Abstract
Background/Objectives: IgG4-related disease (IgG4-RD) is a multi-organ disease with greatly varying therapeutic approaches and a lack of specific treatment algorithms. This systematic review aimed to determine the therapeutic approaches for pediatric IgG4-RD in real-word practice. Methods: We searched PubMed and Google Scholar for articles on pediatric IgG4-RD cases published in English from 2012 to August 2024, focusing on treatments and outcomes. Study type, treatment(s), dose/regimen, age and sex, organ(s) involved, and treatment outcomes were manually extracted from each study. Results: Of the 219 studies identified, we analyzed 81 studies, including 114 pediatric IgG4-RD cases. Fifty-seven percent of patients suffered from multi-organ disease and required several treatment schemes. Around 75% received steroids, alone or in combination, regardless of the organ affected. The treatment outcomes were positive in most cases, although relapses occurred in approximately 30% of patients, usually upon steroid tapering. Other common therapeutic approaches included immunosuppressants, often used as steroid-sparing agents, with azathioprine and mycophenolate mofetil being the most common; surgery for localized disease; and biologics, mainly rituximab, used in more severe/refractory cases. Uncommon but effective therapies included adalimumab and ruxolitinib. Drug combinations seemed to be more efficacious than monotherapies across studies. Patients > 10 years old more frequently received aggressive approaches (surgery and rituximab) and more often experienced relapses. Relapse rates were higher among females. Conclusions: This review highlights the use of systemic steroids as an effective first-line treatment for pediatric IgG4-RD, but also underscores the use of non-steroid-based alternatives in combination with steroids or other immunosuppressants for the effective management of IgG4-RD.
1. Introduction
IgG4-related disease (IgG4-RD) is a relatively recent clinical entity that has emerged as a significant consideration in the field of pediatric medicine. IgG4-RD is characterized by unique histopathological features and commonly by multi-organ involvement, presenting a constellation of challenges, both diagnostic and therapeutic, particularly in pediatric patients. This immune-mediated condition, known for its fibro-inflammatory manifestations, remains an enigma due to its clinical overlap with various other autoimmune and inflammatory diseases. Since its first recognition as a distinct clinical entity in 2001 [1], international teams have made extensive efforts to align regarding its disease recognition, pathology, and patient management. Thus, the first consensus statement on the pathology of IgG4-RD was published in 2012, while an International Consensus Guidance statement on the treatment of IgG4-RD was issued in 2015 [2,3,4]. In 2019, the American College of Rheumatology and European League Against Rheumatism issued classification criteria for IgG4-RD in an effort to improve its disease recognition [5].
Pediatric cases of IgG4-RD are not only less common, but also display clinical presentations that differ markedly from those in adults [6]. This variance in symptomatology and disease progression underscores the importance of a focused inquiry into this unique cohort. Children are not merely small adults; their physiological and immunological systems are in a state of development that influences disease expression and response to treatment. Furthermore, the impact of IgG4-RD and its management on the growth and development of children presents an additional layer of complexity, warranting a thorough investigation.
According to the International Consensus Guidance, all patients with symptomatic, active IgG4-RD should be treated, with some requiring urgent and aggressive intervention to control this highly active disease in specific organs and avoid irreversible damage [4]. Such aggressive therapy may include combinations of corticosteroids at high doses, the use of mechanical devices (e.g., stents and catheters), or the use of biologics [4]. Untreated IgG4-RD may indeed cause irreversible organ damage and can be detrimental for patients [7]. However, evidence to guide the management of IgG4-RD is limited due to the rarity of the disease, especially among children. Systemic steroids serve as the primary treatment option in both adults and children [4], with most experts suggesting maintenance of the initial dosage for at least 2–4 weeks before gradual tapering and eventual discontinuation within 3–6 months of initiation. Importantly, 24–54% of patients relapse after steroid discontinuation and need to be retreated with steroids [8]. In these cases, steroid-sparing agents (SSAs) are the preferred option to avoid long-term steroid exposure. Among SSAs, rituximab, a monoclonal antibody targeting CD20, has shown great promise as a treatment strategy for IgG4-RD [9].
Despite this consensus guidance, treatment schemes and doses vary greatly in real-world clinical practice depending on geographical, epidemiological, and clinical factors, and especially on organ involvement. Although there is an abundance of data from adult patients, pediatric cases are scarce, and treatment approaches have not been captured in real-world scenarios. In this paper, we systematically review the treatment protocols used in pediatric IgG4-RD cases, aiming to uncover treatment patterns according to organ involvement and patients’ age and sex, as well as to highlight the most effective treatments by capturing treatment outcomes. By synthesizing the available data, this review offers a clearer understanding of IgG4-RD in the pediatric context, highlighting current treatment approaches and suggesting directions for future research, thus aiming to enhance the understanding of the management of IgG4-RD in children.
2. Materials and Methods
2.1. Search Strategy
The study design, protocol, and methods were based on the Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) statement and registered in the Open Science Framework (OSF) database with the following Digital Object Identifier (DOI): https://doi.org/10.17605/OSF.IO/SY54A. The following search string was used to retrieve relevant articles published in PubMed and Google Scholar: ((IgG4-related disease) OR (IgG4-RD) OR (IgG4-related sclerosing disease) OR (immunoglobulin G4 related disease)) AND ((pediatric) OR (children) OR (juvenile) OR (adolescent) OR (childhood) OR (infant)) AND ((treatment) OR (corticosteroid) OR (cyclophosphamide) OR (tacrolimus) OR (cyclosporine) OR (methotrexate) OR (azathioprine) OR (prednisone) OR (prednisolone) OR (tocilizumab) OR (rituximab) OR (steroid) OR (belimumab) OR (mycophenolate) OR (surgery) OR (plasmapheresis) OR (outcomes) OR (outcome measures)). The first consensus statement regarding the definition of the IgG4-RD entity and its management was published in 2012. Therefore, we limited our search to articles published from 2012 onwards (search date: 22 August 2024).
The inclusion criteria were studies in English involving pediatric patients (<18 years old) with a probable or definite IgG4-RD diagnosis, in which the regimen used to treat the patients (including drug therapy or surgical intervention) was detailed. The exclusion criteria were randomized placebo-controlled studies, case–control studies, and studies in which the selection criteria for patients focused on a specific treatment, as our goal was to capture the real-word treatment regimens used in pediatric patients with IgG4-RD.
2.2. Data Collection and Handling
The following information was extracted from each study: type of study, number of patients, patients’ age and sex, treatment used, treatment dose and regimen, organ involvement, and treatment outcomes. These data were extracted manually from each study. If the age of patients was reported in months, it was converted to years by dividing by 12.
The mean age of patients and the distribution of patients according to sex were calculated using Microsoft Excel. This was also used to calculate the number of patients for each treatment, the number of patients with a particular organ involvement, and the number of patients with a positive or negative treatment outcome. Diagrams were generated using Microsoft Excel. A pictorial representation of the key study findings was created using Microsoft PowerPoint.
A review management tool was used to assess eligible studies, and a prototype data extraction form was also created to categorize all extracted data. The selection process and data extraction process were performed independently by two investigators (E.S. and L.F.; screening and inclusion based on eligibility criteria). All disagreements were resolved by A.G.-T.
2.3. Risk of Bias Assessment
Since most of the selected studies were case reports or case series, we employed the modified Newcastle–Ottawa Scale to rate the risk of bias for each study. Accordingly, the studies received 0–4 points for the selection of cases, which included 1 point for the representativeness of the cases, i.e., clearly defined cases and cases typical of IgG4-RD; 1 point for the selection of controls, if applicable; 1 point for ascertainment of exposure, i.e., well-documented treatment or risk factors; and 1 point if the outcome of interest was not present at the start of the study. Further, the studies received 0–2 points for comparability, i.e., if they controlled for the major confounding factor (1 point) or additional factors (1 point). The outcome assessment was rated with 0–3 points, 1 for a clear definition and measurement of the outcome, 1 for a sufficient follow-up time to capture the outcome (e.g., >12 months), and 1 if more than 80% of the patients were followed up or if an explanation was provided for patients lost to follow-up.
