
Assessment of the Suitability and Accuracy of Different Methods to Determine the Degree of Photodegradation of High- and Low-Density Polyethylene, Polypropylene, Polyvinyl Chloride, Nylon and Polystyrene Microplastics

 , , and
, , and
Abstract
1. Introduction
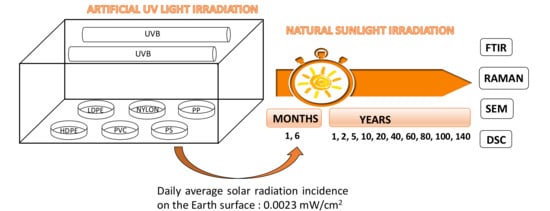
2. Materials and Methods
2.1. Materials and Sample Preparation
2.2. Characterization
3. Results
3.1. SEM Analysis
3.2. FTIR Analysis
3.3. Raman Analysis
3.4. DSC Analysis
3.5. Environmental Relevance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| HDPE | High-density polyethylene |
| LDPE | Low-density polyethylene |
| PP | Polypropylene |
| PVC | Polyvinyl chloride |
| PS | Polystyrene |
| SEM | Scanning Electron Microscopy |
| FTIR | Fourier Transform Infrared spectroscopy |
| DSC | Differential Scanning Calorimetry |
References
- Siracusa, V.; Blanco, I. Bio-Polyethylene (Bio-PE), Bio-Polypropylene (Bio-PP) and Bio-Poly(ethylene terephthalate) (Bio-PET): Recent Developments in Bio-Based Polymers Analogous to Petroleum-Derived Ones for Packaging and Engineering Applications. Polymers 2020, 12, 1641. [Google Scholar] [CrossRef]
- Dai, L.; Karakas, O.; Cheng, Y.; Cobb, K.; Chen, P.; Ruan, R. A review on carbon materials production from plastic wastes. Chem. Eng. J. 2023, 453, 139725. [Google Scholar] [CrossRef]
- Bakir, A.; Desender, M.; Wilkinson, T.; Van Hoytema, N.; Amos, R.; Airahui, S.; Graham, J.; Maes, T. Occurrence and abundance of meso and microplastics in sediment, surface waters, and marine biota from the South Pacific region. Mar. Pollut. Bull. 2020, 160, 111572. [Google Scholar] [CrossRef]
- Yu, Q.; Hu, X.; Yang, B.; Zhang, G.; Wang, J.; Ling, W. Distribution, abundance and risks of microplastics in the environment. Chemosphere 2020, 249, 126059. [Google Scholar] [CrossRef]
- Fotopoulou, K.N.; Karapanagioti, H.K. Degradation of Various Plastics in the Environment. Handb. Environ. Chem. 2019, 78, 71–92. [Google Scholar] [CrossRef]
- Yuan, J.; Ma, J.; Sun, Y.; Zhou, T.; Zhao, Y.; Yu, F. Microbial degradation and other environmental aspects of microplastics/plastics. Sci. Total Environ. 2020, 715, 136968. [Google Scholar] [CrossRef]
- Yu, J.; Sun, L.; Ma, C.; Qiao, Y.; Yao, H. Thermal degradation of PVC: A review. Waste Manag. 2016, 48, 300–314. [Google Scholar] [CrossRef] [PubMed]
- Tamada, J.A.; Langert, R. Erosion kinetics of hydrolytically degradable polymers. Proc. Natl. Acad. Sci. USA 1993, 90, 552–556. [Google Scholar] [CrossRef]
- Pickett, J.E.; Gibson, D.A.; Gardner, M.M. Effects of irradiation conditions on the weathering of engineering thermoplastics. Polym. Degrad. Stab. 2008, 93, 1597–1606. [Google Scholar] [CrossRef]
- White, J.R. Polymer ageing: Physics, chemistry or engineering? Time to reflect. C. R. Chim. 2006, 9, 1396–1408. [Google Scholar] [CrossRef]
- Moezzi, M.; Yekrang, J.; Ghane, M.; Hatami, M. The effects of UV degradation on the physical, thermal, and morphological properties of industrial nylon 66 conveyor belt fabrics. J. Ind. Text. 2020, 50, 240–260. [Google Scholar] [CrossRef]
- Wu, H.; Zhao, Y.; Dong, X.; Su, L.; Wang, K.; Wang, D. Microstructural evolution of isotactic polypropylene during photo-oxidation. Polym. Degrad. Stab. 2021, 183, 109434. [Google Scholar] [CrossRef]
