory response in microglia by inhibiting nuclear fac-tor-kappaB and mitogen-activated protein kinase signalling pathways. J. Pharm. Pharmacol. 2012, 64, 1510–1521. [Google Scholar] [CrossRef]
- Yao, M.; Nguyen, T.V.; Pike, C.J. Beta-amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J. Neurosci. 2005, 25, 1149–1158. [Google Scholar] [CrossRef]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Viña, J. Abeta and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamins E. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Eikelenboom, P.; Van Exel, E.; Hoozemans, J.J.M.; Veerhuis, R.; Rozemuller, A.J.M.; Van Gool, W.A. Neuroinflammation–An early event in both the history and pathogenesis of alzheimer’s disease. Neurodegener. Dis. 2010, 7, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Calvillo, M.; Diaz, A.; Limon, D.I.; Mayoral, M.A.; Chánez-Cárdenas, M.E.; Zenteno, E.; Espinosa, B. Amyloid-beta (25–35) induces a permanent phosphorylation of HSF-1, but a transitory and inflamma-tion-independent overexpression of Hsp-70 in C6 astrocytoma cells. Neuropeptides 2013, 47, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I. Diversity of astroglial responses across human neurodegenerative disorders and brain aging. Brain Pathol. 2017, 27, 645–674. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Agrawal, A.; Singal, C.M.S.; Pandey, H.S.; Seth, P.; Sharma, S.K. Sinomenine inhibits amyloid beta-induced astrocyte activation and protects neurons against indirect toxicity. Mol. Brain 2020, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.; Lee, C.J. Reactive astrocytes in Alzheimer’s disease: A double-edged sword. Neurosci. Res. 2018, 126, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Akama, K.T.; Krafft, G.A.; Chromy, B.A.; Van Eldik, L.J. Amyloid-beta peptide activates cultured astrocytes: Morphological alterations, cytokine induction and nitric oxide release. Brain Res. 1998, 785, 195–206. [Google Scholar] [CrossRef]
- Deng, Y.; Long, L.; Wang, K.; Zhou, J.; Zeng, L.; He, L.; Gong, Q. Icariside II, a broad-spectrum anti-cancer agent, reverses beta-amyloid-induced cognitive impairment through reducing inflammation and apoptosis in rats. Front. Pharmacol. 2017, 8, 39. [Google Scholar] [CrossRef]
- Khan, M.S.; Ali, T.; Abid, M.N.; Jo, M.H.; Khan, A.; Kim, M.W.; Yoon, G.H.; Cheon, E.W.; Rehman, S.U.; Kim, M.O. Lithium ameliorates lipopolysaccharide-induced neurotoxicity in the cortex and hippocampus of the adult rat brain. Neurochem. Int. 2017, 108, 343–354. [Google Scholar] [CrossRef]
- Shi, H.Y.; Yan, S.M.; Guo, Y.M.; Zhang, B.Q.; Guo, X.Y.; Shi, B.L. Vitamin A pretreatment protects NO-induced bovine mammary epithelial cells from oxidative stress by modulating Nrf2 and NF-kappaB signaling pathways. J. Anim. Sci. 2018, 96, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Sandry, J. Working memory and memory loss in neurodegenerative disease. Neurodegener. Dis. Manag. 2015, 5, 1–4. [Google Scholar] [CrossRef]
- Richter, C.; Gogvadze, V.; Laffranchi, R.; Schlapbach, R.; Schweizer, M.; Suter, M.; Walter, P.; Yaffee, M. Oxidants in mitochondria: From physiology to diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1995, 1271, 67–74. [Google Scholar] [CrossRef]
- Bar-Am, O.; Weinreb, O.; Amit, T.; Youdim, M.B.H. Regulation of Bcl-2 family proteins, neurotrophic factors, and APP processing in the neurorescue activity of propargylamine. FASEB J. 2005, 19, 1899–1901. [Google Scholar] [CrossRef]
- Crouch, P.J.; Harding, S.M.E.; White, A.R.; Camakaris, J.; Bush, A.I.; Masters, C.L. Mechanisms of A beta mediated neurodegeneration in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2008, 40, 181–198. [Google Scholar] [CrossRef]
- Sairanen, T.; Szepesi, R.; Karjalainen-Lindsberg, M.-L.; Saksi, J.; Paetau, A.; Lindsberg, P.J. Neuronal caspase-3 and PARP-1 correlate differentially with apoptosis and necrosis in ischemic human stroke. Acta Neuropathol. 2009, 118, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhou, L.; Sun, M.; Zhou, T.; Zhong, K.; Wang, H.; Chui, D. Xylocoside G reduces amyloid-beta induced neurotoxicity by inhibiting NF-kappaB signaling pathway in neuronal cells. J. Alzheimers Dis. 2012, 30, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.-X.; Tao, J.; Zhang, N.; Wang, J.-Z. Neuroprotective properties of vitamin C on equipotent anesthetic concentrations of desflurane, isoflurane, or sevoflurane in high fat diet fed neonatal mice. Int. J. Clin. Exp. Med. 2015, 8, 10444–10458. [Google Scholar]
- Chong, Z.-Z.; Lin, S.-H.; Li, F.; Maiese, K. The sirtuin inhibitor nicotinamide enhances neuronal cell survival during acute anoxic injury through akt, bad, parp, and mitochondrial associated “anti-apoptotic” pathways. Curr. Neurovascular Res. 2005, 2, 271–285. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).