3. Results
In total, 219 articles were identified via a PubMed and Google Scholar search. After removing 41 duplicates, 178 articles were screened further for relevance via titles and abstracts. Sixty-eight articles were excluded because they did not fulfill one of the inclusion criteria, and ten articles were excluded because we could not retrieve the full text. Thirty-one reviews, including systematic reviews, were also screened to retrieve additional articles, but were otherwise excluded from the final list of analyzed articles. Eighteen articles were found based on this screening, one of which was a review and one was a duplicate, so both were excluded. Thus, 85 relevant studies were included in our systematic review. Of these, four more studies were excluded as they described the same patient, as deducted by the list of authors, title, patient’s age, clinical characteristics, treatments, and treatment outcomes. Finally, the information from 81 studies was extracted and analyzed for treatment patterns. The study flow chart is shown in Figure 1. The vast majority of the included analyzed studies were case reports.

Figure 1.
Study selection flow chart.
3.1. Patients
With this systematic literature review, we identified 114 pediatric cases of IgG-RD. The patients’ age ranged from 15 months to 17.5 years (mean 11.5 years; median 12 years), and there were 40 patients younger than 10 years. There was an equal representation of girls and boys in the included studies (58 girls, 55 boys, and 1 not reported). IgG4-RD was commonly diagnosed on the basis of clinical symptoms, imaging, and biopsy results, as well as according to histological evidence of IgG4-positive plasma cell infiltration.
3.2. Organ Manifestation
The cases described in this study showed a spectrum of different organ manifestations. The lymph nodes were the most common organ involved, in 34 of 114 patients (29.8%), often in association with other organs, most commonly including the liver in 10, eye/orbit in 7, lung in 6, and pancreas in 4 patients.
The orbit, including the lacrimal glands, was the next most frequently involved site in the collected cases, observed in 33 of 114 patients (28.9%). Patients with orbital involvement often presented with proptosis, lacrimal gland enlargement, and eyelid swelling, as well as orbital masses. Some also experienced vision changes. Notably, 31 of 33 patients with orbital involvement were girls.
The biliary system, including the liver, was involved in 22 of 114 patients (19.3%), often manifesting as sclerosing cholangitis, hepatosplenomegaly, or intrahepatic duct strictures. The pancreas was involved in 15 of 114 patients (13.2%), typically presenting as autoimmune pancreatitis or a pancreatic mass. In total, 10 of the patients with pancreatic involvement also had biliary system involvement (liver n = 9, bile ducts n = 6, and liver + bile ducts n = 3), while 6 patients also had lymphadenopathy.
The lungs were involved in 18 of 114 patients (15.8%). These patients presented with pleural effusion, inflammatory pseudotumor, mediastinal masses, or bronchiectasis, and often experienced dyspnea, recurrent infections, or hemoptysis.
The CNS, gastrointestinal tract, and kidneys were affected in 10 patients each.
IgG4-RD involved a single organ in 49 patients (43%), while the majority of patients (n = 65, 57%) suffered from multi-organ disease (≥2 organs involved) (Figure 2), in accordance with the multi-system involvement observed in IgG4-RD.
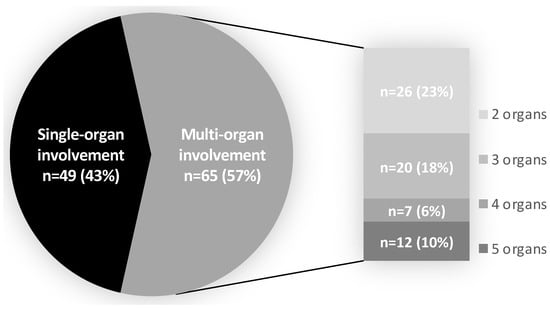
Figure 2.
Organ involvement. Multi-organ involvement was considered if 2 or more organs were involved. The number of patients and percentage having 2, 3, 4, or 5 organs involved are shown on the right of the pie graph.
3.3. Therapeutic Approaches
The treatments used for IgG4-RD varied greatly, reflecting the heterogeneity of organ involvement and disease severity. The most commonly used therapeutic approaches were steroids, immunosuppressants, surgery, and biologics (Figure 3). In several cases, these modalities were used in combination, either simultaneously or sequentially, based on the patient’s response. Figure 4 shows a more detailed diagram of treatment frequencies.
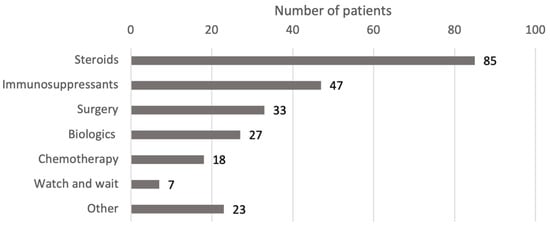
Figure 3.
Treatment modalities by drug class. Steroids include prednisone, prednisolone, methylprednisolone, corticosteroids, steroids, deflazacort, betamethasone, and budesonide. Surgery includes excisions, debulking, resections, ectomies, stent, transplant, and the Whipple procedure. Biologics include rituximab, adalimumab, immunoglobulins, and infliximab. Chemotherapy includes methotrexate, cyclophosphamide, and 6-mercaptopurine. Immunosuppressants include azathioprine, MMF, sirolimus, and cyclosporine. Immunomodulators include tacrolimus, IFNb-1b, and teriflunomide. Antibiotics include ceftriaxone, cefixime, azithromycin, and trimethoprim/sulfamethoxazole. Abbreviations: IFN, interferon; MMF, mycophenolate mofetil.
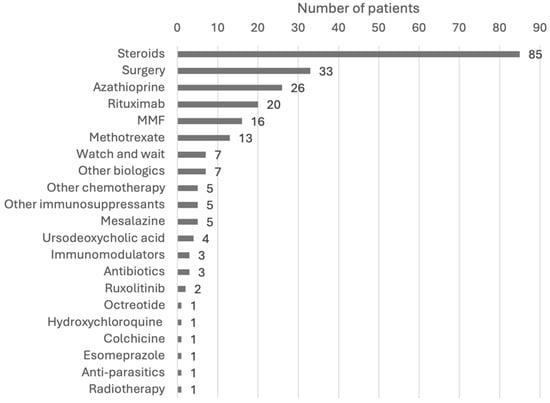
Figure 4.
Breakdown of treatment modalities. Steroids include prednisone, prednisolone, methylprednisolone, corticosteroids, steroids, deflazacort, betamethasone, and budesonide. Surgery includes excisions, debulking, resections, ectomies, stent, transplant, and the Whipple procedure. Other biologics include adalimumab, immunoglobulins, and infliximab. Other chemotherapy includes cyclophosphamide, and 6-mercaptopurine. Other immunosuppressants include sirolimus and cyclosporine. Immunomodulators include tacrolimus, IFNb-1b, and teriflunomide. Antibiotics include ceftriaxone, cefixime, azithromycin, and trimethoprim/sulfamethoxazole. Abbreviations: IFN, interferon; MMF, mycophenolate mofetil.
3.3.1. Steroids
Steroids were the cornerstone of treatment, used in 85 of 115 patients (73.9%), either as a monotherapy or in combination, and usually as a first-line therapy (Table 1). Prednisolone or prednisone was the most commonly administered steroid (in 63 of 85 patients). In the studies that mentioned the steroid dose, these doses ranged from 0.5 to 2 mg/kg/day, with tapering protocols varying between 4 weeks and 6 months. In most cases, steroid therapy resulted in symptom relief and disease control; however, relapses were reported in almost one-third of cases (23 of 85, 27.1%), often upon steroid tapering/discontinuation. In these cases, retreatment with steroids or the use of immunosuppressants was chosen. High-dose steroids such as pulse intravenous methylprednisolone (IVMP) were mostly used following non-response/partial response to a previous treatment or following a relapse. Of six cases with orbital involvement treated with pulse IVMP, the treatment was effective in four cases, leading to minimal clinical improvement in one case [10], and its discontinuation was followed by a relapse in three cases [11,12,13]. No clinical improvement with IVMP was seen in one case of multi-organ disease (five organs involved; [14]), although other cases with multi-organ involvement were more responsive to IVMP.

Table 1.
Identified pediatric cases of IgG4-related disease.
3.3.2. Immunosuppressants
Immunosuppressants were used in 47 of 114 patients (41.2%).
Azathioprine was the most common immunosuppressant used, administered to 26 patients (22.8%) as an SSA. It was used at doses ranging from 0.5 to 2.5 mg/kg/day and commonly in combination with steroids (22 of 26 patients). This combination was effective in maintaining remission and reducing steroid dependency or in achieving response after a relapse. In some patients, however, the combination or azathioprine monotherapy was not effective [89] or induced an initial response, followed by relapse [40,61,64,77].