- Ding, L.; Yu, X.; Guo, X.; Zhang, Y.; Ouyang, Z.; Liu, P.; Zhang, C.; Wang, T.; Jia, H.; Zhu, L. The photodegradation processes and mechanisms of PVC and PET microplastic in aquatic environments. Water Res. 2022, 208, 117879. [Google Scholar] [CrossRef]
- Gardette, M.; Perthue, A.; Gardette, J.-L.; Janecska, T.; Pukánszky, B.; Therias, S. Photo- and thermal-oxidation of polyethylene: Comparison of mechanisms and influence of unsaturation content. Polym. Degrad. Stab. 2013, 98, 2383–2390. [Google Scholar] [CrossRef]
- López-Martínez, E.D.; Martínez-Colunga, J.G.; Ramírez-Vargas, E.; Sanchez-Valdes, S.; Ramos-de Valle, L.F.; Benavides-Cantu, R.; Rodríguez-Gonzalez, J.A.; Mata-Padilla, J.M.; Cruz-Delgado, V.J.; Borjas-Ramos, J.J.; et al. Influence of carbon structures on the properties and photodegradation of LDPE/LLDPE films. Polym. Adv. Technol. 2022, 33, 1727–1741. [Google Scholar] [CrossRef]
- Varghese, A.M.; Rangaraj, V.M.; Luckachan, G.; Mittal, V. UV Aging Behavior of Functionalized Mullite Nanofiber-Reinforced Polypropylene. ACS Omega 2020, 5, 27083–27093. [Google Scholar] [CrossRef]
- Daniloska, V.; Blazevska-Gilev, J.; Dimova, V.; Fajgar, R.; Tomovska, R. UV light induced surface modification of HDPE films with bioactive compounds. Appl. Surf. Sci. 2010, 256, 2276–2283. [Google Scholar] [CrossRef]
- Grigoriadou, I.; Paraskevopoulos, K.M.; Chrissafis, K.; Pavlidou, E.; Stamkopoulos, T.G.; Bikiaris, D. Effect of different nanoparticles on HDPE UV stability. Polym. Degrad. Stab. 2011, 96, 151–163. [Google Scholar] [CrossRef]
- Silva, A.B.; Bastos, A.S.; Justino, C.I.L.; da Costa, J.P.; Duarte, A.C.; Rocha-Santos, T.A.P. Microplastics in the environment: Challenges in analytical chemistry. Anal. Chim. Acta 2018, 1017, 1–19. [Google Scholar] [CrossRef]
- Rafieepour, A.; Ghamari, F.; Mohammadbeigi, A.; Asghari, M. Seasonal variation in exposure level of types A and B ultraviolet radiation. Ann. Med. Health Sci. Res. 2015, 5, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Ainali, N.M.; Bikiaris, D.N.; Lambropoulou, D.A. Aging effects on LDPE, HDPE, PP and PS under UV irradiation: Decomposition mechanism by Py-GC/MS. J. Anal. Appl. Pyrolysis 2021, 158, 105207. [Google Scholar] [CrossRef]
- Cai, L.; Wang, J.; Peng, J.; Wu, Z.; Tan, X. Observation of the degradation of three types of plastic pellets exposed to UV irradiation. Sci. Total Environ. 2018, 628–629, 740–747. [Google Scholar] [CrossRef]
- Dong, M.; Zhang, Q.; Xing, X.; Chen, W.; She, Z.; Luo, Z. Raman spectra and surface changes of microplastics weathered under natural environments. Sci. Total Environ. 2020, 739, 139990. [Google Scholar] [CrossRef] [PubMed]
- Marcilla, A.; Beltrán, M.I.; Navarro, R. TG/FT-IR analysis of HZSM5 and HUSY deactivation during catalytic pyrolysis of polyethylene. J. Anal. Appl. Pyrolysis 2006, 76, 222–229. [Google Scholar] [CrossRef]
- Hedrick, S.A.; Chuang, S.S.C. Temperature programmed decomposition of polypropylene. Thermochim. Acta 1998, 315, 159–168. [Google Scholar] [CrossRef]
- Ramesh, S.; Yi, L.J. FTIR spectra of plasticized high molecular weight PVC–LiCF3SO3 electrolytes. Ionics 2009, 15, 413–420. [Google Scholar] [CrossRef]
- Olmos, D.; Martí, E.V.; González-Benito, J. New molecular-scale information on polystyrene dynamics in PS and PS–BaTiO3 composites. Phys. Chem. Chem. Phys. 2014, 16, 24339. [Google Scholar] [CrossRef]
