Asymmetric Inheritance of Cell Fate Determinants: Focus on RNA


Abstract
1. Introduction
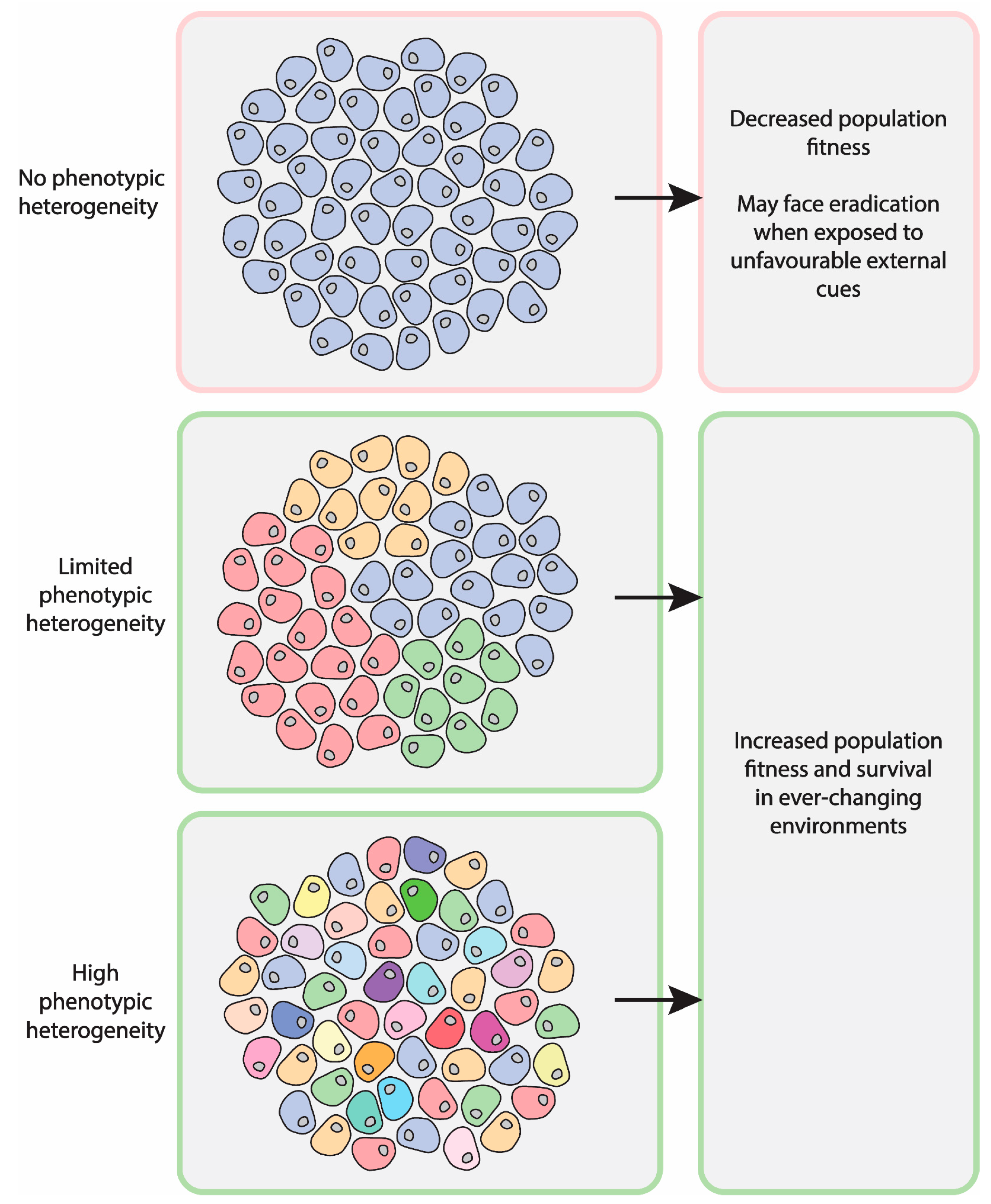
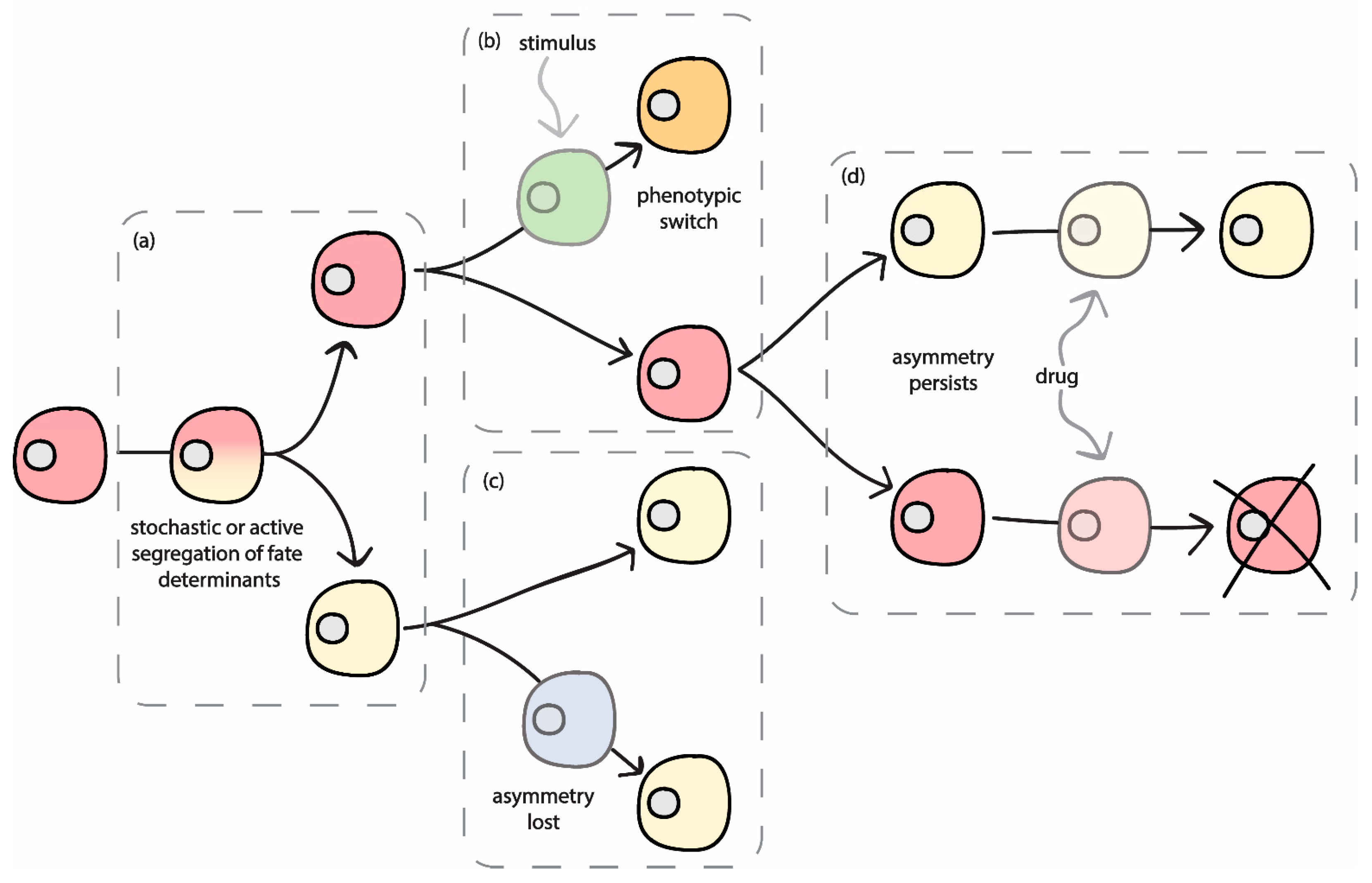
2. Relevance of Asymmetric Segregation
3. Molecular Fate Determinants
3.1. Proteins
3.2. Prions
3.3. DNA
3.4. DNA Modifications and Histone Inheritance
3.5. Organelles
4. Differential RNA Segregation and Inheritance
5. Asymmetric Inheritance of Cell Fate Determinants During Stem Cell Divisions
6. Discussion
Funding
Acknowledgments
Conflicts of Interest
References
- Cheeseman, I.M.; Desai, A. Molecular architecture of the kinetochore-microtubule interface. Nat. Rev. Mol. Cell Biol. 2008, 9, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Theisen, A.; Shaffer, L.G. Disorders caused by chromosome abnormalities. Appl. Clin. Genet. 2010, 3, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Knoblich, J.A. Mechanisms of asymmetric stem cell division. Cell 2008, 132, 583–597. [Google Scholar] [CrossRef]
- Goldstein, B.; Macara, I.G. The PAR proteins: Fundamental players in animal cell polarization. Dev. Cell 2007, 13, 609–622. [Google Scholar] [CrossRef]
- Suzuki, A.; Ohno, S. The PAR-aPKC system: Lessons in polarity. J. Cell Sci. 2006, 119, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Betschinger, J.; Knoblich, J.A. Dare to be different: Asymmetric cell division in Drosophila, C. elegans and vertebrates. Curr. Biol. 2004, 14, R674–R685. [Google Scholar] [CrossRef] [PubMed]
- Dalton, C.M.; Carroll, J. Biased inheritance of mitochondria during asymmetric cell division in the mouse oocyte. J. Cell Sci. 2013, 126, 2955–2964. [Google Scholar] [CrossRef]
- Katajisto, P.; Dohla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef]
- Huh, D.; Paulsson, J. Random partitioning of molecules at cell division. Proc. Natl. Acad. Sci. USA 2011, 108, 15004–15009. [Google Scholar] [CrossRef]
- King, M.L.; Messitt, T.J.; Mowry, K.L. Putting RNAs in the right place at the right time: RNA localization in the frog oocyte. Biol. Cell 2005, 97, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Trovisco, V.; Belaya, K.; Nashchekin, D.; Irion, U.; Sirinakis, G.; Butler, R.; Lee, J.J.; Gavis, E.R.; St Johnston, D. bicoid mRNA localises to the Drosophila oocyte anterior by random Dynein-mediated transport and anchoring. Elife 2016, 5. [Google Scholar] [CrossRef]
- Bergmiller, T.; Andersson, A.M.C.; Tomasek, K.; Balleza, E.; Kiviet, D.J.; Hauschild, R.; Tkacik, G.; Guet, C.C. Biased partitioning of the multidrug efflux pump AcrAB-TolC underlies long-lived phenotypic heterogeneity. Science 2017, 356, 311–315. [Google Scholar] [CrossRef]
- Yang, J.; McCormick, M.A.; Zheng, J.; Xie, Z.; Tsuchiya, M.; Tsuchiyama, S.; El-Samad, H.; Ouyang, Q.; Kaeberlein, M.; Kennedy, B.K.; et al. Systematic analysis of asymmetric partitioning of yeast proteome between mother and daughter cells reveals “aging factors” and mechanism of lifespan asymmetry. Proc. Natl. Acad. Sci. USA 2015, 112, 11977–11982. [Google Scholar] [CrossRef]
- Aguilaniu, H.; Gustafsson, L.; Rigoulet, M.; Nystrom, T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 2003, 299, 1751–1753. [Google Scholar] [CrossRef]
- Conklin, E.G. Mosaic development in ascidian eggs. J. Exp. Zool. 1905, 2, 145–223. [Google Scholar] [CrossRef]
- Cayouette, M.; Raff, M. Asymmetric segregation of Numb: A mechanism for neural specification from Drosophila to mammals. Nat. Neurosci. 2002, 5, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Fickentscher, R.; Weiss, M. Physical determinants of asymmetric cell divisions in the early development of Caenorhabditis elegans. Sci. Rep. 2017, 7, 9369. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, A.D. Oriented cell divisions asymmetrically segregate aPKC and generate cell fate diversity in the early Xenopus embryo. Development 2003, 130, 2657–2668. [Google Scholar] [CrossRef] [PubMed]
- Torres-Padilla, M.E.; Parfitt, D.E.; Kouzarides, T.; Zernicka-Goetz, M. Histone arginine methylation regulates pluripotency in the early mouse embryo. Nature 2007, 445, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Goolam, M.; Scialdone, A.; Graham, S.J.L.; Macaulay, I.C.; Jedrusik, A.; Hupalowska, A.; Voet, T.; Marioni, J.C.; Zernicka-Goetz, M. Heterogeneity in Oct4 and Sox2 Targets Biases Cell Fate in 4-Cell Mouse Embryos. Cell 2016, 165, 61–74. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L.; Feng, G.; Wang, Y.; Li, Y.; Li, X.; Liu, C.; Jiao, G.; Huang, C.; Shi, J.; et al. Asymmetric Expression of LincGET Biases Cell Fate in Two-Cell Mouse Embryos. Cell 2018, 175, 1887–1901. [Google Scholar] [CrossRef]
- Shi, J.; Chen, Q.; Li, X.; Zheng, X.; Zhang, Y.; Qiao, J.; Tang, F.; Tao, Y.; Zhou, Q.; Duan, E. Dynamic transcriptional symmetry-breaking in pre-implantation mammalian embryo development revealed by single-cell RNA-seq. Development 2015, 142, 3468–3477. [Google Scholar] [CrossRef]
