
Complex Interplay of Heme-Copper Oxidases with Nitrite and Nitric Oxide



{kind=link}
{kind=link}
{kind=link}
{kind=link}
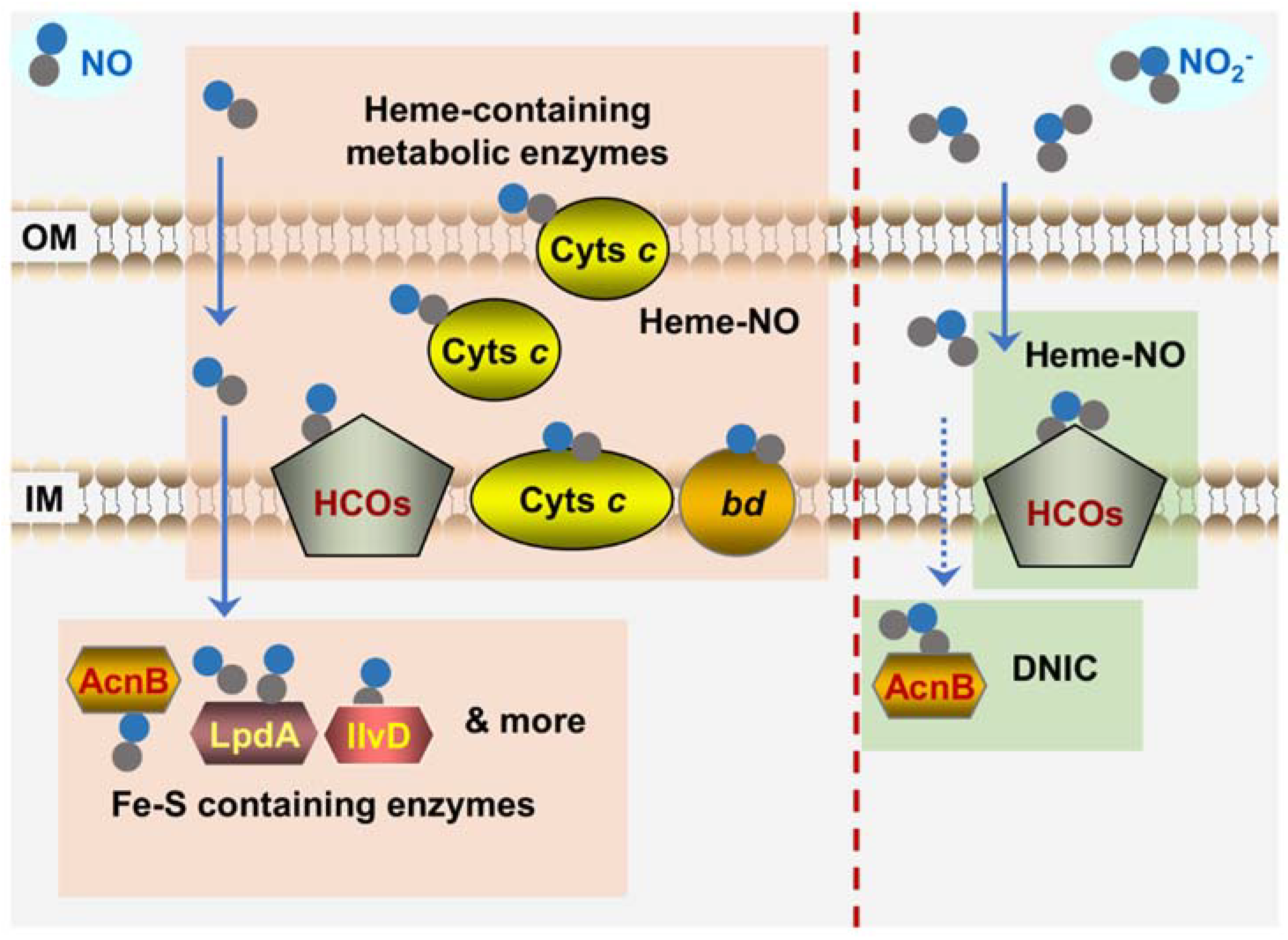{kind=link}
Abstract
1. Introduction
2. Bacterial Terminal Oxidases for pmf Generation
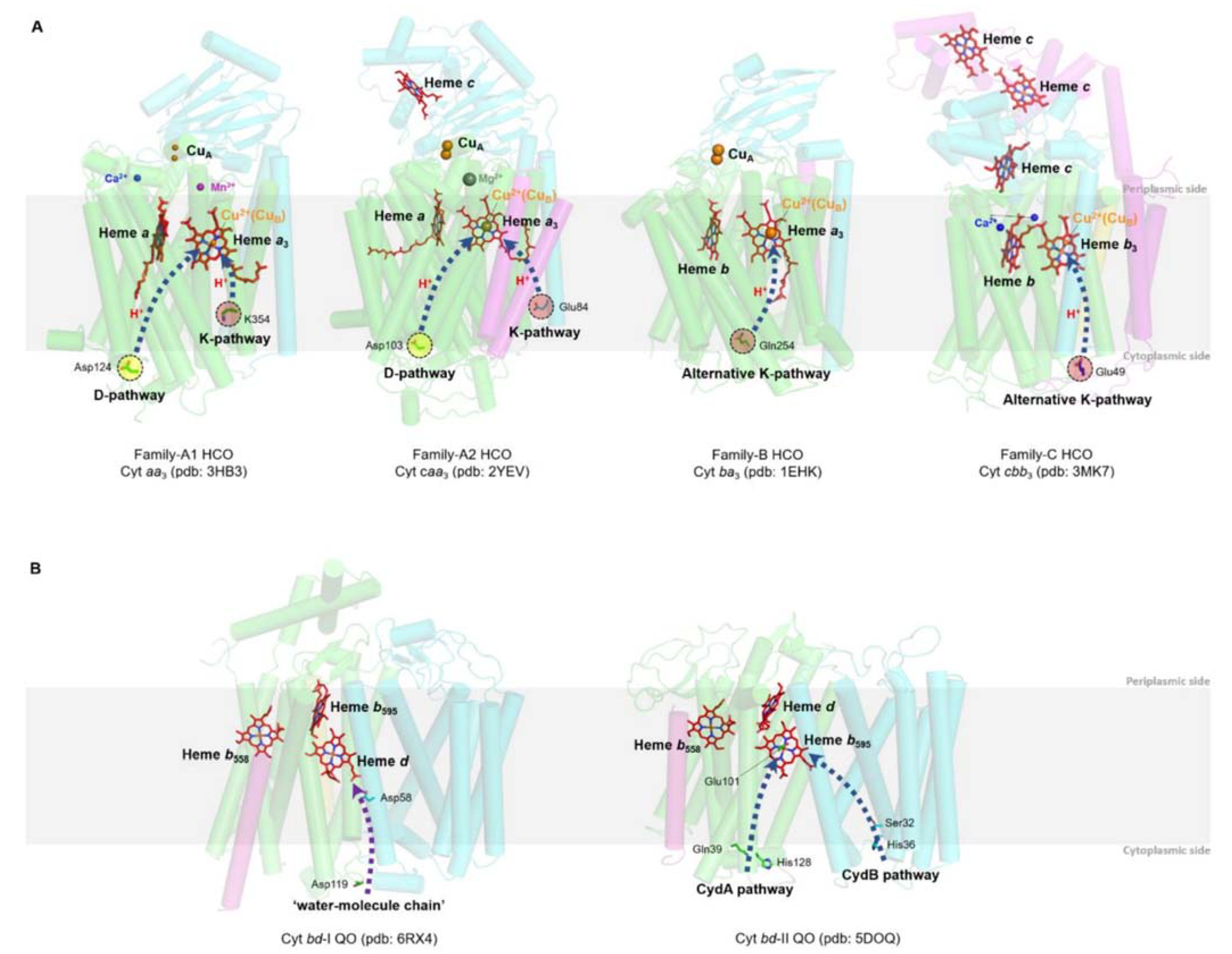
2.1. HCOs
2.2. bd QO
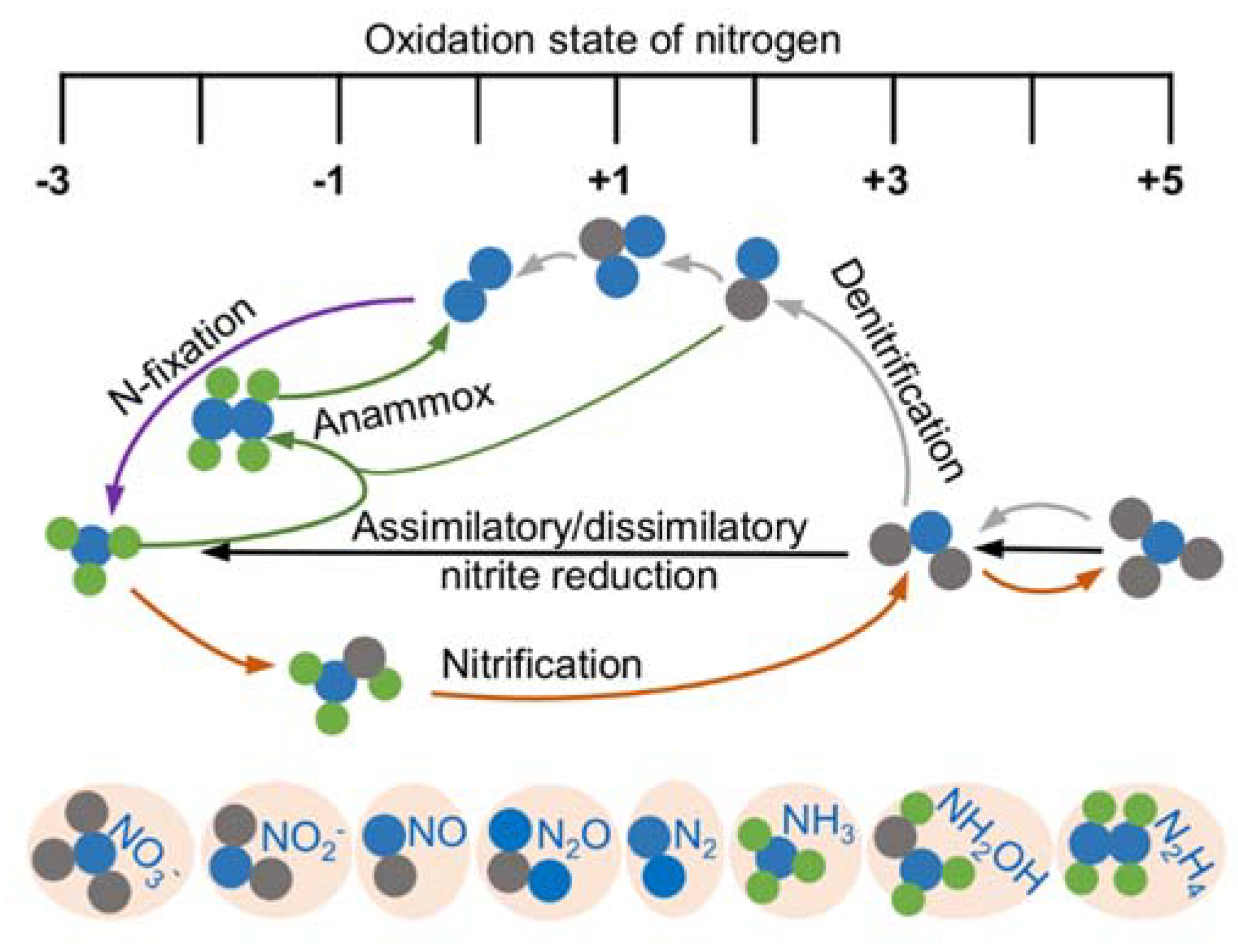
3. Roles of HCOs in the Transformation of Nitrogen Oxides
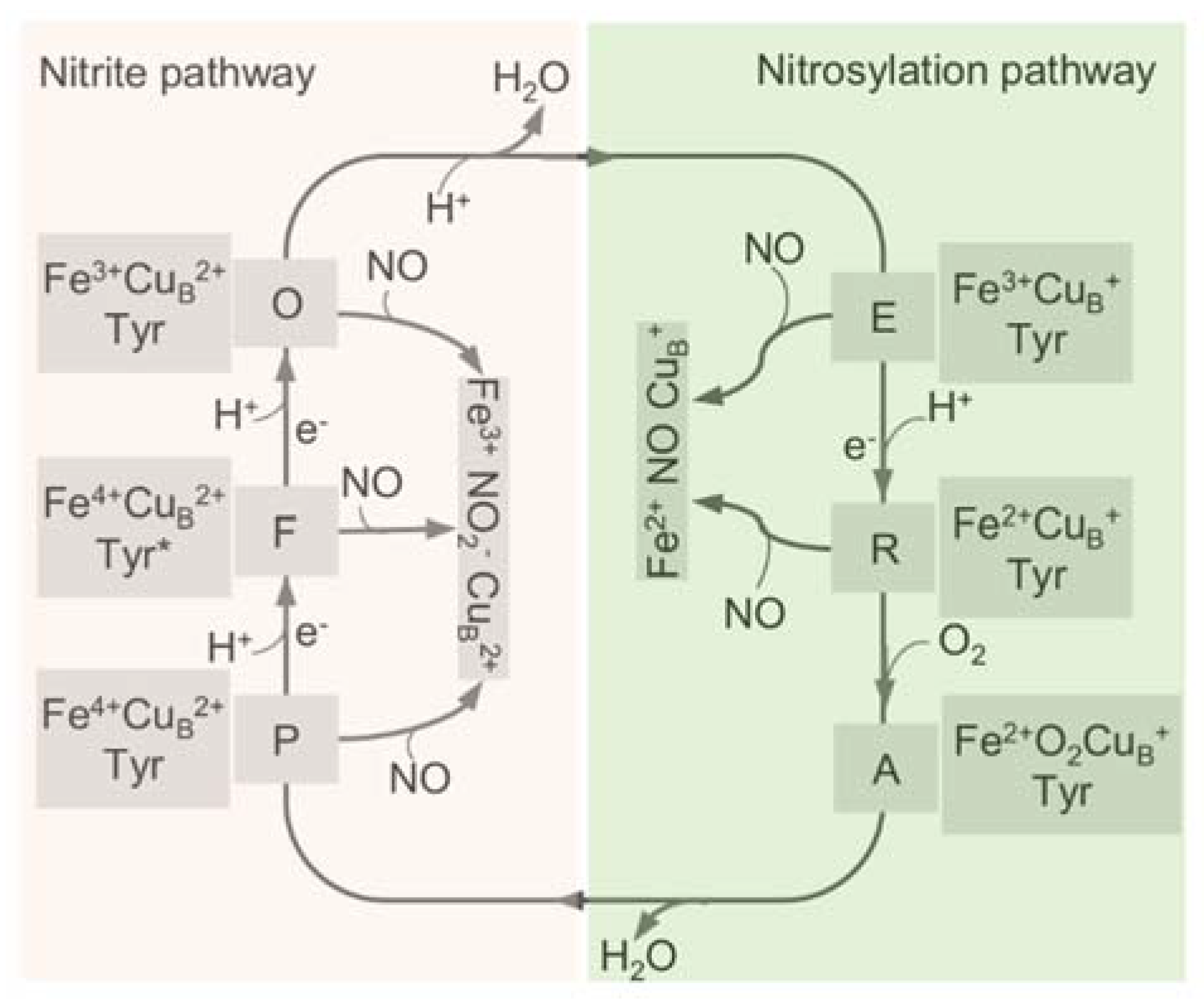
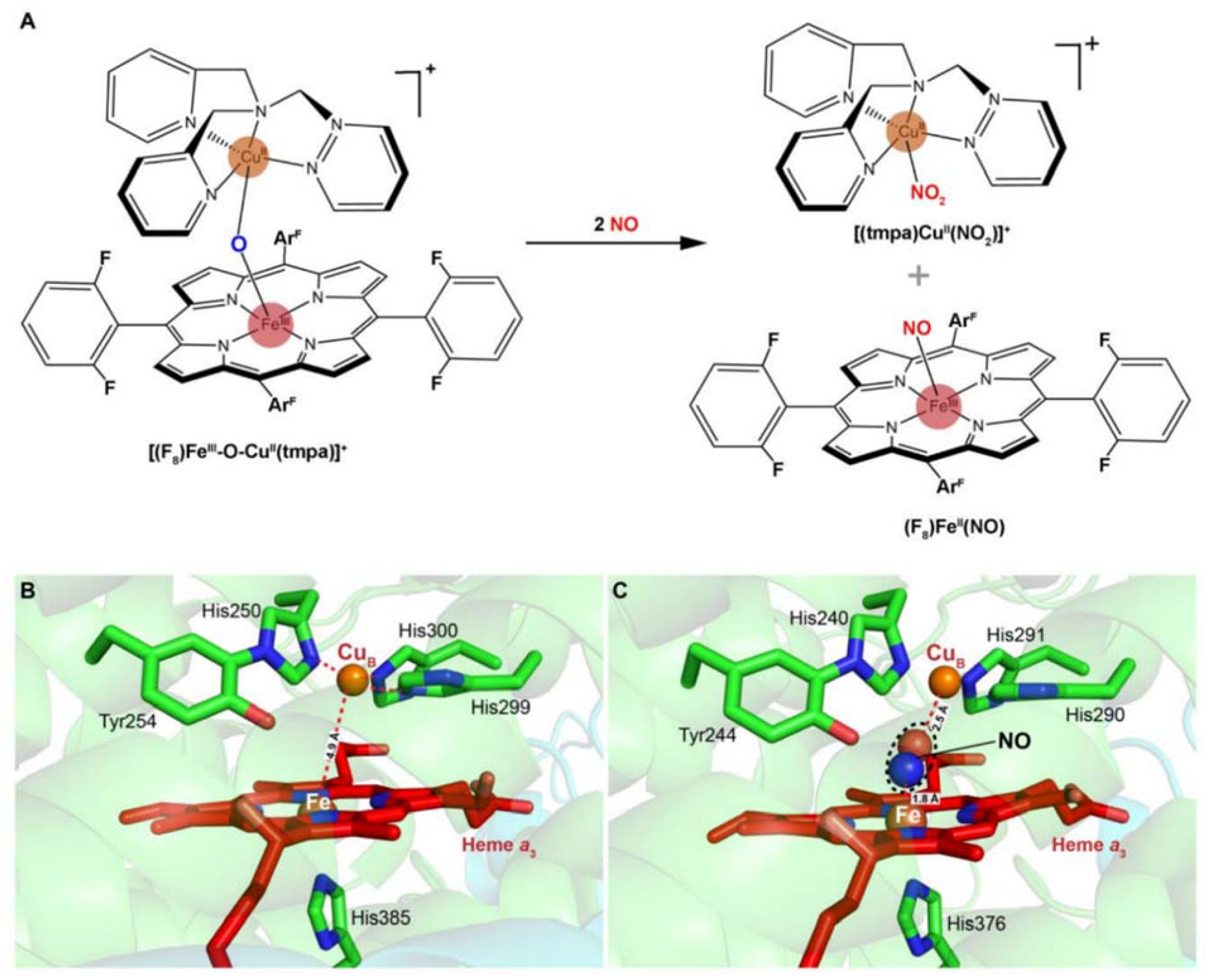
4. Inhibition of HCOs by Nitrite and NO
5. HCOs Are Primary Targets of Nitrite but Not NO In Vivo
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Verstraeten, N.; Knapen, W.J.; Kint, C.I.; Liebens, V.; Van den Bergh, B.; Dewachter, L.; Michiels, J.E.; Fu, Q.; David, C.C.; Fierro, A.C.; et al. Obg and membrane depolarization are part of a microbial bet-hedging strategy that leads to antibiotic tolerance. Mol. Cell. 2015, 59, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chan, E.W.C.; Wan, Y.; Wong, M.H.; Chen, S. Active maintenance of proton motive force mediates starvation-induced bacterial antibiotic tolerance in Escherichia coli. Commun. Biol. 2021, 4, 1068. [Google Scholar] [CrossRef] [PubMed]
- Kaila, V.R.I.; Wikström, M. Architecture of bacterial respiratory chains. Nat. Rev. Microbiol. 2021, 19, 319–330. [Google Scholar] [CrossRef]
- Sousa, F.L.; Alves, R.J.; Ribeiro, M.A.; Pereira-Leal, J.B.; Teixeira, M.; Pereira, M.M. The superfamily of heme-copper oxygen reductases: Types and evolutionary considerations. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 629–637. [Google Scholar] [CrossRef]
- Siletsky, S.A.; Borisov, V.B. Proton pumping and non-pumping terminal respiratory oxidases: Active sites intermediates of these molecular machines and their derivatives. Int. J. Mol. Sci. 2021, 22, 10852. [Google Scholar] [CrossRef]
- Hemp, J.; Gennis, R.B. Diversity of the heme–copper superfamily in archaea: Insights from genomics and structural modeling. In Bioenergetics: Energy Conservation and Conversion; Schäfer, G., Penefsky, H.S., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; Volume 45, pp. 1–31. [Google Scholar]
- Wikström, M.; Krab, K.; Sharma, V. Oxygen activation and energy conservation by cytochrome c oxidase. Chem. Rev. 2018, 118, 2469–2490. [Google Scholar] [CrossRef]
- Abramson, J.; Riistama, S.; Larsson, G.; Jasaitis, A.; Svensson-Ek, M.; Laakkonen, L.; Puustinen, A.; Iwata, S.; Wikström, M. The structure of the ubiquinol oxidase from Escherichia coli and its ubiquinone binding site. Nat. Struct. Biol. 2000, 7, 910–917. [Google Scholar] [PubMed]
- Liu, J.; Hiser, C.; Ferguson-Miller, S. Role of conformational change and K-path ligands in controlling cytochrome c oxidase activity. Biochem. Soc. Trans. 2017, 45, 1087–1095. [Google Scholar] [CrossRef]
- Lyons, J.A.; Aragão, D.; Slattery, O.; Pisliakov, A.V.; Soulimane, T.; Caffrey, M. Structural insights into electron transfer in caa3-type cytochrome oxidase. Nature 2012, 487, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, S.; Warkentin, E.; Xie, H.; Langer, J.D.; Ermler, U.; Michel, H. The structure of cbb3 cytochrome oxidase provides insights into proton pumping. Science 2010, 329, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Faxén, K.; Gilderson, G.; Ädelroth, P.; Brzezinski, P. A mechanistic principle for proton pumping by cytochrome c oxidase. Nature 2005, 437, 286–289. [Google Scholar] [CrossRef]
- Belevich, I.; Bloch, D.A.; Belevich, N.; Wikstrom, M.; Verkhovsky, M.I. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc. Natl. Acad. Sci. USA 2007, 104, 2685–2690. [Google Scholar] [CrossRef]
- Borisov, V.B.; Gennis, R.B.; Hemp, J.; Verkhovsky, M.I. The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 1398–1413. [Google Scholar] [CrossRef] [PubMed]
- Safarian, S.; Rajendran, C.; Müller, H.; Preu, J.; Langer, J.D.; Ovchinnikov, S.; Hirose, T.; Kusumoto, T.; Sakamoto, J.; Michel, H. Structure of a bd oxidase indicates similar mechanisms for membrane-integrated oxygen reductases. Science 2016, 352, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Ostermeier, C.; Ludwig, B.; Michel, H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature 1995, 376, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, S.; Shimada, A. Reaction Mechanism of cytochrome c oxidase. Chem. Rev. 2015, 115, 1936–1989. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Karlin, K.D.; Wikström, M. Computational study of the activated O(H) state in the catalytic mechanism of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2013, 110, 16844–16849. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.B.; Moura, J.J. How biology handles nitrite. Chem. Rev. 2014, 114, 5273–5357. [Google Scholar] [CrossRef]