- Martínez, K.I.; González-Mota, R.; Soto-Bernal, J.J.; Rosales-Candelas, I. Evaluation by IR spectroscopy of the degradation of commercial polyethylene. J. Appl. Polym. Sci. 2021, 138, 50158. [Google Scholar] [CrossRef]
- Dimassi, S.N.; Hahladakis, J.N.; Yahia, M.N.D.; Ahmad, M.I.; Sayadi, S.; Al-Ghouti, M.A. Degradation-fragmentation of marine plastic waste. Arabian J. Chem. 2022, 15, 104262. [Google Scholar] [CrossRef]
- Wang, C.; Xian, Z.; Jin, X.; Liang, S.; Chen, Z.; Pan, B.; Wu, B.; Ok, Y.S.; Gu, C. Photo-aging of PVC microplastic with natural organic acids. Water Res. 2020, 183, 116082. [Google Scholar] [CrossRef]
- Rijavec, T.; Strlič, M.; Cigić, I.K. Plastics in Heritage Collections: PVC degradation and characterization. Acta Chim. Slov. 2020, 67, 993–1013. [Google Scholar] [CrossRef]
- Li, Y.; Fan, Y.; Ma, J. Thermal, physical and chemical stability of porous polystyrene-type beads. Polym. Degrad. Stab. 2001, 73, 163–167. [Google Scholar] [CrossRef]
- Shi, X.; Chen, Z.; Liu, X.; Wei, W.; Ni, B.J. Photochemical behaviors of microplastics via reactive oxygen species. Sci. Total Environ. 2022, 846, 157498. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, H.A. FTIR characterization of neat and UV-stabilized Nylon 6,6 films. J. Pure Appl. Sci. 2011, 24, 86–90. [Google Scholar]
- Holland, B.J.; Hay, J.N. Thermal degradation of nylon polymers. Polym. Int. 2000, 49, 943–948. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, H. A review on photochemical effects of common plastics and their related applications. J. Polym. Sci. 2024, 62, 969–997. [Google Scholar] [CrossRef]
- Chamas, A.; Moon, H.; Zheng, J.; Qiu, Y.; Tabassum, T.; Jang, J.H.; Abu-Omar, M.; Scott, S.L.; Suh, S. Degradation Rates of Plastics in the Environment. ACS Sustain. Chem. Eng 2020, 8, 3494–3511. [Google Scholar] [CrossRef]
- Hirsch, S.G.; Barel, B.; Shpasser, D.; Segal, E.; Gazit, O.M. Correlating chemical and physical changes of photo-oxidized low-density polyethylene to the activation energy of water release. Polym. Test. 2017, 64, 194–199. [Google Scholar] [CrossRef]
- Lala, D.; Rabek, J.F. Destruction of carbonyl and hydroperoxide groups in oxidised polypropylene: Effect on photo-oxidation. Polym. Degrad. Stab. 1981, 3, 327–332. [Google Scholar] [CrossRef]
- Jensen, K.; Aiello, A.; Mitterhofer, S.; Barretta, C.; Oreski, G.; Stafford, C.; Gu, X. Surface photooxidation of polypropylene-based photovoltaic backsheets: A comprehensive spectroscopic investigation. Polym. Degrad. Stab. 2024, 232, 111132. [Google Scholar] [CrossRef]
- Yousif, E.; Haddad, R. Photostabilization of poly(vinyl chloride)–Still on the run. J. Taibah Univ. Sci. 2015, 9, 421–448. [Google Scholar] [CrossRef]
- Mohammed, A.; El-Hiti, G.A.; Yousif, E.; Ahmed, A.A.; Ahmed, D.S.; Alotaibi, M.H. Protection of Poly(Vinyl Chloride) Films against Photodegradation Using Various Valsartan Tin Complexes. Polymers 2020, 12, 969. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.F.A.; Goi, B.E.; Schmitt, C.C.; Neumann, M.G. Photodegradation of Polystyrene Films Containing UV-Visible Sensitizers. J. Res. Updates Polym. Sci. 2013, 2, 39–47. [Google Scholar] [CrossRef]
- Thanki, P.N.; Singh, R.P.; Mathur, G.N. Photo-oxidative degradation of nylon 66 under accelerated weathering. Polymer 1998, 39, 6363–6367. [Google Scholar] [CrossRef]