- Chen, Q.; Shi, J.; Tao, Y.; Zernicka-Goetz, M. Tracing the origin of heterogeneity and symmetry breaking in the early mammalian embryo. Nat. Commun. 2018, 9, 1819. [Google Scholar] [CrossRef]
- Inaba, M.; Yamashita, Y.M. Asymmetric stem cell division: Precision for robustness. Cell Stem Cell 2012, 11, 461–469. [Google Scholar] [CrossRef]
- Yamashita, Y.M.; Fuller, M.T. Asymmetric stem cell division and function of the niche in the Drosophila male germ line. Int. J. Hematol. 2005, 82, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, N.; Garriga, G. Asymmetric cell division: From A to Z. Genes Dev. 1998, 12, 3625–3638. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhong, W.; Chia, W. Neurogenesis and asymmetric cell division. Curr. Opin. Neurobiol. 2008, 18, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Knoblich, J.A. Asymmetric cell division during animal development. Nat. Rev. Mol. Cell Biol. 2001, 2, 11–20. [Google Scholar] [CrossRef]
- Cabernard, C.; Doe, C.Q. Apical/basal spindle orientation is required for neuroblast homeostasis and neuronal differentiation in Drosophila. Dev. Cell 2009, 17, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Caussinus, E.; Gonzalez, C. Induction of tumor growth by altered stem-cell asymmetric division in Drosophila melanogaster. Nat. Genet. 2005, 37, 1125–1129. [Google Scholar] [CrossRef]
- Brand, A.H.; Livesey, F.J. Neural stem cell biology in vertebrates and invertebrates: More alike than different? Neuron 2011, 70, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, R.; Jarosz, D.F.; Jones, S.K.; Chang, A.; Lancaster, A.K.; Lindquist, S. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 2012, 482, 363–368. [Google Scholar] [CrossRef]
- Chernova, T.A.; Wilkinson, K.D.; Chernoff, Y.O. Physiological and environmental control of yeast prions. FEMS Microbiol. Rev. 2014, 38, 326–344. [Google Scholar] [CrossRef] [PubMed]
- True, H.L.; Lindquist, S.L. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 2000, 407, 477–483. [Google Scholar] [CrossRef]
- Liu, B.; Larsson, L.; Franssens, V.; Hao, X.; Hill, S.M.; Andersson, V.; Hoglund, D.; Song, J.; Yang, X.; Oling, D.; et al. Segregation of protein aggregates involves actin and the polarity machinery. Cell 2011, 147, 959–961. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klar, A.J. Differentiated parental DNA strands confer developmental asymmetry on daughter cells in fission yeast. Nature 1987, 326, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Armakolas, A.; Klar, A.J. Cell type regulates selective segregation of mouse chromosome 7 DNA strands in mitosis. Science 2006, 311, 1146–1149. [Google Scholar] [CrossRef]
- Karpowicz, P.; Morshead, C.; Kam, A.; Jervis, E.; Ramunas, J.; Cheng, V.; van der Kooy, D. Support for the immortal strand hypothesis: Neural stem cells partition DNA asymmetrically in vitro. J. Cell Biol. 2005, 170, 721–732. [Google Scholar] [CrossRef]
- Cairns, J. Mutation selection and the natural history of cancer. Nature 1975, 255, 197–200. [Google Scholar] [CrossRef]
- Steinhauser, M.L.; Bailey, A.P.; Senyo, S.E.; Guillermier, C.; Perlstein, T.S.; Gould, A.P.; Lee, R.T.; Lechene, C.P. Multi-isotope imaging mass spectrometry quantifies stem cell division and metabolism. Nature 2012, 481, 516–519. [Google Scholar] [CrossRef]
- Kiel, M.J.; He, S.; Ashkenazi, R.; Gentry, S.N.; Teta, M.; Kushner, J.A.; Jackson, T.L.; Morrison, S.J. Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature 2007, 449, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef]
- Soto, M.; Garcia-Santisteban, I.; Krenning, L.; Medema, R.H.; Raaijmakers, J.A. Chromosomes trapped in micronuclei are liable to segregation errors. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef]
- Cohen, S.; Agmon, N.; Sobol, O.; Segal, D. Extrachromosomal circles of satellite repeats and 5S ribosomal DNA in human cells. Mob. DNA 2010, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Segal, D. Extrachromosomal circular DNA in eukaryotes: Possible involvement in the plasticity of tandem repeats. Cytogenet. Genome Res. 2009, 124, 327–338. [Google Scholar] [CrossRef]
- Shcheprova, Z.; Baldi, S.; Frei, S.B.; Gonnet, G.; Barral, Y. A mechanism for asymmetric segregation of age during yeast budding. Nature 2008, 454, 728–734. [Google Scholar] [CrossRef]
- Denoth-Lippuner, A.; Krzyzanowski, M.K.; Stober, C.; Barral, Y. Role of SAGA in the asymmetric segregation of DNA circles during yeast ageing. Elife 2014, 3. [Google Scholar] [CrossRef]