Mycophenolate mofetil (MMF), the second most common immunosuppressant used, was administered to 16 patients (14.0%). MMF was often employed after initial response to steroids, particularly in cases of orbital involvement. The combination of MMF and steroids provided effective disease control in patients with orbital pseudotumor, proptosis, and lacrimal gland involvement, with minimal relapses [26,48,56,73,75,80,84]. No response to MMF was noted in two patients, one with lymph node and eye involvement [23] and one with orbital involvement [17], both of whom were also not responsive to prednisone and methotrexate. A poor response was noted in a patient with biceps muscle, lymph node, liver, and spleen involvement who had relapsed upon prednisolone tapering [68]. In these two patients, remission was achieved with rituximab.
Other less frequently used immunosuppressants included sirolimus, effective for IgG4-related lymphadenopathy in one patient [55] and for systemic IgG4-RD in combination with rituximab in another patient [53], and cyclosporine, administered alone or in combination with steroids in three patients but showing efficacy only in one with IgG4-related uveitis [64]. In the other two patients, no response and/or relapse was noted [23,73].
3.3.3. Surgical Intervention
Surgery was performed in 33 (28.9%) patients. Surgery was primarily utilized for localized disease, such as orbital pseudotumors, glandular involvement, or soft-tissue masses, or for diagnostic purposes. Surgical intervention often led to complete remission without further pharmacologic intervention. Stent placement, bone marrow transplant, and the Whipple procedure were applied to one patient each.
3.3.4. Biologics
Biologics were administered to 27 patients (23.7%), with rituximab being the most common biologic used (20 patients, 10 females), especially in those with recurrent or resistant IgG4-RD [10,14,23,39,49,57,68]. Most patients treated with rituximab had multi-organ involvement, while all 10 girls who received this therapy had orbital involvement. Rituximab was used as the only therapy in one patient with bilateral orbit and lung involvement with a marked improvement in symptoms after 9 months of follow-up [21]. In some patients, it was used in combination with steroids and immunosuppressants. Most patients who received rituximab achieved complete remission or stable disease, except one patient, who showed resistance to treatment [12], and two patients who showed only partial response [49,89]. One of these latter two patients relapsed after the partial response and was retreated and maintained with rituximab. Similarly, another patient relapsed after the first course of rituximab treatment, but showed the complete resolution of symptoms after a second course, this time in combination with steroids [39]. Relapse on rituximab also occurred in one patient with pericardium and lung involvement who showed an initial improvement in symptoms with rituximab, but relapsed and died 6 months later [77].
Other biologics administered included adalimumab, infliximab, and immunoglobulins. Adalimumab, given to three patients (two girls), led to a complete and rapid resolution of symptoms in all [44,45,61], and remission was maintained for 1 year post-treatment [45,61]. One of these patients, a 16-year-old girl diagnosed with IgG4-sclerosing disease, had received infliximab prior to adalimumab with no response [61]. In a 9-year-old girl with isolated orbit involvement, adalimumab was given off-label at 40 mg twice weekly subcutaneously, leading to a complete response [45].
3.3.5. Chemotherapy
Of the analyzed cases, 18 (15.8%) were treated with chemotherapy agents, with methotrexate being the most frequent one (in 13 patients, 7 girls). All patients who received methotrexate had also received corticosteroids previously or were started on methotrexate while on steroid treatment. Cyclophosphamide was given to four patients (all girls with eye/orbit involvement) in combination with steroids, leading to a partial or complete response, as well as the maintenance of stable disease. Six-mercaptopurine was administered to an 11-year-old female with autoimmune pancreatitis type 1, leading to symptom control and no relapse for up to 5 years of follow-up [36].
3.3.6. Other Treatments
Other treatments used alone, in combination with, or following steroids included mesalazine (n = 5); ursodeoxycholic acid (UDCA; n = 4); the JAK inhibitor ruxolitinib (n = 2); different immunomodulators such as tacrolimus, IFNb-1b, and teriflunomide (n = 1 each); octreotide (n = 1); and radiotherapy (n = 1).
Patients who received mesalazine suffered from ulcerative colitis or inflammatory bowel disease, in agreement with the indications approved for this drug. These patients had multi-organ IgG4-RD (three or more organs involved), involving the pancreas in most cases. Mesalazine was used as a maintenance therapy in three of the patients and as a first-line therapy in two. It led to a complete resolution of symptoms in one patient, in whom it was administered together with IVMP [15].
All four patients that received UDCA suffered from sclerosing cholangitis and had from two to four more organs involved other than the liver. When used as a monotherapy, UDCA induced no or only partial remission [15,20], whereas a complete response or significant improvement was noted when it was given in combination with steroids [20,43]. The patient who showed significant improvement with UDCA had been initially treated with prednisolone, and UDCA was added one month later together with mesalazine and azathioprine [43], indicating that this multi-drug approach may be more efficient than the monotherapy options.
Tacrolimus and IFNb-1b, administered in combination with other regimens, led to clinical improvement or stable disease, whereas teriflunomide was not effective in managing lymph node enlargement in a patient with multi-organ IgG-RD [73].
Two patients were treated with the JAK inhibitor ruxolitinib, which was effective in managing localized disease [53]. Octreotide, a somatostatin analog, was used in a 16-year-old boy with pleural effusion, a mediastinal mass, and mesenteric lymph node involvement. The patient had shown an initial response to prednisone, but relapsed after tapering. Octreotide was administered following a lack of response to prednisone plus azathioprine treatment; however, it also failed to reduce the pleural effusion and mediastinal mass. Surgical intervention was finally needed for this patient, leading to improvement in the pleural effusion [40].
Radiotherapy was applied in a 9.3-year-old girl with orbital swelling and proptosis and fifth cranial nerve involvement. Two courses of radiotherapy were needed in this case for achieving remission after no response to steroids, rituximab, and azathioprine [12].
3.3.7. Watch-and-Wait Strategy
Among the identified cases, there were seven patients that did not receive any treatment for their IgG4-RD. Instead, the selected approach was to watch and wait for the symptoms to resolve. Four of these patients had lymph node involvement only [55,58], one had orbit involvement only [12], one had hepatosplenomegaly in addition to his lymphadenopathy [58], and one patient had kidney and salivary grand involvement in addition to his lymphadenopathy [18]. The latter patient was subjected to renal biopsy, which confirmed the diagnosis of IgG4-RD. Seven months after diagnosis, PET/CT showed the worsening of kidney uptake, which would mandate some treatment, although this was not mentioned by the study authors [18]. For the five patients with lymphadenopathy, the watch-and-wait strategy proved beneficial, as there was no clinical progression noted during follow-up (0.2 up to 8 years). For the patient with orbital involvement, despite an initial spontaneous regression of symptoms, a relapse occurred 38 months after diagnosis, which was treated with pulse methylprednisolone, followed by prednisolone for 1.5 months [12].
3.4. Age and Sex
Since most studies analyzed were cases reports or case series, there was no control for confounders such as age or sex. Keeping this in mind, our analysis showed potential trends in treatment approaches and outcomes. For example, more aggressive approaches such as surgery and rituximab therapy were used more frequently in older children (>10 years) than in younger ones (≤10 years). Specifically, 16 and 10 patients aged more than 10 years were subjected to surgery and received rituximab, respectively, versus only 11 and 4 patients aged 10 years or less, respectively. Such aggressive approaches seemed to be justified, as older children were also more likely to experience a relapse (n = 19 aged >10 years versus n = 11 aged ≤10 years). The relapse rates were also higher among females (n = 18) than among males (n = 12), although it is not clear if this difference is clinically meaningful, as sex has not been reported as a predictor of relapse in IgG4-RD [90,91]
4. Discussion
The primary outcome of this systematic review was to uncover the treatment approaches undertaken in real-world clinical practice for pediatric IgG4-RD. By synthesizing data from 81 studies encompassing 114 pediatric patients, this review provides critical insights into the management of this rare and challenging condition and highlights significant variability in its clinical presentation, organ involvement, and therapeutic strategies. The key findings of this systematic review are summarized in Figure 5.
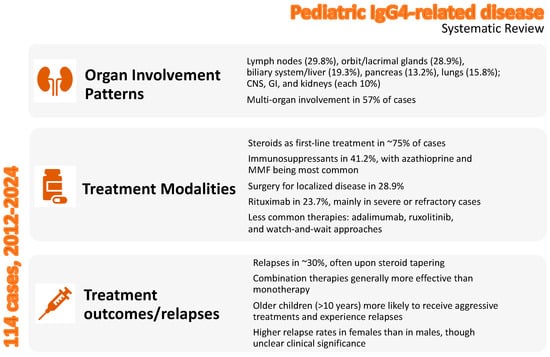
Figure 5.