- Canfield, D.E.; Glazer, A.N.; Falkowski, P.G. The evolution and future of earth’s nitrogen cycle. Science 2010, 330, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Kuypers, M.M.M.; Marchant, H.K.; Kartal, B. The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 2018, 16, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.L.; Gao, H.C. Physiological roles of nitrite and nitric oxide in bacteria: Similar consequences from distinct cell targets, protection, and sensing systems. Adv. Biol. 2021, 5, e2100773. [Google Scholar] [CrossRef]
- Jeney, V.; Ramos, S.; Bergman, M.-L.; Bechmann, I.; Tischer, J.; Ferreira, A.; Oliveira-Marques, V.; Janse, C.J.; Rebelo, S.; Cardoso, S.; et al. Control of disease tolerance to malaria by nitric oxide and carbon monoxide. Cell Rep. 2014, 8, 126–136. [Google Scholar] [CrossRef]
- Mancuso, C.; Navarra, P.; Preziosi, P. Roles of nitric oxide, carbon monoxide, and hydrogen sulfide in the regulation of the hypothalamic–pituitary–adrenal axis. J. Neurochem. 2010, 113, 563–575. [Google Scholar] [CrossRef]
- Mendes, S.S.; Miranda, V.; Saraiva, L.M. Hydrogen sulfide and carbon monoxide tolerance in bacteria. Antioxidants 2021, 10, 729. [Google Scholar] [CrossRef]
- Moustafa, A. Changes in nitric oxide, carbon monoxide, hydrogen sulfide and male reproductive hormones in response to chronic restraint stress in rats. Free Radic. Biol. Med. 2021, 162, 353–366. [Google Scholar] [CrossRef]
- Nowaczyk, A.; Kowalska, M.; Nowaczyk, J.; Grześk, G. Carbon monoxide and nitric oxide as examples of the youngest class of transmitters. Int. J. Mol. Sci. 2021, 22, 6029. [Google Scholar] [CrossRef]
- Stern, A.M.; Zhu, J. An introduction to nitric oxide sensing and response in bacteria. Adv. Appl. Microbiol. 2014, 87, 187–220. [Google Scholar]
- Brudvig, G.W.; Stevens, T.H.; Chan, S.I. Reactions of nitric-oxide with cytochrome-c oxidase. Biochemistry 1980, 19, 5275–5285. [Google Scholar] [CrossRef]
- Sarti, P.; Forte, E.; Mastronicola, D.; Giuffrè, A.; Arese, M. Cytochrome c oxidase and nitric oxide in action: Molecular mechanisms and pathophysiological implications. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 610–619. [Google Scholar] [CrossRef]
- Sarti, P.; Giuffrè, A.; Barone, M.C.; Forte, E.; Mastronicola, D.; Brunori, M. Nitric oxide and cytochrome oxidase: Reaction mechanisms from the enzyme to the cell. Free Radic. Biol. Med. 2003, 34, 509–520. [Google Scholar] [CrossRef]
- Hematian, S.; Siegler, M.A.; Karlin, K.D. Heme/Copper assembly mediated nitrite and nitric oxide interconversion. J. Am. Chem. Soc. 2012, 134, 18912–18915. [Google Scholar] [CrossRef]
- Loullis, A.; Pinakoulaki, E. Probing the nitrite and nitric oxide reductase activity of cbb3 oxidase: Resonance Raman detection of a six-coordinate ferrous heme–nitrosyl species in the binuclear b3/CuB center. Chem. Commun. 2015, 51, 17398–17401. [Google Scholar] [CrossRef]
- Giuffre, A.; Stubauer, G.; Sarti, P.; Brunori, M.; Zumft, W.G.; Buse, G.; Soulimane, T. The heme-copper oxidases of Thermus thermophilus catalyze the reduction of nitric oxide: Evolutionary implications. Proc. Natl. Acad. Sci. USA 1999, 96, 14718–14723. [Google Scholar] [CrossRef]
- Castello, P.R.; David, P.S.; McClure, T.; Crook, Z.; Poyton, R.O. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: Implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab. 2006, 3, 277–287. [Google Scholar] [CrossRef]
- Ford, P.C. Reactions of NO and nitrite with heme models and proteins. Inorg. Chem. 2010, 49, 6226–6239. [Google Scholar] [CrossRef]
- Ford, P.C.; Miranda, K.M. The solution chemistry of nitric oxide and other reactive nitrogen species. Nitric Oxide 2020, 103, 31–46. [Google Scholar] [CrossRef]
- Yoshikawa, S.; Orii, Y. Inhibition mechanism of cytochrome-oxidase reaction 2 classifications of inhibitors based on their modes of action. J. Biochem. 1972, 71, 859–872. [Google Scholar] [CrossRef]
- Rowe, J.J.; Yarbrough, J.M.; Rake, J.B.; Eagon, R.G. Nitrite inhibition of aerobic-bacteria. Curr. Microbiol. 1979, 2, 51–54. [Google Scholar] [CrossRef]
- Stevens, T.H.; Brudvig, G.W.; Bocian, D.F.; Chan, S.I. Structure of cytochrome a3-CuA3 couple in cytochrome c oxidase as revealed by nitric oxide binding studies. Proc. Natl. Acad. Sci. USA 1979, 76, 3320–3324. [Google Scholar] [CrossRef]
- Hori, H.; Tsubaki, M.; Mogi, T.; Anraku, Y. EPR study of NO complexes of bd-type ubiquinol oxidase from Escherichia coli: The proximal axial ligand of heme D is a nitrogenous amino acid residue. J. Biol. Chem. 1996, 271, 9254–9258. [Google Scholar] [CrossRef]
- Mason, M.G.; Shepherd, M.; Nicholls, P.; Dobbin, P.S.; Dodsworth, K.S.; Poole, R.K.; Cooper, C.E. Cytochrome bd confers nitric oxide resistance to Escherichia coli. Nat. Chem. Biol. 2009, 5, 94–96. [Google Scholar] [CrossRef]
- Lyons, J.A.; Hilbers, F.; Caffrey, M. Structure and function of bacterial cytochrome c oxidases. In Cytochrome Complexes: Evolution, Structures, Energy Transduction, and Signaling; Cramer, W.A., Kallas, T., Eds.; Advances in Photosynthesis and Respiration; Springer: Dordrecht, The Netherlands, 2016; Volume 41, pp. 307–329. [Google Scholar]