- Compere, S.J.; Palmiter, R.D. DNA methylation controls the inducibility of the mouse metallothionein-I gene lymphoid cells. Cell 1981, 25, 233–240. [Google Scholar] [CrossRef]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.D. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J. Biol. Chem. 1948, 175, 315–332. [Google Scholar]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2012, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Wilson, G.G.; Kuo, K.C.; Gehrke, C.W. N4-methylcytosine as a minor base in bacterial DNA. J. Bacteriol. 1987, 169, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.Z.; Blanco, M.A.; Greer, E.L.; He, C.; Shi, Y. DNA N (6)-methyladenine: A new epigenetic mark in eukaryotes? Nat. Rev. Mol. Cell Biol. 2015, 16, 705–710. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Pfaffeneder, T.; Hackner, B.; Truss, M.; Munzel, M.; Muller, M.; Deiml, C.A.; Hagemeier, C.; Carell, T. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem. Int. Ed. Engl. 2011, 50, 7008–7012. [Google Scholar] [CrossRef]
- Woodcock, D.M.; Lawler, C.B.; Linsenmeyer, M.E.; Doherty, J.P.; Warren, W.D. Asymmetric methylation in the hypermethylated CpG promoter region of the human L1 retrotransposon. J. Biol. Chem. 1997, 272, 7810–7816. [Google Scholar] [CrossRef]
- Guo, W.; Chung, W.Y.; Qian, M.; Pellegrini, M.; Zhang, M.Q. Characterizing the strand-specific distribution of non-CpG methylation in human pluripotent cells. Nucleic Acids Res. 2013, 42, 3009–3016. [Google Scholar] [CrossRef]
- Ji, H.; Ehrlich, L.I.; Seita, J.; Murakami, P.; Doi, A.; Lindau, P.; Lee, H.; Aryee, M.J.; Irizarry, R.A.; Kim, K.; et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010, 467, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Hodges, E.; Molaro, A.; Dos Santos, C.O.; Thekkat, P.; Song, Q.; Uren, P.J.; Park, J.; Butler, J.; Rafii, S.; McCombie, W.R.; et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol. Cell 2011, 44, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Beerman, I.; Lien, W.H.; Smith, Z.D.; Gu, H.; Boyle, P.; Gnirke, A.; Fuchs, E.; Rossi, D.J.; Meissner, A. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol. Cell 2012, 47, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Jang, H.S.; Shin, W.J.; Lee, J.E.; Do, J.T. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes 2017, 8. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Hermann, A.; Gowher, H.; Jeltsch, A. Biochemistry and biology of mammalian DNA methyltransferases. Cell Mol. Life Sci. 2004, 61, 2571–2587. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, C.M.; Hou, C.; Little, J.A.; Dean, A. Epigenetics of beta-globin gene regulation. Mutat. Res. 2008, 647, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef]
- Dhayalan, A.; Rajavelu, A.; Rathert, P.; Tamas, R.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010, 285, 26114–26120. [Google Scholar] [CrossRef]
- Li, B.-Z.; Huang, Z.; Cui, Q.-Y.; Song, X.-H.; Du, L.; Jeltsch, A.; Chen, P.; Li, G.; Li, E.; Xu, G.-L. Histone tails regulate DNA methylation by allosterically activating de novo methyltransferase. Cell Res. 2011, 21, 1172–1181. [Google Scholar] [CrossRef]
- Cervoni, N.; Szyf, M. Demethylase Activity Is Directed by Histone Acetylation. J. Biol. Chem. 2001, 276, 40778–40787. [Google Scholar] [CrossRef]
- D’Alessio, A.C.; Weaver, I.C.G.; Szyf, M. Acetylation-Induced Transcription Is Required for Active DNA Demethylation in Methylation-Silenced Genes. Mol. Cell. Biol. 2007, 27, 7462–7474. [Google Scholar] [CrossRef]
- Zhang, Y.; Jurkowska, R.; Soeroes, S.; Rajavelu, A.; Dhayalan, A.; Bock, I.; Rathert, P.; Brandt, O.; Reinhardt, R.; Fischle, W.; et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010, 38, 4246–4253. [Google Scholar] [CrossRef] [PubMed]
- Alabert, C.; Groth, A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012, 13, 153–167. [Google Scholar] [CrossRef]
- Xie, J.; Wooten, M.; Tran, V.; Chen, X. Breaking Symmetry − Asymmetric Histone Inheritance in Stem Cells. Trends Cell Biol. 2017, 27, 527–540. [Google Scholar] [CrossRef]
- Tran, V.; Feng, L.; Chen, X. Asymmetric distribution of histones during Drosophila male germline stem cell asymmetric divisions. Chromosome Res. 2013, 21, 255–269. [Google Scholar] [CrossRef][Green Version]
- Xie, J.; Wooten, M.; Tran, V.; Chen, B.C.; Pozmanter, C.; Simbolon, C.; Betzig, E.; Chen, X. Histone H3 Threonine Phosphorylation Regulates Asymmetric Histone Inheritance in the Drosophila Male Germline. Cell 2015, 163, 920–933. [Google Scholar] [CrossRef] [PubMed]
- Pirrotta, V. Histone Marks Direct Chromosome Segregation. Cell 2015, 163, 792–793. [Google Scholar] [CrossRef][Green Version]