Overview of the key findings of the systematic review in pediatric IgG4-related disease.
IgG4-RD is a rare fibroinflammatory condition generally occurring in middle-aged patients, and is even more uncommon in children. Our study confirms the rarity of IgG4-RD in the pediatric population, as we identified only 114 cases occurring between 2012 and 2024, in a span of 12 years. In their systematic review, Karim et al. [92] identified 25 pediatric patients with IgG4-RD over a 5-year span (2010–2015). Considering the 3-year overlap between ours and their systematic review, some of the identified cases overlap. In a descriptive review, Hara et al. [6] reported 135 IgG4-RD cases, many of which also overlap with the cases identified in our study. The authors identified more cases than we did, because they included patients up to the age of 25 years. Considering these two reviews and our findings, less than 150 pediatric cases have been reported since 2010. Nevertheless, considering the diagnostic challenges of this condition, pediatricians should remain alert so as to promptly recognize the disease and provide effective treatments for patients.
IgG4-RD is thought to be more prevalent in men than in women [93]. Among the limited number (N = 25) of pediatric patients reported by Karim et al., sex prevalence followed the opposite trend, with girls being more affected than boys. In contrast, among the 114 identified pediatric IgG4-RD cases in our study, no such trend was found, and both sexes were equally represented, arguing against a sex preference of the disease. The same conclusion was reached by Hara et al. [6]. Further, we found that the pediatric cases demonstrated distinct patterns of organ involvement compared to those in adults. The lymph nodes, orbit, and biliary system were the top three affected sites in our cohort. In an adult cohort of 82 patients, the most frequently affected organs were the lymph nodes (50.6%), pancreas (38.7%), and salivary glands (35.6%) [94]. In a much larger adult cohort, pancreatic, hepatic, and biliary involvement were the most prevalent (in 31%) [95]. These distinct patterns underscore the importance of tailoring diagnostic and therapeutic strategies to pediatric patients, as their disease manifestations and responses to treatment may differ substantially.
In our study, we did not collect information on disease severity from the included studies, because most of them lacked objective measures of disease activity such as the IgG4-RD responder index (RI). Our analysis showed 57% of pediatric patients suffering from multi-organ disease, with 16% having 4–5 organs involved. Serum IgG4 levels, IgG4-RD RI scores, the number of organs involved, and eosinophil count have been reported to reflect IgG4-RD disease activity [91]. However, disease severity may not be uniform across all organs, and some organs may even be asymptomatic [96]. Since disease severity across the involved organs is expected to influence treatment strategies, future studies are warranted to address this point.
Regarding therapeutic approaches, our findings are consistent with the existing literature, which identifies systemic corticosteroids as the cornerstone of IgG4-RD treatment, administered in almost 75% of the analyzed cases. A previous systematic review including 62 studies, encompassing more than 3000 adult patients, also reported glucocorticoid-based regimens in 75% [97]. The use of prednisolone and prednisone as a first-line therapy in most of the analyzed cases of our study led to high rates of symptom relief and disease control. The used doses and tapering protocols are consistent with those reported in the literature for adults, as well as with the 2015 International Consensus Guidance statement for IgG4-RD management [4].
Relapses were commonly seen with steroid monotherapy upon tapering or discontinuation, and still occurred even with the addition of immunosuppressive agents like azathioprine, indicating that some patients may require prolonged or more aggressive therapy. The observed relapse rate of 27.1% after steroid treatment is within the range of that reported in adults [8]. These results reinforce the need for adjunctive or maintenance therapies to mitigate steroid dependency. A high rate of multi-organ involvement across the included cases might explain the observed relapse rate, in line with previous studies [91].
Most included studies did not report side effects with the use of steroids. However, the potential adverse effects associated with long-term steroid use in children, including swelling, weight gain, hair growth, acne, growth delay, and Cushingoid features, should not be overlooked. Among the analyzed cases in our review, there was one case who developed Cushing syndrome after long-term steroid therapy [37]. The risk of side effects depends on the dose used (low dose < 10 mg/day of prednisone, medium dose 10–20 mg/day, and high dose >20 mg/day), type of steroid (long-acting vs short-acting), length of treatment (long-term treatment > 3 months), and other medical problems [98]. Although information on the used steroid doses and duration was not included in several studies, in those that provided this information, the treatment duration ranged from weeks to up to 2 years, while the doses ranged from 0.5 to 2 mg/kg/day, corresponding to low-to-medium dose levels. This might explain the lack of reported side effects. Only two studies reported a rather high dose (up to 25 mg/day [23] and 50 mg/day [30]) used for a very long period (2 years [23] and 9 months [30]), but reported no side effects. Our review suggests that, in real-world practice, physicians seriously consider the long-term effects of steroids in children and try to maintain low doses when treatment is required for longer periods or try to shorten treatment duration when high doses are needed for disease control. The use of SSAs and biologics in the included cases might also have been helpful in mitigating the risk of corticosteroid-associated side effects, as well as aiding in improving disease management.
SSAs such as azathioprine and MMF were the two most commonly used immunosuppressants in the analyzed cohort and were generally effective in maintaining remission or reducing steroid dependence, in agreement with efficacy study results in adult patients [97,99]. However, cases of non-responsiveness or relapse suggest the potential insufficiency of immunosuppressive therapy, underscoring the need for further studies to identify predictive markers of response to these agents. Similarly, rituximab, the most frequently used biologic, demonstrated significant efficacy in patients with multi-organ disease or steroid-refractory cases. Cases where rituximab was used in combination with steroids showed less frequent relapses, suggesting that it may be more effective in preventing recurrence when used in combination with steroids or other immunosuppressants. In agreement, rituximab has been shown to increase remission and reduce relapse rates in IgG4-RD [100] and has been associated with successful outcomes in various organ systems, showing efficacy in cases where glucocorticoids and other immunosuppressive agents fail [101]. Nevertheless, partial responses, relapses, and resistance (in one case) were observed in our analysis even with rituximab, suggesting the potential role of combination therapy, individualized dosing regimens, or repeat therapy protocols to achieve better outcomes.
Our analysis indicated some organ-specific responses to treatment, suggesting that individualized regimens should be tailored to the affected sites. For example, the combination of steroids and MMF was highly effective in managing orbital involvement, including pseudotumors and proptosis, while rituximab was effective for extensive multi-organ disease. Multimodal approaches such as surgery and biologics were required in cases with biliary system and pancreas involvement. Other therapies such as tacrolimus and teriflunomide showed a limited effectiveness in certain organ systems, suggesting that these agents may have a niche role, requiring further evaluation in targeted studies. In contrast, ruxolitinib was effective in localized cases, in agreement with the potential benefit of JAK inhibitors in IgG4-RD [102].
4.1. Limitations
The study results should be interpreted considering certain limitations. First, the vast majority of studies included in this systematic review were case reports or case series, which typically focus on specific, well-defined patient cases and individual patient outcomes and, hence, do not provide broader population-level data, which limits the generalizability of such findings. Moreover, case reports inherently lack a comparison between treatment groups or a control group and, thus, cannot adequately control for confounding factors. Hence, we cannot exclude the possibility of selection bias, and it is difficult to draw definitive conclusions about treatment efficacy beyond individual cases. Second, while the included studies tracked treatment outcomes, many only focused on the short-term efficacy and did not address long-term efficacy, which further limits the conclusions that can be drawn. Thus, although the treatment outcomes were generally positive, the findings must be interpreted with caution due to the limitations of the study designs. Finally, it is possible that some of the reported cases were mimickers of IgG4-RD and not true IgG4-RD, since we relied on the diagnosis made by the authors of each study, which was not necessarily based on the currently accepted diagnostic criteria for the disease. However, even if some cases were not true IgG4-RD, this argues even more for the rare manifestation of the disease in pediatrics. Despite the above limitations, our study captured real-world practices in IgG4-RD management, without attempting to generate any statistical results other than population frequencies.