- Wikström, M.; Verkhovsky, M.I. Mechanism and energetics of proton translocation by the respiratory heme-copper oxidases. Biochim. Biophys. Acta Bioenerg. 2007, 1767, 1200–1214. [Google Scholar] [CrossRef]
- Lu, Y.; Sigman, J.A.; Kim, H.K.; Zhao, X.; Carey, J.R. The role of copper and protons in heme-copper oxidases: Kinetic study of an engineered heme-copper center in myoglobin. J. Inorg. Biochem. 2003, 96, 183. [Google Scholar] [CrossRef]
- Giuffrè, A.; Borisov, V.B.; Arese, M.; Sarti, P.; Forte, E. Cytochrome bd oxidase and bacterial tolerance to oxidative and nitrosative stress. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 1178–1187. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E. Terminal oxidase cytochrome bd protects bacteria against hydrogen sulfide toxicity. Biochemistry 2021, 86, 22–32. [Google Scholar] [CrossRef]
- Borisov, V.B.; Siletsky, S.A.; Nastasi, M.R.; Forte, E. ROS defense systems and terminal oxidases in bacteria. Antioxidants 2021, 10, 839. [Google Scholar] [CrossRef]
- Korshunov, S.; Imlay, K.R.; Imlay, J.A. The cytochrome bd oxidase of Escherichia coli prevents respiratory inhibition by endogenous and exogenous hydrogen sulfide. Mol. Microbiol. 2016, 101, 62–77. [Google Scholar] [CrossRef]
- Yoshikawa, S.; Muramoto, K.; Shinzawa-Itoh, K. Proton-pumping mechanism of cytochrome c oxidase. Annu. Rev. Biophys. 2011, 40, 205–223. [Google Scholar] [CrossRef]
- Pereira, M.M.; Santana, M.; Teixeira, M. A novel scenario for the evolution of haem-copper oxygen reductases. Biochim. Biophys. Acta Bioenerg. 2001, 1505, 185–208. [Google Scholar] [CrossRef]
- Florens, L.; Schmidt, B.; McCracken, J.; Ferguson-Miller, S. Fast deuterium access to the buried magnesium/manganese site in cytochrome c oxidase. Biochemistry 2001, 40, 7491–7497. [Google Scholar] [CrossRef]
- Mills, D.A.; Florens, L.; Hiser, C.; Qian, J.; Ferguson-Miller, S. Where is ′outside′ in cytochrome c oxidase and how and when do protons get there? Biochim. Biophys. Acta Bioenerg. 2000, 1458, 180–187. [Google Scholar] [CrossRef]
- Soulimane, T.; Buse, G.; Bourenkov, G.P.; Bartunik, H.D.; Huber, R.; Than, M.E. Structure and mechanism of the aberrant ba(3)-cytochrome c oxidase from thermus thermophilus. EMBO J. 2000, 19, 1766–1776. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Zeng, H.; Zhang, S.; Juli, J.; Tai, L.; Zhang, D.; Pang, X.; Zhang, Y.; Lam, S.M.; Zhu, Y.; et al. The unusual homodimer of a heme-copper terminal oxidase allows itself to utilize two electron donors. Angew. Chem. Int. Ed. 2021, 60, 13323–13330. [Google Scholar] [CrossRef]
- Pinakoulaki, E.; Stavrakis, S.; Urbani, A.; Varotsis, C. Resonance Raman Detection of a Ferrous Five-Coordinate Nitrosylheme b3 Complex in Cytochrome cbb3 Oxidase from Pseudomonas stutzeri. J. Am. Chem. Soc. 2002, 124, 9378–9379. [Google Scholar] [CrossRef] [PubMed]
- Kohlstaedt, M.; Buschmann, S.; Langer, J.D.; Xie, H.; Michel, H. Subunit CcoQ is involved in the assembly of the cbb(3)-type cytochrome c oxidases from Pseudomonas stutzeri ZoBell but not required for their activity. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Agmon, N. The grotthuss mechanism. Chem. Phys. Lett. 1995, 244, 456–462. [Google Scholar] [CrossRef]
- Harrenga, A.; Michel, H. The cytochrome c oxidase from Paracoccus denitrificans does not change the metal center ligation upon reduction. J. Biol. Chem. 1999, 274, 33296–33299. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Wikström, M.; Hummer, G. Kinetic gating of the proton pump in cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2009, 106, 13707–13712. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.M.; Santana, M.; Soares, C.M.; Mendes, J.; Carita, J.N.; Fernandes, A.S.; Saraste, M.; Carrondo, M.A.; Teixeira, M. The caa3 terminal oxidase of the thermohalophilic bacterium Rhodothermus marinus: A HiPIP:oxygen oxidoreductase lacking the key glutamate of the D-channel. Biochim. Biophys. Acta Bioenerg. 1999, 1413, 1–13. [Google Scholar] [CrossRef]
- Qin, L.; Mills, D.A.; Buhrow, L.; Hiser, C.; Ferguson-Miller, S. A conserved steroid binding site in cytochrome c oxidase. Biochemistry 2008, 47, 9931–9933. [Google Scholar] [CrossRef][Green Version]
- Hiser, C.; Buhrow, L.; Liu, J.; Kuhn, L.; Ferguson-Miller, S. A conserved amphipathic ligand binding region influences k-path-dependent activity of cytochrome c oxidase. Biochemistry 2013, 52, 1385–1396. [Google Scholar] [CrossRef][Green Version]
- Rauhamäki, V.; Baumann, M.; Soliymani, R.; Puustinen, A.; Wikström, M. Identification of a histidine-tyrosine cross-link in the active site of the cbb3-type cytochrome c oxidase from Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA 2006, 103, 16135–16140. [Google Scholar] [CrossRef] [PubMed]
- Drosou, V.; Malatesta, F.; Ludwig, B. Mutations in the docking site for cytochrome c on the Paracoccus heme aa3 oxidase. Electron entry and kinetic phases of the reaction. Eur. J. Biochem. 2002, 269, 2980–2988. [Google Scholar] [CrossRef] [PubMed]