- Valls, E.; Sanchez-Molina, S.; Martinez-Balbas, M.A. Role of histone modifications in marking and activating genes through mitosis. J. Biol. Chem. 2005, 280, 42592–42600. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Tseng, B.S.; Dormann, H.L.; Ueberheide, B.M.; Garcia, B.A.; Shabanowitz, J.; Hunt, D.F.; Funabiki, H.; Allis, C.D. Regulation of HP1–chromatin binding by histone H3 methylation and phosphorylation. Nature 2005, 438, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Kouskouti, A.; Talianidis, I. Histone modifications defining active genes persist after transcriptional and mitotic inactivation. EMBO J. 2005, 24, 347–357. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial inheritance in yeast. Biochim. Biophys. Acta 2014, 1837, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Böckler, S.; Chelius, X.; Hock, N.; Klecker, T.; Wolter, M.; Weiss, M.; Braun, R.J.; Westermann, B. Fusion, fission, and transport control asymmetric inheritance of mitochondria and protein aggregates. J. Cell Biol. 2017, 216, 2481–2498. [Google Scholar] [CrossRef]
- Rapp, S.; Saffrich, R.; Anton, M.; Jakle, U.; Ansorge, W.; Gorgas, K.; Just, W.W. Microtubule-based peroxisome movement. J. Cell Sci. 1996, 109(Pt. 4), 837–849. [Google Scholar]
- Fagarasanu, A.; Fagarasanu, M.; Rachubinski, R.A. Maintaining peroxisome populations: A story of division and inheritance. Annu. Rev. Cell Dev. Biol. 2007, 23, 321–344. [Google Scholar] [CrossRef]
- Asare, A.; Levorse, J.; Fuchs, E. Coupling organelle inheritance with mitosis to balance growth and differentiation. Science 2017, 355. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; de Boer, R.; van der Klei, I.J. Yeast cells contain a heterogeneous population of peroxisomes that segregate asymmetrically during cell division. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Robbins, E.; Jentzsch, G.; Micali, A. The centriole cycle in synchronized HeLa cells. J. Cell Biol. 1968, 36, 329–339. [Google Scholar] [CrossRef]
- Tsou, M.F.; Stearns, T. Mechanism limiting centrosome duplication to once per cell cycle. Nature 2006, 442, 947–951. [Google Scholar] [CrossRef]
- Yamashita, Y.M.; Mahowald, A.P.; Perlin, J.R.; Fuller, M.T. Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science 2007, 315, 518–521. [Google Scholar] [CrossRef] [PubMed]
- Conduit, P.T.; Raff, J.W. Cnn dynamics drive centrosome size asymmetry to ensure daughter centriole retention in Drosophila neuroblasts. Curr. Biol. 2010, 20, 2187–2192. [Google Scholar] [CrossRef]
- Januschke, J.; Llamazares, S.; Reina, J.; Gonzalez, C. Drosophila neuroblasts retain the daughter centrosome. Nat. Commun. 2011, 2, 243. [Google Scholar] [CrossRef]
- Wang, X.; Tsai, J.W.; Imai, J.H.; Lian, W.N.; Vallee, R.B.; Shi, S.H. Asymmetric centrosome inheritance maintains neural progenitors in the neocortex. Nature 2009, 461, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Babour, A.; Bicknell, A.A.; Tourtellotte, J.; Niwa, M. A surveillance pathway monitors the fitness of the endoplasmic reticulum to control its inheritance. Cell 2010, 142, 256–269. [Google Scholar] [CrossRef]
- Estrada, P.; Kim, J.; Coleman, J.; Walker, L.; Dunn, B.; Takizawa, P.; Novick, P.; Ferro-Novick, S. Myo4p and She3p are required for cortical ER inheritance inSaccharomyces cerevisiae. J. Cell Biol. 2003, 163, 1255–1266. [Google Scholar] [CrossRef]
- Poteryaev, D.; Squirrell, J.M.; Campbell, J.M.; White, J.G.; Spang, A. Involvement of the actin cytoskeleton and homotypic membrane fusion in ER dynamics in Caenorhabditis elegans. Mol. Biol. Cell 2005, 16, 2139–2153. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.T.; Schoborg, T.A.; Bergman, Z.J.; Riggs, B.; Rusan, N.M. Proper symmetric and asymmetric endoplasmic reticulum partitioning requires astral microtubules. Open Biol. 2015, 5. [Google Scholar] [CrossRef]
- Eritano, A.S.; Altamirano, A.; Beyeler, S.; Gaytan, N.; Velasquez, M.; Riggs, B. The endoplasmic reticulum is partitioned asymmetrically during mitosis before cell fate selection in proneuronal cells in the early Drosophila embryo. Mol. Biol. Cell 2017, 28, 1530–1538. [Google Scholar] [CrossRef]
- Jeffery, W.R.; Tomlinson, C.R.; Brodeur, R.D. Localization of actin messenger RNA during early ascidian development. Dev. Biol. 1983, 99, 408–417. [Google Scholar] [CrossRef]
- Salles, F.J.; Lieberfarb, M.E.; Wreden, C.; Gergen, J.P.; Strickland, S. Coordinate initiation of Drosophila development by regulated polyadenylation of maternal messenger RNAs. Science 1994, 266, 1996–1999. [Google Scholar] [CrossRef] [PubMed]
- Forrest, K.M.; Clark, I.E.; Jain, R.A.; Gavis, E.R. Temporal complexity within a translational control element in the nanos mRNA. Development 2004, 131, 5849–5857. [Google Scholar] [CrossRef] [PubMed]
- Dahanukar, A.; Walker, J.A.; Wharton, R.P. Smaug, a novel RNA-binding protein that operates a translational switch in Drosophila. Mol. Cell 1999, 4, 209–218. [Google Scholar] [CrossRef]
- Nelson, M.R.; Leidal, A.M.; Smibert, C.A. Drosophila Cup is an eIF4E-binding protein that functions in Smaug-mediated translational repression. EMBO J. 2004, 23, 150–159. [Google Scholar] [CrossRef]