4.2. Conclusions and Future Perspectives
The findings of this systematic review underscore the importance of a multidisciplinary approach in managing pediatric IgG4-RD. While systemic steroids remain the first-line treatment, prolonged use can lead to significant side effects, especially in children (e.g., growth delay and bone thinning). Therefore, future research should focus on developing strategies to reduce steroid dependence. Immunosuppressive drugs like azathioprine, MMF, and methotrexate are already used in some cases. Investigating their efficacy in combination with or as alternatives to steroids in children will be important for reducing side effects. Further, adjunctive therapies should be considered early in patients at high risk of relapse or those with severe multi-organ involvement. The emergence of biologics like rituximab offers a promising alternative, particularly for refractory cases, but long-term safety and efficacy data are needed. Other biologics such as infliximab and adalimumab, both of which target tumor necrosis factor-alpha, have been used in refractory cases of autoimmune diseases. Research is needed to assess their effectiveness in pediatric IgG4-RD, particularly in cases with high disease activity or organ involvement. Moreover, the potential role of emerging therapies, such as JAK inhibitors (ruxolitinib) and targeted biologics, should be explored in larger pediatric cohorts. Ruxolitinib has shown potential in managing inflammatory conditions and, thus, investigating its role in pediatric IgG4-RD could provide an option for steroid-sparing therapy. Importantly, our review highlights the need to develop strategies to prevent relapses, including understanding the role of maintenance therapies and developing biomarkers or advanced imaging techniques. for relapse prediction, which could allow for timely interventions, thus reducing long-term damage. Considering the heterogeneity of IgG4-RD, personalized treatment plans based on the child’s specific disease manifestation will be crucial. Research into identifying genetic or immunological markers that predict response to therapy could help clinicians to choose the most effective treatment. The development of pediatric-specific treatment guidelines is critical to standardize care and improve outcomes. Global research collaborations across centers and countries will help to accumulate more data on the efficacy of various treatments and enable the development of evidence-based guidelines for treating pediatric IgG4-RD. This systematic review provides a basis towards this direction, additionally to informing physicians of current practices in pediatric IgG4-RD.
Author Contributions
Conceptualization, E.S. and E.P.K.; methodology, E.S., V.-R.T. and L.F. (Liana Fidani); software, E.S., L.F. (Lampros Fotis) and L.F. (Liana Fidani); validation, E.S.; formal analysis, E.S. and E.P.K.; investigation, E.S. and V.-R.T.; resources, E.S. and L.F. (Lampros Fotis); data extraction and curation, E.S. and L.F. (Lampros Fotis); writing—original draft preparation, E.S., E.P.K., V.-R.T., L.F. (Lampros Fotis), L.F. (Liana Fidani) and A.G.-T.; writing—review and editing, E.S., E.P.K., V.-R.T., L.F. (Lampros Fotis), L.F. (Liana Fidani) and A.G.-T.; visualization, E.S.; supervision, A.G.-T.; project administration, E.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hamano, H.; Kawa, S.; Horiuchi, A.; Unno, H.; Furuya, N.; Akamatsu, T.; Fukushima, M.; Nikaido, T.; Nakayama, K.; Usuda, N.; et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N. Engl. J. Med. 2001, 344, 732–738. [Google Scholar] [CrossRef]
- Deshpande, V.; Zen, Y.; Chan, J.K.; Yi, E.E.; Sato, Y.; Yoshino, T.; Klöppel, G.; Heathcote, J.G.; Khosroshahi, A.; Ferry, J.A.; et al. Consensus statement on the pathology of IgG4-related disease. Mod. Pathol. 2012, 25, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Wallace, Z.; Khosroshahi, A.; Carruthers, M.D.; Perugino, C.A.; Choi, H.; Campochiaro, C.; Culver, E.L.; Cortazar, F.; Della-torre, E.; Ebbo, M.; et al. An international multispecialty validation study of the IgG4-related disease responder index. Arthritis Care Res. 2018, 70, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Khosroshahi, A.; Wallace, Z.; Crowe, J.; Akamizu, T.; Azumi, A.; Carruthers, M.N.; Chari, S.T.; Della-Torre, E.; Frulloni, L.; Goto, H.; et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. 2015, 67, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Wallace, Z.S.; Naden, R.P.; Chari, S.; Choi, H.; Della-Torre, E.; Dicaire, J.; Hart, P.A.; Inoue, D.; Kawano, M.; Khosroshahi, A.; et al. The 2019 American College of Rheumatology/European League against Rheumatism classification criteria for IgG4-related disease. Arthritis Rheumatol. 2019, 72, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Hara, S.; Yoshida, M.; Sanada, H.; Suzuki, Y.; Sato, Y.; Mizushima, I.; Kawano, M. Pediatric IgG4-related disease: A descriptive review. Expert. Rev. Clin. Immunol. 2024, 20, 97–119. [Google Scholar] [CrossRef] [PubMed]
- Floreani, A.; Okazaki, K.; Uchida, K.; Gershwin, M.E. IgG4-related disease: Changing epidemiology and new thoughts on a multisystem disease. J. Transl. Autoimmun. 2020, 4, 100074. [Google Scholar] [CrossRef]
- Zongfei, J.; Lingli, C.; Ying, S.; Lingying, M.; Lijuan, Z.; Dongmei, L.; Xiaomin, D.; Yingyong, H.; Huiyong, C.; Lili, M.; et al. Clinical and pathological predictors of relapse in IgG4-related disease. Arthritis Res. Ther. 2022, 24, 106. [Google Scholar] [CrossRef]
- Patel, U.; Saxena, A.; Patel, D.; Ayesha, I.E.; Monson, N.R.; Klair, N.; Yu, A.K. Therapeutic uses of rituximab and clinical features in immunoglobulin G4-related disease: A systematic review. Cureus 2023, 15, e45044. [Google Scholar] [CrossRef] [PubMed]
- Sane, M.; Chelnis, J.; Kozielski, R.; Fasiuddin, A. Immunoglobulin G4-related sclerosing disease with orbital inflammation in a 12-year-old girl. J. AAPOS 2013, 17, 548–550. [Google Scholar] [CrossRef]
- Jariwala, M.P.; Agarwal, M.; Mulay, K.; Sawhney, S. IgG4-related orbital inflammation presenting as unilateral pseudotumor. Indian. J. Pediatr. 2014, 81, 1108–1110. [Google Scholar] [CrossRef] [PubMed]
- Kaya Akca, Ü.; Atalay, E.; Kasap Cüceoğlu, M.; Şener, S.; Balık, Z.; Başaran, Ö.; Batu, E.D.; Karadağ, Ö.; Özen, S.; Bilginer, Y. IgG4-related disease in pediatric patients: A single-center experience. Rheumatol. Int. 2022, 42, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Mulay, K.; Aggarwal, E.; Jariwala, M.; Honavar, S.G. Orbital immunoglobulin-G4-related disease: Case series and literature review. Clin. Exp. Ophthalmol. 2014, 42, 682–687. [Google Scholar] [CrossRef]
- Demir, A.M.; Aydin, F.; Acar, B.; Kurt, T.; Poyraz, A.; Kiremitci, S.; Gülleroglu, B.; Azili, M.N.; Bayrakci, U.S. IgG4-related disease and ANCA positive vasculitis in childhood: A case-based review. Clin. Rheumatol. 2021, 40, 3817–3825. [Google Scholar] [CrossRef] [PubMed]