- Muresanu, L.; Pristovsek, P.; Löhr, F.; Maneg, O.; Mukrasch, M.D.; Rüterjans, H.; Ludwig, B.; Lücke, C. The electron transfer complex between cytochrome c552 and the CuA domain of the Thermus thermophilus ba3 oxidase. A combined NMR and computational approach. J. Biol. Chem. 2006, 281, 14503–14513. [Google Scholar] [CrossRef] [PubMed]
- Gray, H.B. Electron flow through metalloproteins. J. Inorg. Biochem. 2001, 86, 1. [Google Scholar] [CrossRef]
- Maneg, O.; Malatesta, F.; Ludwig, B.; Drosou, V. Interaction of cytochrome c with cytochrome oxidase: Two different docking scenarios. Biochim. Biophys. Acta Bioenerg. 2004, 1655, 274–281. [Google Scholar] [CrossRef]
- Chance, B.; Saronio, C.; Leigh, J.S., Jr. Functional intermediates in the reaction of membrane-bound cytochrome oxidase with oxygen. J. Biol. Chem. 1975, 250, 9226–9237. [Google Scholar] [CrossRef]
- Fabian, M.; Wong, W.W.; Gennis, R.B.; Palmer, G. Mass spectrometric determination of dioxygen bond splitting in the "peroxy" intermediate of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 1999, 96, 13114–13117. [Google Scholar] [CrossRef]
- Giuffrè, A.; Barone, M.C.; Mastronicola, D.; D′Itri, E.; Sarti, P.; Brunori, M. Reaction of nitric oxide with the turnover intermediates of cytochrome c oxidase: Reaction pathway and functional effects. Biochemistry 2000, 39, 15446–15453. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, M.R.A. Activation of O(2) and NO in heme-copper oxidases—Mechanistic insights from computational modelling. Chem. Soc. Rev. 2020, 49, 7301–7330. [Google Scholar] [CrossRef]
- Michel, H. Cytochrome c oxidase: Catalytic cycle and mechanisms of proton pumping—A discussion. Biochemistry 1999, 38, 15129–15140. [Google Scholar] [CrossRef] [PubMed]
- Kaila, V.R.; Sharma, V.; Wikström, M. The identity of the transient proton loading site of the proton-pumping mechanism of cytochrome c oxidase. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 80–84. [Google Scholar] [CrossRef]
- Kaila, V.R.; Johansson, M.P.; Sundholm, D.; Laakkonen, L.; Wiström, M. The chemistry of the CuB site in cytochrome c oxidase and the importance of its unique His-Tyr bond. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Murali, R.; Gennis, R.B.; Hemp, J. Evolution of the cytochrome bd oxygen reductase superfamily and the function of CydAA′ in Archaea. ISME J. 2021, 15, 3534–3548. [Google Scholar] [CrossRef] [PubMed]
- Azarkina, N.; Siletsky, S.; Borisov, V.; von Wachenfeldt, C.; Hederstedt, L.; Konstantinov, A.A. A cytochrome bb′-type quinol oxidase in Bacillus subtilis strain 168. J. Biol. Chem. 1999, 274, 32810–32817. [Google Scholar] [CrossRef]
- Williams, C.; McColl, K.E. Review article: Proton pump inhibitors and bacterial overgrowth. Aliment. Pharmacol. Ther. 2006, 23, 3–10. [Google Scholar] [CrossRef]
- Hoeser, J.; Hong, S.; Gehmann, G.; Gennis, R.B.; Friedrich, T. Subunit CydX of Escherichia coli cytochrome bd ubiquinol oxidase is essential for assembly and stability of the di-heme active site. FEBS Lett. 2014, 588, 1537–1541. [Google Scholar] [CrossRef]
- Chen, H.; Luo, Q.; Yin, J.; Gao, T.; Gao, H. Evidence for the requirement of CydX in function but not assembly of the cytochrome bd oxidase in Shewanella oneidensis. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 318–328. [Google Scholar] [CrossRef]
- Thesseling, A.; Rasmussen, T.; Burschel, S.; Wohlwend, D.; Kaegi, J.; Mueller, R.; Boettcher, B.; Friedrich, T. Homologous bd oxidases share the same architecture but differ in mechanism. Nat. Commun. 2019, 10, 5138. [Google Scholar] [CrossRef]
- Osborne, J.P.; Gennis, R.B. Sequence analysis of cytochrome bd oxidase suggests a revised topology for subunit I. Biochim. Biophys. Acta Bioenerg. 1999, 1410, 32–50. [Google Scholar] [CrossRef]
- Bertsova, Y.V.; Bogachev, A.V.; Skulachev, V.P. Generation of protonic potential by the bd-type quinol oxidase of Azotobacter vinelandii. FEBS Lett. 1997, 414, 369–372. [Google Scholar]
- Forte, E.; Urbani, A.; Saraste, M.; Sarti, P.; Brunori, M.; Giuffrè, A. The cytochrome cbb3 from Pseudomonas stutzeri displays nitric oxide reductase activity. Eur. J. Biochem. 2001, 268, 6486–6491. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.S.; Forte, E.; Maria Scandurra, F.; Arese, M.; Giuffré, A.; Greenwood, C.; Sarti, P. Cytochrome bo3 from Escherichia coli: The binding and turnover of nitric oxide. Biochem. Biophys. Res. Commun. 2002, 296, 1272–1278. [Google Scholar] [CrossRef]
- Hematian, S.; Garcia-Bosch, I.; Karlin, K.D. Synthetic heme/copper assemblies: Toward an understanding of cytochrome c oxidase interactions with dioxygen and nitrogen oxides. Acc. Chem. Res. 2015, 48, 2462–2474. [Google Scholar] [CrossRef]
- Hematian, S.; Kenkel, I.; Shubina, T.E.; Dürr, M.; Liu, J.J.; Siegler, M.A.; Ivanovic-Burmazovic, I.; Karlin, K.D. Nitrogen oxide atom-transfer redox chemistry; mechanism of NO(g) to nitrite conversion utilizing μ-oxo Heme-FeIII–O–CuII(L) constructs. J. Am. Chem. Soc. 2015, 137, 6602–6615. [Google Scholar] [CrossRef]