- Smibert, C.A.; Lie, Y.S.; Shillinglaw, W.; Henzel, W.J.; Macdonald, P.M. Smaug, a novel and conserved protein, contributes to repression of nanos mRNA translation in vitro. RNA 1999, 5, 1535–1547. [Google Scholar] [CrossRef]
- Holt, C.E.; Bullock, S.L. Subcellular mRNA localization in animal cells and why it matters. Science 2009, 326, 1212–1216. [Google Scholar] [CrossRef]
- Ephrussi, A.; Dickinson, L.K.; Lehmann, R. Oskar organizes the germ plasm and directs localization of the posterior determinant nanos. Cell 1991, 66, 37–50. [Google Scholar] [CrossRef]
- Gavis, E.R.; Lehmann, R. Localization of nanos RNA controls embryonic polarity. Cell 1992, 71, 301–313. [Google Scholar] [CrossRef]
- Lecuyer, E.; Yoshida, H.; Parthasarathy, N.; Alm, C.; Babak, T.; Cerovina, T.; Hughes, T.R.; Tomancak, P.; Krause, H.M. Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell 2007, 131, 174–187. [Google Scholar] [CrossRef]
- Heym, R.G.; Niessing, D. Principles of mRNA transport in yeast. Cell Mol. Life Sci. 2012, 69, 1843–1853. [Google Scholar] [CrossRef]
- Haber, J.E. Mating-type genes and MAT switching in Saccharomyces cerevisiae. Genetics 2012, 191, 33–64. [Google Scholar] [CrossRef]
- Gonsalvez, G.B. RNA-protein interactions promote asymmetric sorting of the ASH1 mRNA ribonucleoprotein complex. RNA 2003, 9, 1383–1399. [Google Scholar] [CrossRef]
- Gonzalez, I.; Buonomo, S.B.; Nasmyth, K.; von Ahsen, U. ASH1 mRNA localization in yeast involves multiple secondary structural elements and Ash1 protein translation. Curr. Biol. 1999, 9, 337–340. [Google Scholar] [CrossRef]
- Edelmann, F.T.; Schlundt, A.; Heym, R.G.; Jenner, A.; Niedner-Boblenz, A.; Syed, M.I.; Paillart, J.C.; Stehle, R.; Janowski, R.; Sattler, M.; et al. Molecular architecture and dynamics of ASH1 mRNA recognition by its mRNA-transport complex. Nat. Struct. Mol. Biol. 2017, 24, 152–161. [Google Scholar] [CrossRef]
- Pokrywka, N.J.; Stephenson, E.C. Microtubules mediate the localization of bicoid RNA during Drosophila oogenesis. Development 1991, 113, 55–66. [Google Scholar]
- Clark, I.; Giniger, E.; Ruohola-Baker, H.; Jan, L.Y.; Jan, Y.N. Transient posterior localization of a kinesin fusion protein reflects anteroposterior polarity of the Drosophila oocyte. Curr. Biol. 1994, 4, 289–300. [Google Scholar] [CrossRef]
- Roth, S.; Schupbach, T. The relationship between ovarian and embryonic dorsoventral patterning in Drosophila. Development 1994, 120, 2245–2257. [Google Scholar] [PubMed]
- St Johnston, D. Moving messages: The intracellular localization of mRNAs. Nat. Rev. Mol. Cell Biol. 2005, 6, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, A.M.; Weil, T.T.; Goodhouse, J.; Gavis, E.R.; Schupbach, T. The dynamics of fluorescently labeled endogenous gurken mRNA in Drosophila. J. Cell Sci. 2008, 121, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Zimyanin, V.L.; Belaya, K.; Pecreaux, J.; Gilchrist, M.J.; Clark, A.; Davis, I.; St Johnston, D. In vivo imaging of oskar mRNA transport reveals the mechanism of posterior localization. Cell 2008, 134, 843–853. [Google Scholar] [CrossRef]
- Pellettieri, J.; Seydoux, G. Anterior-posterior polarity in C. elegans and Drosophila—PARallels and differences. Science 2002, 298, 1946–1950. [Google Scholar] [CrossRef]
- Gonzalez-Reyes, A.; Elliott, H.; St Johnston, D. Polarization of both major body axes in Drosophila by gurken-torpedo signalling. Nature 1995, 375, 654–658. [Google Scholar] [CrossRef]
- Forrest, K.M.; Gavis, E.R. Live Imaging of Endogenous RNA Reveals a Diffusion and Entrapment Mechanism for nanos mRNA Localization in Drosophila. Curr. Biol. 2003, 13, 1159–1168. [Google Scholar] [CrossRef]
- Weil, T.T.; Parton, R.M.; Herpers, B.; Soetaert, J.; Veenendaal, T.; Xanthakis, D.; Dobbie, I.M.; Halstead, J.M.; Hayashi, R.; Rabouille, C.; et al. Drosophila patterning is established by differential association of mRNAs with P bodies. Nat. Cell Biol. 2012, 14, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.M.; Davidson, A.; Davis, I.; Weil, T.T. Subcellular mRNA localisation at a glance. J. Cell Sci. 2014, 127, 2127–2133. [Google Scholar] [CrossRef]
- Little, S.C.; Sinsimer, K.S.; Lee, J.J.; Wieschaus, E.F.; Gavis, E.R. Independent and coordinate trafficking of single Drosophila germ plasm mRNAs. Nat. Cell Biol. 2015, 17, 558–568. [Google Scholar] [CrossRef]
- Updike, D.; Strome, S. P granule assembly and function in Caenorhabditis elegans germ cells. J. Androl. 2010, 31, 53–60. [Google Scholar] [CrossRef]