- Akkelle, B.Ş.; Tutar, E.; Ergelen, R.; Çelikel, Ç.A.; Ertem, D. IgG4 related disease in a seven-year-old girl with multiple organ involvement: A rare presentation. Turk. Pediatri Ars. 2020, 55, 191–194. [Google Scholar] [CrossRef]
- Basu, S.; Utpat, K.; Joshi, J. 18F-FDG PET/CT imaging features of IgG4-related pulmonary inflammatory pseudotumor at initial diagnosis and during early treatment monitoring. J. Nucl. Med. Technol. 2016, 44, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Batu, E.D.; Arici, Z.S.; Orhan, D.; Kiratli, H.; Özen, S. Immunoglobulin G4-related orbital disease: Report of two pediatric cases. Clin. Exp. Rheumatol. 2015, 33, 409–410. [Google Scholar]
- Bélissant, O., Jr.; Guernou, M.; Rouvier, P.; Compain, C.; Bonardel, G. IgG4-related tubulointerstitial nephritis pattern in 18F-FDG PET/CT. Clin. Nucl. Med. 2015, 40, 808–809. [Google Scholar] [CrossRef]
- Bloria, S.D.; Kataria, K.; Luthra, A.; Bloria, P. Anesthetic considerations in a child with IgG4 disease. MAMC J. Med. Sci. 2018, 4, 142–144. [Google Scholar] [CrossRef]
- Bolia, R.; Chong, S.Y.; Coleman, L.; MacGregor, D.; Hardikar, W.; Oliver, M.R. Autoimmune pancreatitis and IgG4-related disease in three children. ACG Case Rep. J. 2016, 3, e115. [Google Scholar] [CrossRef] [PubMed]
- Bu, F.; Koo, S.C. Clinicopathologic characterization of IgG4-rich pediatric head and neck lesions. Arch. Pathol. Lab. Med. 2022, 146, 611–618. [Google Scholar] [CrossRef]
- Bullock, D.R.; Miller, B.S.; Clark, H.B.; Hobday, P.M. Rituximab treatment for isolated IgG4-related hypophysitis in a teenage female. Endocrinol. Diabetes Metab. Case Rep. 2018, 2018, 18-0135. [Google Scholar] [CrossRef]
- Caso, F.; Fiocco, U.; Costa, L.; Sfriso, P.; Punzi, L.; Doria, A. Successful use of rituximab in a young patient with immunoglobulin G4-related disease and refractory scleritis. Jt. Bone Spine 2014, 81, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Pal, P.; Dutta, S.; Nada, R. IgG4-related disease at rectovesical pouch mimicking inflammatory myofibroblastic tumor. Indian. Pediatr. 2019, 56, 1059–1061. [Google Scholar] [CrossRef]
- Chen, C.; Chen, K.; Huang, X.; Wang, K.; Qian, S. Concurrent eosinophilia and IgG4-related disease in a child: A case report and review of the literature. Exp. Ther. Med. 2018, 15, 2739–2748. [Google Scholar] [CrossRef] [PubMed]
- Cinar, O.K.; Khaosut, P.; Sebire, N.; Eleftheriou, D.; Al-Obaidi, M. P43 Immunoglobulin G4-related disease in a 10 year-old girl with multisystem involvement. Rheumatology 2019, 58 (Suppl. S4), kez416.010. [Google Scholar] [CrossRef]
- Corujeira, S.; Ferraz, C.; Nunes, T.; Fonseca, E.; Vaz, L.G. Severe IgG4-related disease in a young child: A diagnosis challenge. Case Rep. Pediatr. 2015, 2015, 140753. [Google Scholar] [CrossRef]
- Decker, L.; Crawford, A.M.; Lorenzo, G.; Stippler, M.; Konstantinov, K.N.; SantaCruz, K. IgG4-related hypophysitis: Case report and literature review. Cureus 2016, 8, e907. [Google Scholar] [CrossRef]
- Samundeeswari, D.; Warrier, K.; Camina, N.; Eveleigh, C. Orbital mass in a 9-year-old girl. Rheumatology 2018, 57 (Suppl. S8), key273.024. [Google Scholar] [CrossRef][Green Version]
- Díaz-Ramírez, G.S.; Medina-Quintero, L.F.; Salinas-César, A.; Zea-Vera, A.F. Dacryoadenitis associated with IgG4-related disease in an Afro-Colombian adolescent. Reumatol. Clin. (Engl. Ed.) 2018, 14, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Dincă, O.M.; Bucur, A.; Zurac, S.A.; Niţă, T.; Jugulete, G.; Vlădan, G.C.; Pădurariu, L.C. Chronic sclerosing sialadenitis of the bilateral submandibular glands in childhood—A diagnostic dilemma. Rom. J. Morphol. Embryol. 2024, 65, 113–118. [Google Scholar] [CrossRef]
- Dylewska, K.; Kobusińska, K.; Kurylak, A. Tumor of the orbit and pterygopalatine fossa: Delayed recognition of possible IgG4-related disease. Contemp. Oncol. 2020, 24, 136–139. [Google Scholar] [CrossRef]
- Emiroglu, M.; Bozkurt, B.; Emiroglu, H.H. IgG4-related disease and ligneous conjunctivitis in a girl: A coexistence or a relationship? Eur. J. Allergy 2019, 74, 379. [Google Scholar]
- Ewing, D.; Hammer, R. IgG4-related disease simulating Hodgkin lymphoma in a child. Am. J. Clin. Pathol. 2014, 142 (Suppl. S1), A111. [Google Scholar] [CrossRef][Green Version]
- Ferreira da Silva, R.C.; Lieberman, S.M.; Hoffman, H.T.; Policeni, B.; Bashir, A.; Smith, R.J.; Sato, T.S. IgG4-related disease in an adolescent with radiologic-pathologic correlation. Radiol. Case Rep. 2016, 12, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, J.; Quiros, J.A.; Morgan, T.; Zhang, Z.; Tian, W.; Kehr, E.; Shackleton, D.V.; Zigman, A.; Stenzel, P. Diagnosis of autoimmune pancreatitis vs neoplasms in children with pancreatic mass and biliary obstruction. Clin. Gastroenterol. Hepatol. 2012, 10, 1051–1055.e1. [Google Scholar] [CrossRef]
- Gabrovska, N.; Velizarova, S.; Spasova, A.; Kostadinov, D.; Yanev, N.; Shivachev, H.; Rangelov, E.; Pahnev, Y.; Antonova, Z.; Kartulev, N.; et al. A case of tracheal stenosis as an isolated form of immunoproliferative hyper-IgG4 disease in a 17-year-old girl. Children 2021, 8, 589. [Google Scholar] [CrossRef] [PubMed]
- Galloway, D.P.; Wallihan, D.; Smith, M.T.; Abu-El-Haija, M. An unusual presentation of pediatric autoimmune pancreatitis. Pancreas 2016, 45, e1–e2. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gillispie, M.C.; Thomas, R.D.; Hennon, T.R. Successful treatment of IgG-4 related sclerosing disease with rituximab: A novel case report. Clin. Exp. Rheumatol. 2015, 33, 549–550. [Google Scholar]
- Goag, E.K.; Park, J.E.; Lee, E.H.; Park, Y.M.; Kim, C.Y.; Lee, J.M.; Kim, Y.J.; Kim, Y.S.; Kim, S.K.; Chang, J.; et al. A case of extensive IgG4-related disease presenting as massive pleural effusion, mediastinal mass, and mesenteric lymphadenopathy in a 16-year-old male. Tuberc. Respir. Dis. 2015, 78, 396–400. [Google Scholar] [CrossRef]
- Griepentrog, G.J.; Vickers, R.W.; Karesh, J.W.; Azari, A.A.; Albert, D.M.; Bukat, C.N. A clinicopathologic case study of two patients with pediatric orbital IgG4-related disease. Orbit 2013, 32, 389–391. [Google Scholar] [CrossRef]
- Hasosah, M.Y.; Satti, M.B.; Yousef, Y.A.; Alzahrani, D.M.; Almutairi, S.A.; Alsahafi, A.F.; Sukkar, G.A.; Alzaben, A.A. IgG4-related sclerosing mesenteritis in a 7-year-old Saudi girl. Saudi J. Gastroenterol. 2014, 20, 385–388. [Google Scholar] [CrossRef]
- Hsu, C.-T.; Jeng, Y.-M.; Wu, J.-F. Immunoglobulin G4-related sclerosing cholangitis in a 3-year-old boy. Adv. Dig. Med. 2021, 8, 59–63. [Google Scholar] [CrossRef]
- Huelsen, A.; Bailey, W.; Whitehead, M.; Chalmers-Watson, T. Autoimmune pancreatitis and primary sclerosing cholangitis in a 16-year-old boy with inflammatory bowel disease. Clin. J. Gastroenterol. 2012, 5, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Jalaj, S.; Dunbar, K.; Campbell, A.; Kazim, M. Treatment of pediatric IgG4-related orbital disease with TNF-α inhibitor. Ophthalmic Plast. Reconstr. Surg. 2018, 34, e10–e12. [Google Scholar] [CrossRef]