- Torres, J.; Cooper, C.E.; Wilson, M.T. A common mechanism for the interaction of nitric oxide with the oxidized binuclear centre and oxygen intermediates of cytochromec oxidase. J. Biol. Chem. 1998, 273, 8756–8766. [Google Scholar] [CrossRef]
- Loullis, A.; Noor, M.R.; Soulimane, T.; Pinakoulaki, E. The structure of a ferrous heme-nitro species in the binuclear heme a3/CuB center of ba3-cytochrome c oxidase as determined by resonance Raman spectroscopy. Chem. Commun. 2015, 51, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Pinakoulaki, E.; Ohta, T.; Soulimane, T.; Kitagawa, T.; Varotsis, C. Detection of the His-Heme Fe2+−NO species in the reduction of NO to N2O by ba3-oxidase from Thermus thermophilus. J. Am. Chem. Soc. 2005, 127, 15161–15167. [Google Scholar] [CrossRef]
- Daskalakis, V.; Ohta, T.; Kitagawa, T.; Varotsis, C. Structure and properties of the catalytic site of nitric oxide reductase at ambient temperature. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1240–1244. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Soulimane, T.; Kitagawa, T.; Varotsis, C. Nitric oxide activation by caa3 oxidoreductase from Thermus thermophilus. Phys. Chem. Chem. Phys. 2015, 17, 10894–10898. [Google Scholar] [CrossRef]
- Varotsis, C.; Ohta, T.; Kitagawa, T.; Soulimane, T.; Pinakoulaki, E. The structure of the hyponitrite species in a heme Fe-Cu binuclear center. Angew. Chem. Int. Ed. 2007, 46, 2210–2214. [Google Scholar] [CrossRef]
- Forte, E.; Giuffrè, A.; Huang, L.-s.; Berry, E.A.; Borisov, V.B. Nitric oxide does not inhibit but is metabolized by the cytochrome bcc-aa3 supercomplex. Int. J. Mol. Sci. 2020, 21, 8521. [Google Scholar] [CrossRef]
- Girsch, P.; de Vries, S. Purification and initial kinetic and spectroscopic characterization of NO reductase from Paracoccus denitrificans. Biochim. Biophys. Acta Bioenerg. 1997, 1318, 202–216. [Google Scholar] [CrossRef]
- Hasenoehrl, E.J.; Wiggins, T.J.; Berney, M. Bioenergetic inhibitors: Antibiotic efficacy and mechanisms of action in Mycobacterium tuberculosis. Front. Cell. Infect. Microbiol. 2021, 10, 611683. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.R.; Markert, T.; Ross, L.J.; White, E.L.; Rasmussen, L.; Zhang, W.; Everts, M.; Moellering, D.R.; Bailey, S.M.; Suto, M.J.; et al. Identification of small molecule inhibitors of human cytochrome c oxidase that target chemoresistant glioma cells. J. Biol. Chem. 2016, 291, 24188–24199. [Google Scholar] [CrossRef] [PubMed]
- Pinakoulaki, E.; Vamvouka, M.; Varotsis, C. Resonance Raman detection of the Fe2+−C−N Modes in heme−copper oxidases: a probe of the active site. Inorg. Chem. 2004, 43, 4907–4910. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; Hatano, K.; Tadehara, H.; Yano, N.; Shinzawa-Itoh, K.; Yamashita, E.; Muramoto, K.; Tsukihara, T.; Yoshikawa, S. X-ray structural analyses of azide-bound cytochrome c oxidases reveal that the H-pathway is critically important for the proton-pumping activity. J. Biol. Chem. 2018, 293, 14868–14879. [Google Scholar] [CrossRef]
- Alleyne, T.; Ignacio, D.N.; Sampson, V.B.; Ashe, D.; Wilson, M. Simulating the slow to fast switch in cytochrome c oxidase catalysis by introducing a loop flip near the enzyme′s cytochrome c (substrate) binding site. Biotechnol. Appl. Biochem. 2017, 64, 677–685. [Google Scholar] [CrossRef]
- Moody, A.J.; Cooper, C.E.; Rich, P.R. Characterisation of ‘fast’ and ‘slow’ forms of bovine heart cytochrome-c oxidase. Biochim. Biophys. Acta Bioenerg. 1991, 1059, 189–207. [Google Scholar] [CrossRef]
- Bloch, D.; Belevich, I.; Jasaitis, A.; Ribacka, C.; Puustinen, A.; Verkhovsky, M.I.; Wikström, M. The catalytic cycle of cytochrome c oxidase is not the sum of its two halves. Proc. Natl. Acad. Sci. USA 2004, 101, 529–533. [Google Scholar] [CrossRef]
- Andersson, R.; Safari, C.; Dods, R.; Nango, E.; Tanaka, R.; Yamashita, A.; Nakane, T.; Tono, K.; Joti, Y.; Båth, P.; et al. Serial femtosecond crystallography structure of cytochrome c oxidase at room temperature. Sci. Rep. 2017, 7, 4518. [Google Scholar] [CrossRef]
- Aoyama, H.; Muramoto, K.; Shinzawa-Itoh, K.; Hirata, K.; Yamashita, E.; Tsukihara, T.; Ogura, T.; Yoshikawa, S. A peroxide bridge between Fe and Cu ions in the O2 reduction site of fully oxidized cytochrome c oxidase could suppress the proton pump. Proc. Natl. Acad. Sci. USA 2009, 106, 2165–2169. [Google Scholar] [CrossRef]
- Koepke, J.; Olkhova, E.; Angerer, H.; Müller, H.; Peng, G.; Michel, H. High resolution crystal structure of Paracoccus denitrificans cytochrome c oxidase: New insights into the active site and the proton transfer pathways. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 635–645. [Google Scholar] [CrossRef]
- Kruse, F.; Nguyen, A.D.; Dragelj, J.; Heberle, J.; Hildebrandt, P.; Mroginski, M.A.; Weidinger, I.M. A Resonance Raman marker band characterizes the slow and fast form of cytochrome c oxidase. J. Am. Chem. Soc. 2021, 143, 2769–2776. [Google Scholar] [CrossRef] [PubMed]