- Fechine, G.J.M.; Rabello, M.S.; Souto-Maior, R.M. Photo-irradiation induced morphological changes in nylon 66. Polymer 2001, 42, 535–538. [Google Scholar] [CrossRef]
- Sang, C.; Orimoto, Y.; Aoki, Y. Photodegradation Pathways of Aliphatic Polyamide through Conical Intersection between Ground and Excited States. J. Phys. Chem. A 2024, 128, 8865–8877. [Google Scholar] [CrossRef]
- Manoharan, R.; Chadha, S.; Ghiamati, E.; Nelson, W.H. UV-Excited Raman and Resonance Raman Spectra of Synthetic Polymers. Appl. Spectrosc. 1992, 46, 1176–1181. [Google Scholar] [CrossRef]
- Guo, X.; Lin, Z.; Wang, Y.; He, Z.; Wang, M.; Jin, G. Monitoring PP degradation under multiple extrusions by Raman spectroscopy. Polymers 2019, 11, 1698. [Google Scholar] [CrossRef]
- Stroe, M.; Cristea, M.; Matei, E.; Galatanu, A.; Cotet, L.C.; Pop, L.C.; Baia, M.; Danciu, V.; Anghel, I.; Baia, L.; et al. Optical Properties of Graphene Oxide/Polystyrene Composites. Molecules 2020, 25, 2419. [Google Scholar] [CrossRef]
- Kaczmarek, H.; Buffeteau, T.; Sourisseau, C. Photo- and bio-degradation in polyethylene, cellulose and blends studied by ATR-FTIR and Raman. J. Mater. Sci. 2005, 40, 4189–4198. [Google Scholar] [CrossRef]
- Menchaca, C.; Álvarez-Castillo, A.; Martínez-Barrera, G.; López-Valdivia, H.; Carrasco, H.; Castaño, V.M. Modification of nylon 6,12 by gamma irradiation. Int. J. Mater. Prod. Technol. 2003, 19, 521–529. [Google Scholar] [CrossRef]
- Matsui, H.; Schehr, C.A.; Valentini, J.J.; Weber, J.N. Resonance Raman investigation of Nylon 6,6 model compound degradation. Polymer 2001, 42, 5625–5632. [Google Scholar] [CrossRef]
- Ye, L.; Li, T.; Hong, L. Enhanced char formation in PVC thermal decomposition. Mater. Today Commun. 2021, 26, 102186. [Google Scholar] [CrossRef]
- Chrissafis, K.; Paraskevopoulos, K.M.; Pavlidou, E.; Bikiaris, D. Thermal degradation mechanism of HDPE nanocomposites. Thermochim. Acta 2009, 485, 65–71. [Google Scholar] [CrossRef]
- Valadez-Gonzalez, A.; Cervantes-Uc, J.M.; Veleva, L. Mineral filler influence on HDPE photo-oxidation. Polym. Degrad. Stab. 1999, 63, 253–260. [Google Scholar] [CrossRef]
- Kaci, M.; Sadoun, T.; Cimmino, S. Crystallinity measurements of LDPE under natural weathering. Int. J. Polym. Anal. Charact. 2001, 6, 455–464. [Google Scholar] [CrossRef]
- Rwei, S.-P.; Ranganathan, P.; Lee, Y.-H. Isothermal Crystallization Kinetics Study of Fully Aliphatic PA6 Copolyamides: Effect of Novel Long-Chain Polyamide Salt as a Comonomer. Polymers 2019, 11, 472. [Google Scholar] [CrossRef]
- Yano, S.; Murayama, M. Photo-oxidation of nylon 6 above 300 nm. Polym. Photochem. 1981, 1, 177–190. [Google Scholar] [CrossRef]
- Shackleford, A.S.D.; Williams, R.J.; Brown, R.; Wingham, J.R.; Majewski, C. Degradation of Laser Sintered polyamide 12 parts due to accelerated exposure to ultraviolet radiation. Addit. Manuf. 2021, 46, 102132. [Google Scholar] [CrossRef]
- Campanale, C.; Savino, I.; Massarelli, C.; Uricchio, V.F. Fourier Transform Infrared Spectroscopy to Assess the Degree of Alteration of Artificially Aged and Environmentally Weathered Microplastics. Polymers 2023, 15, 911. [Google Scholar] [CrossRef] [PubMed]
- Fernández-González, V.; Andrade-Garda, J.M.; López-Mahía, P.; Muniategui-Lorenzo, S. Impact of weathering on the chemical identification of microplastics from usual packaging polymers in the marine environment. Anal. Chim. Acta 2021, 1142, 179–188. [Google Scholar] [CrossRef]