- Voronina, E.; Seydoux, G.; Sassone-Corsi, P.; Nagamori, I. RNA granules in germ cells. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Gallo, C.M.; Wang, J.T.; Motegi, F.; Seydoux, G. Cytoplasmic partitioning of P granule components is not required to specify the germline in C. elegans. Science 2010, 330, 1685–1689. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, I.; Amiri, A.; Fan, Y.; Meyer, N.; Dunkelbarger, S.; Motohashi, T.; Karashima, T.; Bossinger, O.; Strome, S. The PGL family proteins associate with germ granules and function redundantly in Caenorhabditis elegans germline development. Genetics 2004, 167, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Spike, C.; Meyer, N.; Racen, E.; Orsborn, A.; Kirchner, J.; Kuznicki, K.; Yee, C.; Bennett, K.; Strome, S. Genetic analysis of the Caenorhabditis elegans GLH family of P-granule proteins. Genetics 2008, 178, 1973–1987. [Google Scholar] [CrossRef]
- Spike, C.A.; Bader, J.; Reinke, V.; Strome, S. DEPS-1 promotes P-granule assembly and RNA interference in C. elegans germ cells. Development 2008, 135, 983–993. [Google Scholar] [CrossRef]
- Batista, P.J.; Ruby, J.G.; Claycomb, J.M.; Chiang, R.; Fahlgren, N.; Kasschau, K.D.; Chaves, D.A.; Gu, W.; Vasale, J.J.; Duan, S.; et al. PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Mol. Cell 2008, 31, 67–78. [Google Scholar] [CrossRef]
- Das, P.P.; Bagijn, M.P.; Goldstein, L.D.; Woolford, J.R.; Lehrbach, N.J.; Sapetschnig, A.; Buhecha, H.R.; Gilchrist, M.J.; Howe, K.L.; Stark, R.; et al. Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline. Mol. Cell 2008, 31, 79–90. [Google Scholar] [CrossRef]
- Wang, G.; Reinke, V.A. C. elegans Piwi, PRG-1, regulates 21U-RNAs during spermatogenesis. Curr. Biol. 2008, 18, 861–867. [Google Scholar] [CrossRef]
- Ruby, J.G.; Jan, C.; Player, C.; Axtell, M.J.; Lee, W.; Nusbaum, C.; Ge, H.; Bartel, D.P. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell 2006, 127, 1193–1207. [Google Scholar] [CrossRef]
- Ohara, T.; Sakaguchi, Y.; Suzuki, T.; Ueda, H.; Miyauchi, K.; Suzuki, T. The 3’ termini of mouse Piwi-interacting RNAs are 2’-O-methylated. Nat. Struct. Mol. Biol. 2007, 14, 349–350. [Google Scholar] [CrossRef]
- Saito, K.; Nishida, K.M.; Mori, T.; Kawamura, Y.; Miyoshi, K.; Nagami, T.; Siomi, H.; Siomi, M.C. Specific association of Piwi with rasiRNAs derived from retrotransposon and heterochromatic regions in the Drosophila genome. Genes Dev. 2006, 20, 2214–2222. [Google Scholar] [CrossRef] [PubMed]
- Horwich, M.D.; Li, C.; Matranga, C.; Vagin, V.; Farley, G.; Wang, P.; Zamore, P.D. The Drosophila RNA methyltransferase, DmHen1, modifies germline piRNAs and single-stranded siRNAs in RISC. Curr. Biol. 2007, 17, 1265–1272. [Google Scholar] [CrossRef]
- Kirino, Y.; Mourelatos, Z. The mouse homolog of HEN1 is a potential methylase for Piwi-interacting RNAs. RNA 2007, 13, 1397–1401. [Google Scholar] [CrossRef]
- Bagijn, M.P.; Goldstein, L.D.; Sapetschnig, A.; Weick, E.M.; Bouasker, S.; Lehrbach, N.J.; Simard, M.J.; Miska, E.A. Function, targets, and evolution of Caenorhabditis elegans piRNAs. Science 2012, 337, 574–578. [Google Scholar] [CrossRef]
- Lee, H.C.; Gu, W.; Shirayama, M.; Youngman, E.; Conte, D., Jr.; Mello, C.C. C. elegans piRNAs mediate the genome-wide surveillance of germline transcripts. Cell 2012, 150, 78–87. [Google Scholar] [CrossRef]
- Conine, C.C.; Moresco, J.J.; Gu, W.; Shirayama, M.; Conte, D., Jr.; Yates, J.R., 3rd; Mello, C.C. Argonautes promote male fertility and provide a paternal memory of germline gene expression in C. elegans. Cell 2013, 155, 1532–1544. [Google Scholar] [CrossRef]
- Conine, C.C.; Batista, P.J.; Gu, W.; Claycomb, J.M.; Chaves, D.A.; Shirayama, M.; Mello, C.C. Argonautes ALG-3 and ALG-4 are required for spermatogenesis-specific 26G-RNAs and thermotolerant sperm in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2010, 107, 3588–3593. [Google Scholar] [CrossRef]
- Hyman, A.A.; Weber, C.A.; Julicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Julicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Mitchison, T.J.; Hyman, A.A. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4334–4339. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.C.; Brangwynne, C.P. Getting RNA and protein in phase. Cell 2012, 149, 1188–1191. [Google Scholar] [CrossRef] [PubMed]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef]
- Li, P.; Banjade, S.; Cheng, H.C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef]
- Lin, Y.; Protter, D.S.; Rosen, M.K.; Parker, R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Elbaum-Garfinkle, S.; Langdon, E.M.; Taylor, N.; Occhipinti, P.; Bridges, A.A.; Brangwynne, C.P.; Gladfelter, A.S. RNA Controls PolyQ Protein Phase Transitions. Mol. Cell 2015, 60, 220–230. [Google Scholar] [CrossRef]
- Lee, C.; Zhang, H.; Baker, A.E.; Occhipinti, P.; Borsuk, M.E.; Gladfelter, A.S. Protein aggregation behavior regulates cyclin transcript localization and cell-cycle control. Dev. Cell 2013, 25, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Occhipinti, P.; Gladfelter, A.S. PolyQ-dependent RNA-protein assemblies control symmetry breaking. J. Cell Biol. 2015, 208, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.D.; Nagy, L.M. Asymmetric inheritance of centrosomally localized mRNAs during embryonic cleavages. Nature 2002, 420, 682–686. [Google Scholar] [CrossRef]