- Thörnström, P.-E.; Nilsson, T.; Malmström, B.G. The possible role of the closed-open transition in proton pumping by cytochrome c oxidase: The pH dependence of cyanide inhibition. Biochim. Biophys. Acta Bioenerg. 1988, 935, 103–108. [Google Scholar] [CrossRef]
- Cooper, C.E.; Torres, J.; Sharpe, M.A.; Wilson, M.T. Nitric oxide ejects electrons from the binuclear centre of cytochrome c oxidase by reacting with oxidised copper: A general mechanism for the interaction of copper proteins with nitric oxide? FEBS Lett. 1997, 414, 281–284. [Google Scholar]
- Muramoto, K.; Ohta, K.; Shinzawa-Itoh, K.; Kanda, K.; Taniguchi, M.; Nabekura, H.; Yamashita, E.; Tsukihara, T.; Yoshikawa, S. Bovine cytochrome c oxidase structures enable O2 reduction with minimization of reactive oxygens and provide a proton-pumping gate. Proc. Natl. Acad. Sci. USA 2010, 107, 7740–7745. [Google Scholar] [CrossRef]
- Stubauer, G.; Giuffrè, A.; Brunori, M.; Sarti, P. Cytochrome c oxidase does not catalyze the anaerobic reduction of NO. Biochem. Biophys. Res. Commun. 1998, 245, 459–465. [Google Scholar] [CrossRef]
- Sarti, P.; Giuffré, A.; Forte, E.; Mastronicola, D.; Barone, M.C.; Brunori, M. Nitric oxide and cytochrome c oxidase: Mechanisms of inhibition and no degradation. Biochem. Biophys. Res. Commun. 2000, 274, 183–187. [Google Scholar] [CrossRef]
- Antonini, E.; Brunori, M.; Colosimo, A.; Greenwood, C.; Wilson, M.T. Oxygen ′pulsed′ cytochrome c oxidase: Functional properties and catalytic relevance. Proc. Natl. Acad. Sci. USA 1977, 74, 3128–3132. [Google Scholar] [CrossRef]
- Baker, G.M.; Noguchi, M.; Palmer, G. The reaction of cytochrome oxidase with cyanide. Preparation of the rapidly reacting form and its conversion to the slowly reacting form. J. Biol. Chem. 1987, 262, 595–604. [Google Scholar] [CrossRef]
- Giuffrè, A.; Stubauer, G.; Brunori, M.; Sarti, P.; Torres, J.; Wilson, M.T. Chloride bound to oxidized cytochrome c oxidase controls the reaction with nitric oxide. J. Biol. Chem. 1998, 273, 32475–32478. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Antunes, F.; Cadenas, E. The mechanism of cytochrome C oxidase inhibition by nitric oxide. Front. Biosci. 2007, 12, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Sarti, P.; Arese, M.; Forte, E.; Giuffrè, A.; Mastronicola, D. Mitochondria and Nitric oxide: Chemistry and pathophysiology. In Advances in Mitochondrial Medicine; Scatena, R., Bottoni, P., Giardina, B., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 75–92. [Google Scholar]
- Mason, M.G.; Nicholls, P.; Wilson, M.T.; Cooper, C.E. Nitric oxide inhibition of respiration involves both competitive (heme) and noncompetitive (copper) binding to cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2006, 103, 708–713. [Google Scholar] [CrossRef]
- Moncada, S.; Erusalimsky, J.D. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat. Rev. Mol. Cell Biol. 2002, 3, 214–220. [Google Scholar] [CrossRef]
- Ford, P.C.; Wink, D.A.; Stanbury, D.M. Autoxidation kinetics of aqueous nitric-oxide. FEBS Lett. 1993, 326, 1–3. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E.; Sarti, P.; Brunori, M.; Konstantinov, A.A.; Giuffrè, A. Redox control of fast ligand dissociation from Escherichia coli cytochrome bd. Biochem. Biophys. Res. Commun. 2007, 355, 97–102. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E.; Giuffrè, A.; Konstantinov, A.; Sarti, P. Reaction of nitric oxide with the oxidized di-heme and heme–copper oxygen-reducing centers of terminal oxidases: Different reaction pathways and end-products. J. Inorg. Biochem. 2009, 103, 1185–1187. [Google Scholar] [CrossRef]
- Ohta, K.; Muramoto, K.; Shinzawa-Itoh, K.; Yamashita, E.; Yoshikawa, S.; Tsukihara, T. X-ray structure of the NO-bound CuB in bovine cytochrome c oxidase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 251–253. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E.; Konstantinov, A.A.; Poole, R.K.; Sarti, P.; Giuffrè, A. Interaction of the bacterial terminal oxidase cytochrome bd with nitric oxide. FEBS Lett. 2004, 576, 201–204. [Google Scholar] [CrossRef]
- Reddy, D.; Lancaster, J.R., Jr.; Cornforth, D.P. Nitrite inhibition of Clostridium botulinum: Electron spin resonance detection of iron-nitric oxide complexes. Science 1983, 221, 769–770. [Google Scholar] [CrossRef]
- Tonzetich, Z.J.; McQuade, L.E.; Lippard, S.J. Detecting and understanding the roles of nitric oxide in biology. Inorg. Chem. 2010, 49, 6338–6348. [Google Scholar] [CrossRef]