- Binda, G.; Kalčíková, G.; Allan, I.J.; Hurley, R.; Rødland, E.; Spanu, D.; Nizzetto, L. Microplastic aging processes: Environmental relevance and analytical implications. TrAC Trends Anal. Chem. 2024, 172, 117566. [Google Scholar] [CrossRef]
- Phan, S.; Padilla-Gamiño, J.L.; Luscombe, C.K. The effect of weathering environments on microplastic chemical identification with Raman and IR spectroscopy: Part I. Polyethylene and polypropylene. Polym. Test. 2022, 116, 107752. [Google Scholar] [CrossRef]
- De Frond, H.; Rubinovitz, R.; Rochman, C.M. μATR-FTIR Spectral Libraries of Plastic Particles (FLOPP and FLOPP-e) for the Analysis of Microplastics. Anal. Chem. 2021, 93, 15878–15885. [Google Scholar] [CrossRef]
- Anshari, R.; Tsuboi, M.; Sato, H.; Tashiro, K.; Ozaki, Y. Raman and ATR-FTIR unmask crystallinity changes and carboxylate group and vinyl group accumulation in natural weathering polypropylene microplastics. Sci. Rep. 2025, 15, 2518. [Google Scholar] [CrossRef] [PubMed]
- Marica, I.; Pînzaru, S.C. A Raman spectral database of naturally aged plastics: A proof-of-concept study for waste plastic sorting. J. Raman Spectrosc. 2023, 54, 305–313. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Band (cm−1) | Assignment |
|---|---|---|
| HDPE [20] | 2915 | Asymmetric stretching vibration CH2 |
| 2848 | Symmetric stretching vibration CH2 | |
| 1462 | Bending vibrations CH2 | |
| 713 | Rocking vibrations CH2 | |
| LDPE [23] | 2900 | Asymmetric stretching vibration CH2 |
| 2820 | Symmetric stretching vibration CH2 | |
| 1462 | Bending vibration CH2 | |
| 1365 | Bending mode CH3 terminal groups | |
| 722 | Rocking vibration CH2 | |
| PP [24] | 3000–2800 | Asymmetric stretching vibration CH3; asymmetric stretching vibration CH2; symmetric stretching vibration CH3; symmetric stretching vibration CH2 |
| 1470 | Asymmetric bending vibration CH3 | |
| 1370 | Symmetric bending vibration CH3 | |
| 1162 | Rotation vibration C-H | |
| 985 | Rocking vibration CH3 | |
| 970, 835 | Deformation C-C bonding | |
| PVC [25] | 2972 | Asymmetric stretching vibration CH2 |
| 2910 | Symmetric stretching vibration CH2 | |
| 1440 | Bending vibrations CH | |
| 1250 | Bending vibrations CH (near Cl) | |
| 1100–930 | C-H stretching bond backbone chain | |
| 680–600 | Bending vibration C-Cl | |
| PS [26] | 3026 | Aromatic CH stretching vibration |
| 2920 | Asymmetric stretching vibration CH2 | |
| 2850 | Symmetric stretching vibration CH2 | |
| 1598 | Aromatic C=C vinyl group stretching | |
| 1492 | Bending vibration benzene ring | |
| 752 | Bending vibration substituted benzene derivative | |
| Nylon [27] | 3420 | NH2 stretching vibration |
| 3300 | N-H stretching vibration | |
| 2940 | Asymmetric stretching vibration CH2 | |
| 2865 | Symmetric stretching vibration CH2 | |
| 1640 | Amide I | |
| 1545, 1455 | Amide II | |
| 1265 | Amide III |
| Polymer | Band (cm−1) | Assignment |
|---|---|---|
| HDPE, LDPE [35] | 2881, 2846 | CH2 asymmetric stretching, CH2 symmetric stretching |
| 1438, 1293 | CH2 bending | |
| 1127, 1060 | C-C stretching | |
| PP [47] | 1454 | CH2 bending, CH3 asymmetric bending |
| 1335 | CH stretching, CH3 bending | |
| 1155 | C-C stretching, CH bending | |
| 977 | C-C stretching | |
| 813 | C-C stretching | |
| PVC [22] | 1440 | CH2 bending |
| 700 | C-Cl stretching | |