- Johnson, J.S.; Saltzman, A.F.; Treece, A.L.; Cost, N.G. A case of IgG4-related renal pseudotumor in a child with history of Wilms tumor. Urol. Case Rep. 2018, 21, 107–109. [Google Scholar] [CrossRef]
- Jordan, V.A.; Herrera Hernandez, L.P.; Cofer, S.A.; Roby, B.B. Pediatric laryngeal expression and surgical treatment of IgG4-related disease. JAMA Otolaryngol. Head. Neck Surg. 2018, 144, 1183–1184. [Google Scholar] [CrossRef] [PubMed]
- Kalapesi, F.B.; Garrott, H.M.; Moldovan, C.; Williams, M.; Ramanan, A.; Herbert, H.M. IgG4 orbital inflammation in a 5-year-old child presenting as an orbital mass. Orbit 2013, 32, 137–140. [Google Scholar] [CrossRef]
- Karim, A.F.; Bansie, R.D.; Rombach, S.M.; Paridaens, D.; Verdijk, R.M.; van Hagen, P.M.; Van Laar, J.A.M. The treatment outcomes in IgG4-related disease. Neth. J. Med. 2018, 76, 275–285. [Google Scholar]
- Kato, D.; Uchida, H.; Hinoki, A.; Sumida, W.; Shirota, C.; Makita, S.; Okamoto, M.; Takimoto, A.; Takada, S.; Nakagawa, Y. IgG4-related disease of duodenal obstruction due to multiple ulcers in a 12-year-old girl. BMC Pediatr. 2023, 23, 376. [Google Scholar] [CrossRef]
- Kaur, R.; Mitra, S.; Rajwanshi, A.; Das, A.; Nahar Saikia, U.; Dey, P. Fine needle aspiration cytology of IgG4-related disease: A potential diagnostic pitfall? Diagn. Cytopathol. 2016, 45, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Kemp, W.; Majeed, A.; Mitchell, J.; Majumdar, A.; Tse, E.; Skoien, R.; Croagh, D.; Dev, A.; Gao, H.; Weltman, M.; et al. Management, outcomes, and survival of an Australian IgG4-SC cohort: The MOSAIC study. Liver Int. 2021, 41, 2934–2943. [Google Scholar] [CrossRef] [PubMed]
- Kozlova, A.; Burlakov, V.; Abramov, D.; Laberko, A.; Sagoyan, G.; Shcherbina, A. AB1082 Characterisation of a group of patients with Igg4-related disease: Single centre experience. Ann. Rheum. Dis. 2018, 77, 1651. [Google Scholar] [CrossRef]
- La Porta, E.; Pierri, F.; Pisani, I.; Pilato, F.P.; Lanino, E.; Lanino, L.; Sementa, A.R.; Verrina, E.E. IGG4-related disease: Nephropathy and bone marrow failure in a 2-year-old child. Nephrol. Dial. Transplant. 2020, 35 (Suppl. S3), gfaa142.P1832. [Google Scholar] [CrossRef]
- Mainardi, C.; Pizzi, M.; Marzollo, A.; Carraro, E.; Boaro, M.P.; Mussolin, L.; Massano, D.; Tazzoli, S.; Onofrillo, D.; Cesaro, S.; et al. Pediatric IgG4-related lymphadenopathy: A rare condition associated with autoimmunity and lymphoproliferative disorders. Pediatr. Allergy Immunol. 2020, 31, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Manojlović, M.; Lazarević, D.; Ratković Janković, M.; Stanković, S.; Golubović, E.; Vojinović, J. Immunoglobulin G4 (IgG4) related disease and granulomatosis with polyangiitis (Gpa) in childhood: A case report of new overlap syndrome. Acta Medica Median. 2023, 62, 103–107. [Google Scholar] [CrossRef]
- Marissen, J.; Pagel, J.; Steinmetz, A.; Härtel, C.; Lauten, M. IgG4-related disease and successful treatment with rituximab in a three-year-old boy. Ann. Hematol. Oncol. 2021, 8, 1334. [Google Scholar]
- Meli, M.; Arrabito, M.; Salvatorelli, L.; Soma, R.; Presti, S.; Licciardello, M.; Miraglia, V.; Scuderi, M.G.; Belfiore, G.; Magro, G.; et al. Report of two cases of pediatric IgG4-related lymphadenopathy (IgG4-LAD): IgG4-related disease (IgG4-RD) or a distinct clinical pathological entity? Children 2022, 9, 1472. [Google Scholar] [CrossRef]
- Melo, J.C.; Kitsko, D.; Reyes-Múgica, M. Pediatric chronic sclerosing sialadenitis: Küttner tumor. Pediatr. Dev. Pathol. 2012, 15, 165–169. [Google Scholar] [CrossRef]
- Mittal, R.; Ganguly, A.; Rath, S.; Das, B.; Mishra, A. IgG4-related orbital inflammation presenting as bilateral proptosis in a child. Eye 2014, 28, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Naghibi, M.; Ahmed, A.; al Badri, A.M.; Bateman, A.C.; Shepherd, H.A.; Gordon, J.N. The successful treatment of IgG4-positive colitis with adalimumab in a patient with IgG4-related sclerosing disease—A new subtype of aggressive colitis? J. Crohns Colitis 2013, 7, e81–e84. [Google Scholar] [CrossRef]
- Nair, N.; Abraham, S.; Ganesh, S.K. Recurrent posterior scleritis with secondary choroidal osteoma in a child. Indian. J. Ophthalmol. 2020, 68, 2509–2511. [Google Scholar] [CrossRef] [PubMed]
- Nambirajan, A.; Sharma, M.C.; Garg, K.; Sriram, S.; Boorgula, M.T.; Suri, V. Large dural-based mass with bony hyperostosis in a 16-year-old male: IgG4-related disease mimicking lymphoplasmacyte-rich meningioma. Childs Nerv. Syst. 2019, 35, 1423–1427. [Google Scholar] [CrossRef]
- Nastri, M.M.F.; Novak, G.V.; Sallum, A.E.M.; Campos, L.M.A.; Teixeira, R.A.P.; Silva, C.A. Immunoglobulin G4-related disease with recurrent uveitis and kidney tumor mimicking childhood polyarteritis nodosa: A rare case report. Acta Reumatol. Port. 2018, 43, 226–229. [Google Scholar] [PubMed]
- Nayak, H.K.; Bhat, S.J.; Panigrahi, M.K.; Chouhan, I.; Kumar, C.; Samal, S.C. IgG4-related sclerosing cholangitis: A great mimicker. Indian. J. Gastroenterol. 2020, 39, 614–618. [Google Scholar] [CrossRef]
- Nelson, M.B.; Schocken, D.; De Guzman, M. Is pediatric IgG4-related disease distinct from its adult counterpart? A single center case series. In Proceedings of the Pediatric Research Symposium, Baylor College of Medicine—Texas Children’s Hospital, Jan and Dan Duncan Neurological Research Institute, Houston, TX, USA, 24 March 2020. [Google Scholar]
- Orlicka, J.; Turowska-Heydel, D.; Żuber, K.A. IgG4-related disease (IgG4-RD) in developmental age—Diagnostic and therapeutic problems. Forum Reumatol. 2019, 5, 149–153. [Google Scholar] [CrossRef]
- Özdel, S.; Ekim, M.; Kaygusuz, G.; Çelikel, E.; Vatansever, G.; Taçyıldız, N. A new location for pediatric immunoglobulin G4-related disease: The biceps muscle. Turk. J. Pediatr. 2020, 62, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Chatterjee, D.; Singla, R.; Jain, N.; Bhansali, A.; Dutta, P. Co-occurrence of craniopharyngioma and IgG4-related hypophysitis: An epiphenomenon or a mere coincidence? World Neurosur. 2020, 136, 193–197. [Google Scholar] [CrossRef]
- Pasic, S.; Ristic, G.; Djuricic, S. PReS-FINAL-2276: IgG4 related disease in a 10-year-old girl. Pediatr. Rheumatol. Online J. 2013, 11 (Suppl. S2), P266. [Google Scholar] [CrossRef]
- Pifferi, M.; Di Cicco, M.; Bush, A.; Caramella, D.; Chilosi, M.; Boner, A.L. Uncommon pulmonary presentation of IgG4-related disease in a 15-year-old boy. Chest 2013, 144, 669–671. [Google Scholar] [CrossRef]
- Prabhu, S.M.; Yadav, V.; Irodi, A.; Mani, S.; Varghese, A.M. IgG4-related disease with sinonasal involvement: A case series. Indian J. Radiol. Imaging 2014, 24, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Qing, P.; Lu, C.; Yan, B.; Liu, C.; Fox, D.A.; Zhao, Y.; Liu, Y.; Tan, C. Case report: IgG4-related intracranial lesions mimicking multiple sclerosis in a 14-year-old girl. Front. Neurol. 2022, 13, 1007153. [Google Scholar] [CrossRef] [PubMed]