- Xu, N.; Yi, J.; Richter-Addo, G.B. Linkage isomerization in heme-NOx compounds: Understanding NO, nitrite, and hyponitrite interactions with iron porphyrins. Inorg. Chem. 2010, 49, 6253–6266. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Grubina, R.; Doyle, M.P. The new chemical biology of nitrite reactions with hemoglobin: R-state catalysis, oxidative denitrosylation, and nitrite reductase/anhydrase. Acc. Chem. Res. 2009, 42, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Dent, M.R.; DeMartino, A.W.; Tejero, J.; Gladwin, M.T. Endogenous hemoprotein-dependent signaling pathways of nitric oxide and nitrite. Inorg. Chem. 2021, 60, 15918–15940. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Grubina, R.; Huang, J.; Conradie, J.; Huang, Z.; Jeffers, A.; Jiang, A.; He, X.; Azarov, I.; Seibert, R.; et al. Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nat. Chem. Biol. 2007, 3, 785–794. [Google Scholar] [CrossRef]
- Keszler, A.; Piknova, B.; Schechter, A.N.; Hogg, N. The reaction between nitrite and oxyhemoglobin: A mechanistic study. J. Biol. Chem. 2008, 283, 9615–9622. [Google Scholar] [CrossRef]
- Tompkin, R. Nitrite. In Antimicrobials in Food, 3rd ed.; Davidson, P., Sofos, J., Branen, A., Eds.; CRC Press, Taylor&Frances Group: Boca Raton, FL, USA, 2005; pp. 169–236. [Google Scholar]
- Yang, T.Y. Mechanism of nitrite inhibition of cellular respiration in Pseudomonas aeruginosa. Curr. Microbiol. 1985, 12, 35–39. [Google Scholar] [CrossRef]
- Chabrière, E.; Charon, M.H.; Volbeda, A.; Pieulle, L.; Hatchikian, E.C.; Fontecilla-Camps, J.C. Crystal structures of the key anaerobic enzyme pyruvate: Ferredoxin oxidoreductase, free and in complex with pyruvate. Nat. Struct. Biol. 1999, 6, 182–190. [Google Scholar]
- Woods, L.F.; Wood, J.M. A note on the effect of nitrite inhibition on the metabolism of Clostridium botulinum. J. Appl. Bacteriol. 1982, 52, 109–110. [Google Scholar] [CrossRef]
- Bourret, T.J.; Boylan, J.A.; Lawrence, K.A.; Gherardini, F.C. Nitrosative damage to free and zinc-bound cysteine thiols underlies nitric oxide toxicity in wild-type Borrelia burgdorferi. Mol. Microbiol. 2011, 81, 259–273. [Google Scholar] [CrossRef]
- Husain, M.; Bourret, T.J.; McCollister, B.D.; Jones-Carson, J.; Laughlin, J.; Vázquez-Torres, A. Nitric oxide evokes an adaptive response to oxidative stress by arresting respiration. J. Biol. Chem. 2008, 283, 7682–7689. [Google Scholar] [CrossRef] [PubMed]
- Hyduke, D.R.; Jarboe, L.R.; Tran, L.M.; Chou, K.J.; Liao, J.C. Integrated network analysis identifies nitric oxide response networks and dihydroxyacid dehydratase as a crucial target in Escherichia coli. Proc. Natl. Acad. Sci. USA 2007, 104, 8484–8489. [Google Scholar] [CrossRef]
- Richardson, A.R.; Payne, E.C.; Younger, N.; Karlinsey, J.E.; Thomas, V.C.; Becker, L.A.; Navarre, W.W.; Castor, M.E.; Libby, S.J.; Fang, F.C. Multiple targets of nitric oxide in the tricarboxylic acid cycle of Salmonella enterica serovar typhimurium. Cell Host Microbe 2011, 10, 33–43. [Google Scholar] [CrossRef]
- Vine, C.E.; Cole, J.A. Unresolved sources, sinks, and pathways for the recovery of enteric bacteria from nitrosative stress. FEMS Microbiol. Lett. 2011, 325, 99–107. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Butler, C.S.; Seward, H.E.; Greenwood, C.; Thomson, A.J. Fast cytochrome bo from Escherichia coli binds two molecules of nitric oxide at CuB. Biochemistry 1997, 36, 16259–16266. [Google Scholar] [CrossRef]
- Giuffre, A.; Forte, E.; Brunori, M.; Sarti, P. Nitric oxide, cytochrome c oxidase and myoglobin: Competition and reaction pathways. FEBS Lett. 2005, 579, 2528–2532. [Google Scholar] [CrossRef][Green Version]
- Jones-Carson, J.; Husain, M.; Liu, L.; Orlicky, D.J.; Vázquez-Torres, A. Cytochrome bd-dependent bioenergetics and antinitrosative defenses in Salmonella pathogenesis. mBio 2016, 7, e02052-16. [Google Scholar] [CrossRef]
- Meyer, T.E.; Tsapin, A.I.; Vandenberghe, I.; de Smet, L.; Frishman, D.; Nealson, K.H.; Cusanovich, M.A.; van Beeumen, J.J. Identification of 42 possible cytochrome c genes in the Shewanella oneidensis genome and characterization of six soluble cytochromes. Omics 2004, 8, 57–77. [Google Scholar] [CrossRef]
- Meng, Q.; Sun, Y.; Gao, H. Cytochromes c constitute a layer of protection against nitric oxide but not nitrite. Appl. Environ. Microbiol. 2018, 84, e01255-18. [Google Scholar] [CrossRef]
- Fredrickson, J.K.; Romine, M.F.; Beliaev, A.S.; Auchtung, J.M.; Driscoll, M.E.; Gardner, T.S.; Nealson, K.H.; Osterman, A.L.; Pinchuk, G.; Reed, J.L.; et al. Towards environmental systems biology of Shewanella. Nat. Rev. Microbiol. 2008, 6, 592–603. [Google Scholar] [CrossRef]
- Kranz, R.G.; Richard-Fogal, C.; Taylor, J.S.; Frawley, E.R. Cytochrome c biogenesis: Mechanisms for covalent modifications and trafficking of heme and for heme-iron redox control. Microbiol. Mol. Biol. Rev. 2009, 73, 510–528. [Google Scholar] [CrossRef]
- Meng, Q.; Yin, J.; Jin, M.; Gao, H. Distinct nitrite and nitric oxide physiologies in Escherichia coli and Shewanella oneidensis. Appl. Environ. Microbiol. 2018, 84, e00559-18. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Chen, H.; Wang, J.; Zhou, G.; Zhang, H.; Zhang, L.; Gao, H. Crp-dependent cytochrome bd oxidase confers nitrite resistance to Shewanella oneidensis. Environ. Microbiol. 2013, 15, 2198–2212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fu, H.; Wang, J.; Sun, L.; Jiang, Y.; Zhang, L.; Gao, H. Impacts of nitrate and nitrite on physiology of Shewanella oneidensis. PLoS ONE 2013, 8, e62629. [Google Scholar] [CrossRef]