| 640 | C-Cl stretching | |
| PS [48] | 1445 | CH bending |
| 1180 | C-C asymmetric stretching | |
| 1154 | C-C symmetric stretching | |
| 1028 | C-C symmetric stretching in the benzene ring | |
| 999 | benzene ring breathing | |
| 789 | C-H bending (out of plane) | |
| Nylon [49] | 1630 | N-H stretching amide I |
| 1440 | CH2 bending | |
| 1380 | CH2 wagging | |
| 1300 | CH2 twisting | |
| 1213 | N-H wagging | |
| 1125, 1077, 1060 | C-C stretching | |
| 930 | C-CO stretching |
| Sample | LDPE | PP | Nylon | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Tm | ∆Hm | Xc | Tm | ∆Hm | Xc | Tm | ∆Hm | Xc | |
| °C | J/g | % | °C | J/g | % | °C | J/g | % | |
| Reference | 110.81 | 98.1 | 33.37 | 162.36 | 63.83 | 30.82 | 223.64 | 62.64 | 33.32 |
| 1 month | 114.09 | 96.99 | 32.99 | 162.84 | 83.18 | 40.16 | 222.92 | 62.09 | 33.03 |
| 6 month | 113.84 | 95.62 | 32.52 | 162.96 | 84.56 | 40.83 | 222.75 | 60.69 | 32.28 |
| 1 year | 111.47 | 105.3 | 35.82 | 161.39 | 81.77 | 39.48 | 223.13 | 60.98 | 32.44 |
| 2 years | 111.3 | 78.71 | 26.77 | 165.62 | 77.81 | 37.57 | 222.92 | 61.79 | 32.87 |
| 5 years | 110.68 | 100.6 | 34.22 | 161.68 | 82.11 | 39.65 | 223.19 | 62.76 | 33.38 |
| 10 years | 110.85 | 100.7 | 34.25 | 161.80 | 79.13 | 38.21 | 223.05 | 65.66 | 34.93 |
| 20 years | 111.17 | 91.89 | 31.26 | 161.05 | 81.28 | 39.25 | 224.03 | 64.65 | 34.39 |
| 40 years | 111.36 | 106.7 | 36.29 | 162.28 | 78.86 | 38.08 | - | - | - |
| 60 years | 111.18 | 102.7 | 34.93 | 161.38 | 77.21 | 37.28 | 223.17 | 65.98 | 35.10 |
| 80 years | 114.27 | 110.5 | 37.59 | 161.15 | 80.25 | 38.75 | 222.75 | 64.91 | 34.53 |
| 100 years | 111.36 | 117.5 | 39.97 | 160.92 | 91.56 | 44.21 | 223.36 | 66.35 | 35.29 |
| 130 years | 114.14 | 126.4 | 42.99 | 157.49 | 102.3 | 49.40 | 222.38 | 66.00 | 35.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Yeste, M.P.; Bergaliyeva, S.; Cauqui, M.Á.; Cajaraville, M.P.; Sendra, M. Assessment of the Suitability and Accuracy of Different Methods to Determine the Degree of Photodegradation of High- and Low-Density Polyethylene, Polypropylene, Polyvinyl Chloride, Nylon and Polystyrene Microplastics. Microplastics 2026, 5, 62. https://doi.org/10.3390/microplastics5020062
Yeste MP, Bergaliyeva S, Cauqui MÁ, Cajaraville MP, Sendra M. Assessment of the Suitability and Accuracy of Different Methods to Determine the Degree of Photodegradation of High- and Low-Density Polyethylene, Polypropylene, Polyvinyl Chloride, Nylon and Polystyrene Microplastics. Microplastics. 2026; 5(2):62. https://doi.org/10.3390/microplastics5020062
Chicago/Turabian StyleYeste, María Pilar, Saltanat Bergaliyeva, Miguel Ángel Cauqui, Miren P. Cajaraville, and Marta Sendra. 2026. "Assessment of the Suitability and Accuracy of Different Methods to Determine the Degree of Photodegradation of High- and Low-Density Polyethylene, Polypropylene, Polyvinyl Chloride, Nylon and Polystyrene Microplastics" Microplastics 5, no. 2: 62. https://doi.org/10.3390/microplastics5020062
APA StyleYeste, M. P., Bergaliyeva, S., Cauqui, M. Á., Cajaraville, M. P., & Sendra, M. (2026). Assessment of the Suitability and Accuracy of Different Methods to Determine the Degree of Photodegradation of High- and Low-Density Polyethylene, Polypropylene, Polyvinyl Chloride, Nylon and Polystyrene Microplastics. Microplastics, 5(2), 62. https://doi.org/10.3390/microplastics5020062