- Rakesh, Κ.; Rawat, A.; Gupta, A.; Guleria, S.; Nameirakpam, J.; Suri, D.; Gupta, A.; Nada, R.; Singh, S. SAT0504 IgG4 related disease in children: A single centre experience from North-West India. Ann. Rheum. Dis. 2019, 78 (Suppl. S2), 1341. [Google Scholar] [CrossRef]
- Rojas-Ramirez, O.; Nunez-Velazquez, M.; Acosta-Jimenez, E.; Vargas-Caro, A. IgG4-related ophthalmic disease in children: A case report. Ann. Allergy Asthma Immunol. 2016, 117 (Suppl. S5), S6. [Google Scholar] [CrossRef]
- Rosen, D.; Thung, S.; Sheflin-Findling, S.; Lai, J.; Rosen, A.; Arnon, R.; Chu, J. IgG4-sclerosing cholangitis in a pediatric patient. Semin. Liver Dis. 2015, 35, 89–94. [Google Scholar] [CrossRef]
- Saad, M.A.; Ahmed, H.; Elgohary, R.; El Gendy, H.I. IgG4-related pericardium and lung disease in pediatric patient complicated with fatal massive hemoptysis: A case report and review of literature. Pediatr. Rheumatol. Online J. 2023, 21, 16. [Google Scholar] [CrossRef] [PubMed]
- Salama, O.H.; Ibrahim, E.N.A.; Hussein, M.O.; Alkady, A.M.M.; Abd El-Salam, M.E.; Ghanem, S. IgG4-related dacryoadenitis in Egyptian patients: A retrospective study. Clin. Ophthalmol. 2022, 16, 2765–2773. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, A.; Kindley, K.J.; Noland, M.M.; Gru, A.A. IgG4-related skin disease presenting as a pseudolymphoma in a white adolescent girl. Am. J. Dermatopathol. 2019, 41, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Sozeri, B.; Kalin, S.; Cakan, M. Pachymeningitis in a pediatric case of IgG4-related disease successfully treated with mycophenolate mofetil. North. Clin. Istanb. 2024, 11, 359–360. [Google Scholar] [CrossRef] [PubMed]
- Szczawinska-Poplonyk, A.; Wojsyk-Banaszak, I.; Jonczyk-Potoczna, K.; Breborowicz, A. Pulmonary manifestation of immunoglobulin G4-related disease in a 7-year-old immunodeficient boy with Epstein-Barr virus infection: A case report. Ital. J. Pediatr. 2016, 42, 58. [Google Scholar] [CrossRef]
- Tille, L.; Schnabel, A.; Laass, M.W.; Hahn, G.; Taut, H.; Leszczynska, A.; Pablik, J.; Berner, R.; Brück, N.; Hedrich, C.M. Orbital inflammation and colitis in pediatric IgG4-related disease: A case report and review of the literature. Eur. J. Rheumatol. 2020, 7 (Suppl. S1), S21–S27. [Google Scholar] [CrossRef]
- Timeus, F.; Calvo, M.M.; Caci, A.M.; Gallone, G.O.; Vittone, F. IgG4-related chronic sclerosing sialadenitis in a child with recurrent parotitis: A case report. BMC Pediatr. 2021, 21, 586. [Google Scholar] [CrossRef]
- Tong, J.Y.; Leahy, K.E.; Wong, M.; Krivanek, M.; Tumuluri, K. IgG4-related disease of the orbit in an infant. J. AAPOS 2021, 25, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Vakrakou, A.G.; Evangelopoulos, M.E.; Boutzios, G.; Tzanetakos, D.; Tzartos, J.; Velonakis, G.; Toulas, P.; Anagnostouli, M.; Andreadou, E.; Koutsis, G.; et al. Recurrent myelitis and asymptomatic hypophysitis in IgG4-related disease: Case-based review. Rheumatol. Int. 2020, 40, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, A.K.; Kumar, G.A.; Rajesh, S.; Ahamed, M.Z. IgG4-related coronary aneurysm in a child. Indian J. Pediatr. 2021, 88, 593. [Google Scholar] [CrossRef]
- Wan, Q.; Xu, Z.; Liu, X.; Wu, Z.; Zhong, Q.; Wu, C. A case report of IgG4-related hepatic inflammatory pseudotumor in a 3-year-old boy. Front. Immunol. 2024, 15, 1376276. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.C.; Tien, P.T.; Li, Y.H.; Chen, R.Y.; Cho, D.Y. IgG4-related cerebral pseudotumor with perineural spreading along branches of the trigeminal nerves causing compressive optic neuropathy: A case report. Medicine 2017, 96, e8709. [Google Scholar] [CrossRef]
- Karim, A.F.; Eurelings, L.E.M.; Bansie, R.D.; van Hagen, P.M.; van Laar, J.A.M.; Dik, W.A. Soluble interleukin-2 receptor: A potential marker for monitoring disease activity in IgG4-related disease. Mediat. Inflamm. 2018, 2018, 6103064. [Google Scholar] [CrossRef]
- Wallace, Z.S.; Mattoo, H.; Mahajan, V.S.; Kulikova, M.; Lu, L.; Deshpande, V.; Choi, H.K.; Pillai, S.; Stone, J.H. Predictors of disease relapse in IgG4-related disease following rituximab. Rheumatology 2016, 55, 1000–1008. [Google Scholar] [CrossRef]
- Peng, Y.; Li, J.Q.; Zhang, P.P.; Zhang, X.; Peng, L.Y.; Chen, H.; Zhou, J.X.; Zhang, S.Z.; Yang, H.X.; Liu, J.J.; et al. Clinical outcomes and predictive relapse factors of IgG4-related disease following treatment: A long-term cohort study. J. Intern. Med. 2019, 286, 542–552. [Google Scholar] [CrossRef]
- Karim, F.; Loeffen, J.; Bramer, W.; Westenberg, L.; Verdijk, R.; van Hagen, M.; van Laar, J. IgG4-related disease: A systematic review of this unrecognized disease in pediatrics. Pediatr. Rheumatol. Online J. 2016, 14, 18. [Google Scholar] [CrossRef]
- Kamisawa, T.; Zen, Y.; Pillai, S.; Stone, J.H. IgG4-related disease. Lancet 2015, 385, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Zhou, L.; Zheng, L.; Wang, J.; Zhai, Y.; Zhou, X.; Zhao, P. Clinical patterns and risk factors for multiorgan involvement in IgG4-related disease patients. Heliyon 2023, 10, e23433. [Google Scholar] [CrossRef]
- Wallace, Z.S.; Zhang, Y.; Perugino, C.A.; Naden, R.; Choi, H.K.; Stone, J.H.; ACR/EULAR IgG4-RD Classification Criteria Committee. Clinical phenotypes of IgG4-related disease: An analysis of two international cross-sectional cohorts. Ann. Rheum. Dis. 2019, 78, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.; Khosroshahi, A. Diagnostic and treatment workup for IgG4-related disease. Expert. Rev. Clin. Immunol. 2017, 13, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Brito-Zerón, P.; Kostov, B.; Bosch, X.; Acar-Denizli, N.; Ramos-Casals, M.; Stone, J.H. Therapeutic approach to IgG4-related disease: A systematic review. Medicine 2016, 95, e4002. [Google Scholar] [CrossRef]
- Deshmukh, C.T. Minimizing side effects of systemic corticosteroids in children. Indian. J. Dermatol. Venereol. Leprol. 2007, 73, 218–221. [Google Scholar] [CrossRef]
- Yunyun, F.; Yu, P.; Panpan, Z.; Xia, Z.; Linyi, P.; Jiaxin, Z.; Li, Z.; Shangzhu, Z.; Jinjing, L.; Di, W.; et al. Efficacy and safety of low-dose mycophenolate mofetil treatment for immunoglobulin G4-related disease: A randomized clinical trial. Rheumatology 2019, 58, 52–60. [Google Scholar] [CrossRef]
- Omar, D.; Chen, Y.; Cong, Y.; Dong, L. Glucocorticoids and steroid-sparing medications monotherapies or in combination for IgG4-RD: A systematic review and network meta-analysis. Rheumatology 2020, 59, 718–726. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, P.; Wang, M.; Feng, R.; Lai, Y.; Peng, L.; Fei, Y.; Zhang, X.; Zhao, Y.; Zeng, X.; et al. Failure of remission induction by glucocorticoids alone or in combination with immunosuppressive agents in IgG4-related disease: A prospective study of 215 patients. Arthritis Res. Ther. 2018, 20, 65. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Gordins, P.; Durairaj, S. JAK inhibition as a therapeutic strategy for IgG4-RD. J. Investig. Allergol. Clin. Immunol. 2021, 31, 280–281. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).