- Zhou, G.; Yin, J.; Chen, H.; Hua, Y.; Sun, L.; Gao, H. Combined effect of loss of the caa3 oxidase and Crp regulation drives Shewanella to thrive in redox-stratified environments. ISME J. 2013, 7, 1752–1763. [Google Scholar] [CrossRef]
- Jin, M.; Fu, H.; Yin, J.; Yuan, J.; Gao, H. Molecular underpinnings of nitrite effect on Cyma-dependent respiration in Shewanella oneidensis. Front. Microbiol. 2016, 7, 1154. [Google Scholar] [CrossRef]
- Yin, J.; Jin, M.; Zhang, H.; Ju, L.; Zhang, L.; Gao, H. Regulation of nitrite resistance of the cytochrome cbb3 oxidase by cytochrome c ScyA in Shewanella oneidensis. Microbiologyopen 2015, 4, 84–99. [Google Scholar] [CrossRef]
- Zemke, A.C.; Gladwin, M.T.; Bomberger, J.M. Sodium nitrite blocks the activity of aminoglycosides against Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2015, 59, 3329–3334. [Google Scholar] [CrossRef]
- Zemke, A.C.; Kocak, B.R.; Bomberger, J.M. Sodium nitrite inhibits killing of Pseudomonas aeruginosa biofilms by ciprofloxacin. Antimicrob. Agents Chemother. 2017, 61, e00448-16. [Google Scholar] [CrossRef]
- Zemke, A.C.; Shiva, S.; Burns, J.L.; Moskowitz, S.M.; Pilewski, J.M.; Gladwin, M.T.; Bomberger, J.M. Nitrite modulates bacterial antibiotic susceptibility and biofilm formation in association with airway epithelial cells. Free. Radic. Biol. Med. 2014, 77, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, K.; Meng, Q.; Gao, H. Nitrite modulates aminoglycoside tolerance by inhibiting cytochrome heme-copper oxidase in bacteria. Commun. Biol. 2020, 3, 269. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Wang, J.; Mora, A.L.; Gladwin, M.T. Nitrite signaling in pulmonary hypertension: Mechanisms of bioactivation, signaling, and therapeutics. Antioxid. Redox Signal. 2013, 18, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Dunn, A.K.; Karr, E.A.; Wang, Y.; Batton, A.R.; Ruby, E.G.; Stabb, E.V. The alternative oxidase (AOX) gene in Vibrio fischeri is controlled by NsrR and upregulated in response to nitric oxide. Mol. Microbiol. 2010, 77, 44–55. [Google Scholar] [CrossRef]
- Yoon, S.S.; Coakley, R.; Lau, G.W.; Lymar, S.V.; Gaston, B.; Karabulut, A.C.; Hennigan, R.F.; Hwang, S.H.; Buettner, G.; Schurr, M.J.; et al. Anaerobic killing of mucoid Pseudomonas aeruginosa by acidified nitrite derivatives under cystic fibrosis airway conditions. J. Clin. Investig. 2006, 116, 436–446. [Google Scholar] [CrossRef]
- Feelisch, M.; Akaike, T.; Griffiths, K.; Ida, T.; Prysyazhna, O.; Goodwin, J.J.; Gollop, N.D.; Fernandez, B.O.; Minnion, M.; Cortese-Krott, M.M.; et al. Long-lasting blood pressure lowering effects of nitrite are NO-independent and mediated by hydrogen peroxide, persulfides, and oxidation of protein kinase G1α redox signalling. Cardiovasc. Res. 2019, 116, 51–62. [Google Scholar] [CrossRef]
- Williams, D.E.; Nisbett, L.M.; Bacon, B.; Boon, E. Bacterial heme-based sensors of nitric oxide. Antioxid. Redox Signal. 2018, 29, 1872–1887. [Google Scholar] [CrossRef]
- Anand, C.R.; Bhavya, B.; Jayakumar, K.; Harikrishnan, V.S.; Gopala, S. Inorganic nitrite alters mitochondrial dynamics without overt changes in cell death and mitochondrial respiration in cardiomyoblasts under hyperglycemia. Toxicol. In Vitro 2021, 70, 105048. [Google Scholar] [CrossRef]
- Bailey, J.C.; Feelisch, M.; Horowitz, J.D.; Frenneaux, M.P.; Madhani, M. Pharmacology and therapeutic role of inorganic nitrite and nitrate in vasodilatation. Pharmacol. Ther. 2014, 144, 303–320. [Google Scholar] [CrossRef]
- Rix, P.J.; Vick, A.; Attkins, N.J.; Barker, G.E.; Bott, A.W.; Alcorn, H., Jr.; Gladwin, M.T.; Shiva, S.; Bradley, S.; Hussaini, A.; et al. Pharmacokinetics, pharmacodynamics, safety, and tolerability of nebulized sodium nitrite (AIR001) following repeat-dose inhalation in healthy subjects. Clin. Pharmacokinet. 2015, 54, 261–272. [Google Scholar] [CrossRef][Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Xie, P.; Huang, Y.; Gao, H. Complex Interplay of Heme-Copper Oxidases with Nitrite and Nitric Oxide. Int. J. Mol. Sci. 2022, 23, 979. https://doi.org/10.3390/ijms23020979
Chen J, Xie P, Huang Y, Gao H. Complex Interplay of Heme-Copper Oxidases with Nitrite and Nitric Oxide. International Journal of Molecular Sciences. 2022; 23(2):979. https://doi.org/10.3390/ijms23020979
Chicago/Turabian StyleChen, Jinghua, Peilu Xie, Yujia Huang, and Haichun Gao. 2022. "Complex Interplay of Heme-Copper Oxidases with Nitrite and Nitric Oxide" International Journal of Molecular Sciences 23, no. 2: 979. https://doi.org/10.3390/ijms23020979
APA StyleChen, J., Xie, P., Huang, Y., & Gao, H. (2022). Complex Interplay of Heme-Copper Oxidases with Nitrite and Nitric Oxide. International Journal of Molecular Sciences, 23(2), 979. https://doi.org/10.3390/ijms23020979