- Kingsley, E.P.; Chan, X.Y.; Duan, Y.; Lambert, J.D. Widespread RNA segregation in a spiralian embryo. Evol. Dev. 2007, 9, 527–539. [Google Scholar] [CrossRef]
- Alliegro, M.C.; Alliegro, M.A.; Palazzo, R.E. Centrosome-associated RNA in surf clam oocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 9034–9038. [Google Scholar] [CrossRef]
- Fuentealba, L.C.; Eivers, E.; Geissert, D.; Taelman, V.; De Robertis, E.M. Asymmetric mitosis: Unequal segregation of proteins destined for degradation. Proc. Natl. Acad. Sci. USA 2008, 105, 7732–7737. [Google Scholar] [CrossRef]
- Fuentealba, L.C.; Eivers, E.; Ikeda, A.; Hurtado, C.; Kuroda, H.; Pera, E.M.; De Robertis, E.M. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 2007, 131, 980–993. [Google Scholar] [CrossRef]
- Macara, I.G.; Mili, S. Polarity and differential inheritance--universal attributes of life? Cell 2008, 135, 801–812. [Google Scholar] [CrossRef]
- Dionne, L.K.; Wang, X.J.; Prekeris, R. Midbody: From cellular junk to regulator of cell polarity and cell fate. Curr. Opin. Cell Biol. 2015, 35, 51–58. [Google Scholar] [CrossRef]
- Schweisguth, F. Regulation of notch signaling activity. Curr. Biol. 2004, 14, R129–R138. [Google Scholar] [CrossRef] [PubMed]
- Le Borgne, R.; Bardin, A.; Schweisguth, F. The roles of receptor and ligand endocytosis in regulating Notch signaling. Development 2005, 132, 1751–1762. [Google Scholar] [CrossRef]
- Kawaguchi, D.; Furutachi, S.; Kawai, H.; Hozumi, K.; Gotoh, Y. Dll1 maintains quiescence of adult neural stem cells and segregates asymmetrically during mitosis. Nat. Commun. 2013, 4, 1880. [Google Scholar] [CrossRef]
- Shen, Q.; Zhong, W.; Jan, Y.N.; Temple, S. Asymmetric Numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development 2002, 129, 4843–4853. [Google Scholar]
- Li, P.; Yang, X.; Wasser, M.; Cai, Y.; Chia, W. Inscuteable and Staufen mediate asymmetric localization and segregation of prospero RNA during Drosophila neuroblast cell divisions. Cell 1997, 90, 437–447. [Google Scholar] [CrossRef]
- Broadus, J.; Fuerstenberg, S.; Doe, C.Q. Staufen-dependent localization of prospero mRNA contributes to neuroblast daughter-cell fate. Nature 1998, 391, 792–795. [Google Scholar] [CrossRef]
- Schuldt, A.J.; Adams, J.H.; Davidson, C.M.; Micklem, D.R.; Haseloff, J.; St Johnston, D.; Brand, A.H. Miranda mediates asymmetric protein and RNA localization in the developing nervous system. Genes Dev. 1998, 12, 1847–1857. [Google Scholar] [CrossRef] [PubMed]
- Kusek, G.; Campbell, M.; Doyle, F.; Tenenbaum, S.A.; Kiebler, M.; Temple, S. Asymmetric segregation of the double-stranded RNA binding protein Staufen2 during mammalian neural stem cell divisions promotes lineage progression. Cell Stem Cell 2012, 11, 505–516. [Google Scholar] [CrossRef]
- Neumuller, R.A.; Knoblich, J.A. Dividing cellular asymmetry: Asymmetric cell division and its implications for stem cells and cancer. Genes Dev. 2009, 23, 2675–2699. [Google Scholar] [CrossRef] [PubMed]
- Habib, S.J.; Chen, B.C.; Tsai, F.C.; Anastassiadis, K.; Meyer, T.; Betzig, E.; Nusse, R. A localized Wnt signal orients asymmetric stem cell division in vitro. Science 2013, 339, 1445–1448. [Google Scholar] [CrossRef]
- Biddle, A.; Gammon, L.; Liang, X.; Costea, D.E.; Mackenzie, I.C. Phenotypic Plasticity Determines Cancer Stem Cell Therapeutic Resistance in Oral Squamous Cell Carcinoma. EBioMedicine 2016, 4, 138–145. [Google Scholar] [CrossRef]
- Mathis, R.A.; Sokol, E.S.; Gupta, P.B. Cancer cells exhibit clonal diversity in phenotypic plasticity. Open Biol. 2017, 7. [Google Scholar] [CrossRef]
- Besse, F.; Lopez de Quinto, S.; Marchand, V.; Trucco, A.; Ephrussi, A. Drosophila PTB promotes formation of high-order RNP particles and represses oskar translation. Genes Dev. 2009, 23, 195–207. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef]
- Kussell, E.; Kishony, R.; Balaban, N.Q.; Leibler, S. Bacterial persistence: A model of survival in changing environments. Genetics 2005, 169, 1807–1814. [Google Scholar] [CrossRef]
- Flusberg, D.A.; Roux, J.; Spencer, S.L.; Sorger, P.K. Cells surviving fractional killing by TRAIL exhibit transient but sustainable resistance and inflammatory phenotypes. Mol. Biol. Cell 2013, 24, 2186–2200. [Google Scholar] [CrossRef] [PubMed]
- Pavet, V.; Shlyakhtina, Y.; He, T.; Ceschin, D.G.; Kohonen, P.; Perala, M.; Kallioniemi, O.; Gronemeyer, H. Plasminogen activator urokinase expression reveals TRAIL responsiveness and supports fractional survival of cancer cells. Cell Death Dis. 2014, 5, e1043. [Google Scholar] [CrossRef]
- Shlyakhtina, Y.; Pavet, V.; Gronemeyer, H. Dual role of DR5 in death and survival signaling leads to TRAIL resistance in cancer cells. Cell Death Dis 2017, 8, e3025. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Kurata, T.; Tamura, K.; Kaneda, H.; Nogami, T.; Uejima, H.; Asai Go, G.; Nakagawa, K.; Fukuoka, M. Effect of re-treatment with gefitinib (‘Iressa’, ZD1839) after acquisition of resistance. Ann. Oncol. 2004, 15, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Nakataki, E.; Ohtsuka, S.; Inayama, M.; Tomimoto, H.; Edakuni, N.; Kakiuchi, S.; Nishikubo, N.; Muguruma, H.; Sone, S. Retreatment of lung adenocarcinoma patients with gefitinib who had experienced favorable results from their initial treatment with this selective epidermal growth factor receptor inhibitor: A report of three cases. Oncol. Res. 2005, 15, 107–111. [Google Scholar] [CrossRef]
- Cara, S.; Tannock, I.F. Retreatment of patients with the same chemotherapy: Implications for clinical mechanisms of drug resistance. Ann. Oncol. 2001, 12, 23–27. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Nature of Cell Fate Determinant | ID | Function | Reference |
|---|---|---|---|
| mRNA | ASH1 | Asymmetrically segregates into daughter cell and prevents mating type switching in budding yeast | [114,115,116,117,118] |
| Gurken (grk), Bicoid (bcd) | Asymmetrically localizes to the anterior pole of oocytes and early embryos. Essential for axis formation and primary patterning in Drosophila and C. elegans | [119,123,126,128] | |
| Oskar (osk), Nanos (nos) | Asymmetrically localizes to the posterior pole of oocytes and early embryos. Essential for axis formation and primary patterning in Drosophila | [112,120,124,125,127] | |
| BNI1 | Potential crucial role in P-granules formation through phase separation. Participates in the establishment of polarity | [156] | |
| Prospero | Asymmetrically inherited during stem cell differentiation | [171,172] | |
| Glh-1, rde-4 | Contributes to germ cell proliferation and fertility in C. elegans | [135] | |
| Non-coding RNA | 21U-RNA | Co-localizes with the P-granules that are asymmetrically segregated into the germ line of C. elegans. They may promote specific silencing programs | [140,141,142,143,144,145,146,147] |
| 26G-RNA | Co-localizes with the P-granules at late pachytene stage of male gametogenesis in C. elegans. They may promote specific silencing programs | [148] | |
| Proteins | Numb, Prospero, Brat | Asymmetrically inherited during stem cell differentiation | [29] |
| Par-3, Par-6, aPKC, Inscuteable, Pins, GαI, Mud | Asymmetrically inherited during stem cell differentiation | [4] | |
| DEPS-1, GLH-1, PGL-1 | Participate in oocyte and sperm production at restrictive temperatures | [134,135,136] | |
| PRG1 | Participates in temperature-dependent germline processes, such as fertility in C. elegans | [137,140,141,142,143,144] | |
| ALG-3, ALG-4 | Required for the localization of 26G-RNAs to P-granules at late pachytene stage of male gametogenesis | [148] | |
| H3 histone | Asymmetrically inherited during male germline stem cell divisions in Drosophila | [81,82] | |
| Prions | [PSI1+] | Increased nonsense suppression | [34] |
| DNA | Extra-chromosomal DNA circles | Yeast ageing | [48,49] |
| Micronuclei | Potential mutagenesis pathway that results in pathological states (e.g., cancer) | [43,44,45] | |
| Organelles | Mitochondria | Yeast ageing Asymmetrically inherited during stem cell differentiation | [8,87,88] |
| Peroxisomes | Asymmetric segregation in mammalian epidermal stem cells required for differentiation of daughters. Also contribute to cellular ageing in yeast | [91,92] | |
| Centrosomes | Asymmetric inheritance in stem cells. Also required for asymmetric segregation of protein and RNA molecules | [96,97,98,113,160,163,164] | |
| Endoplasmic reticulum | Segregated to mother cell upon stress in yeast to promote survival. Asymmetric inheritance during Drosophila embryogenesis required for proper neuroblast division. | [99,103] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shlyakhtina, Y.; Moran, K.L.; Portal, M.M. Asymmetric Inheritance of Cell Fate Determinants: Focus on RNA. Non-Coding RNA 2019, 5, 38. https://doi.org/10.3390/ncrna5020038
Shlyakhtina Y, Moran KL, Portal MM. Asymmetric Inheritance of Cell Fate Determinants: Focus on RNA. Non-Coding RNA. 2019; 5(2):38. https://doi.org/10.3390/ncrna5020038
Chicago/Turabian StyleShlyakhtina, Yelyzaveta, Katherine L. Moran, and Maximiliano M. Portal. 2019. "Asymmetric Inheritance of Cell Fate Determinants: Focus on RNA" Non-Coding RNA 5, no. 2: 38. https://doi.org/10.3390/ncrna5020038
APA StyleShlyakhtina, Y., Moran, K. L., & Portal, M. M. (2019). Asymmetric Inheritance of Cell Fate Determinants: Focus on RNA. Non-Coding RNA, 5(2), 38. https://doi.org/10.3390/ncrna5020038