
Hypothetical Mechanism of Exercise-Induced Acute Kidney Injury Associated with Renal Hypouricemia


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. EIAKI Due to Increased Urate Excretion after Exhaustive Exercise
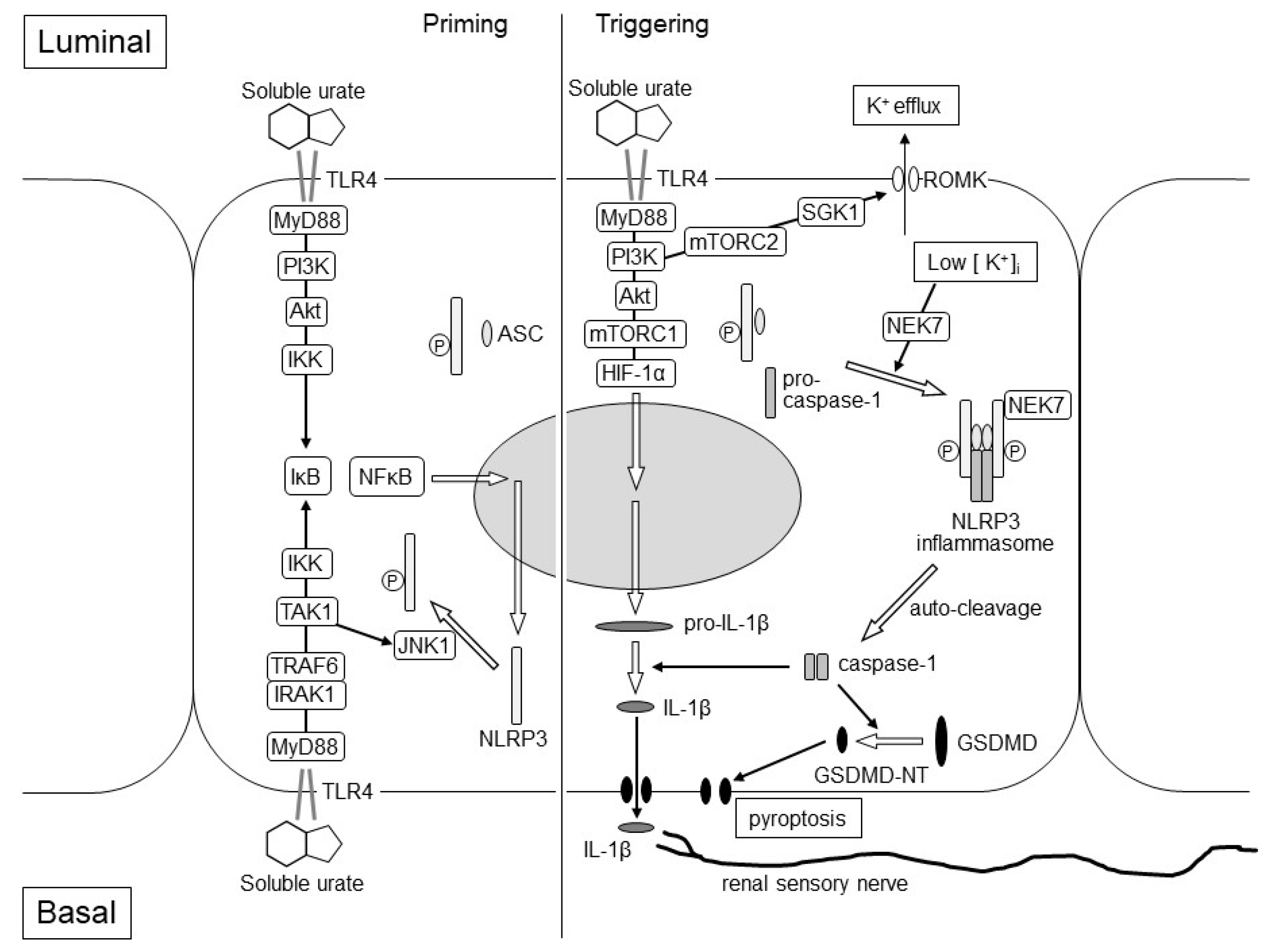
3. TLR4 in the Kidney
4. IL-1β Production and Acute Kidney Injury
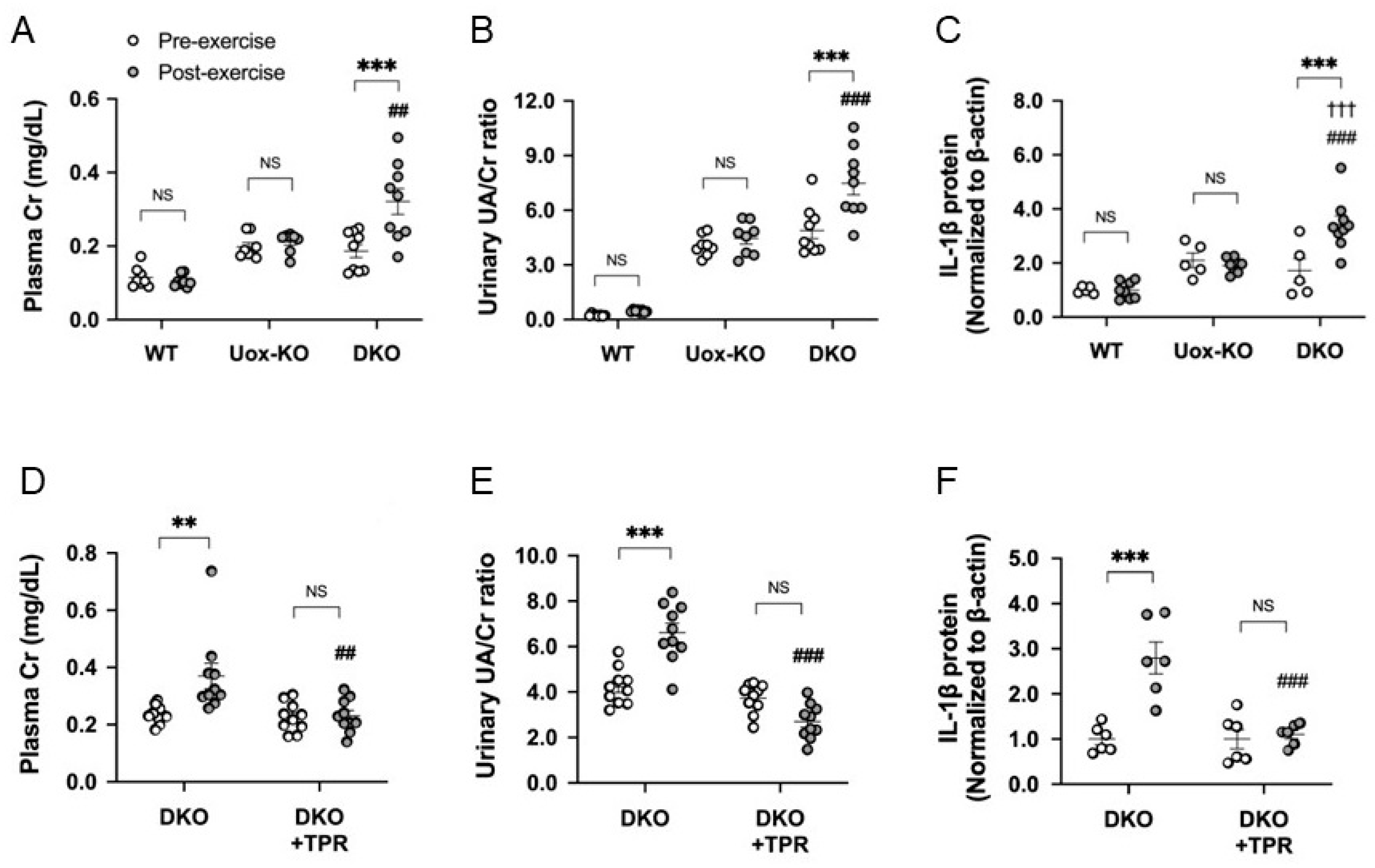
5. Increased Urinary Urate Excretion Due to Exercise
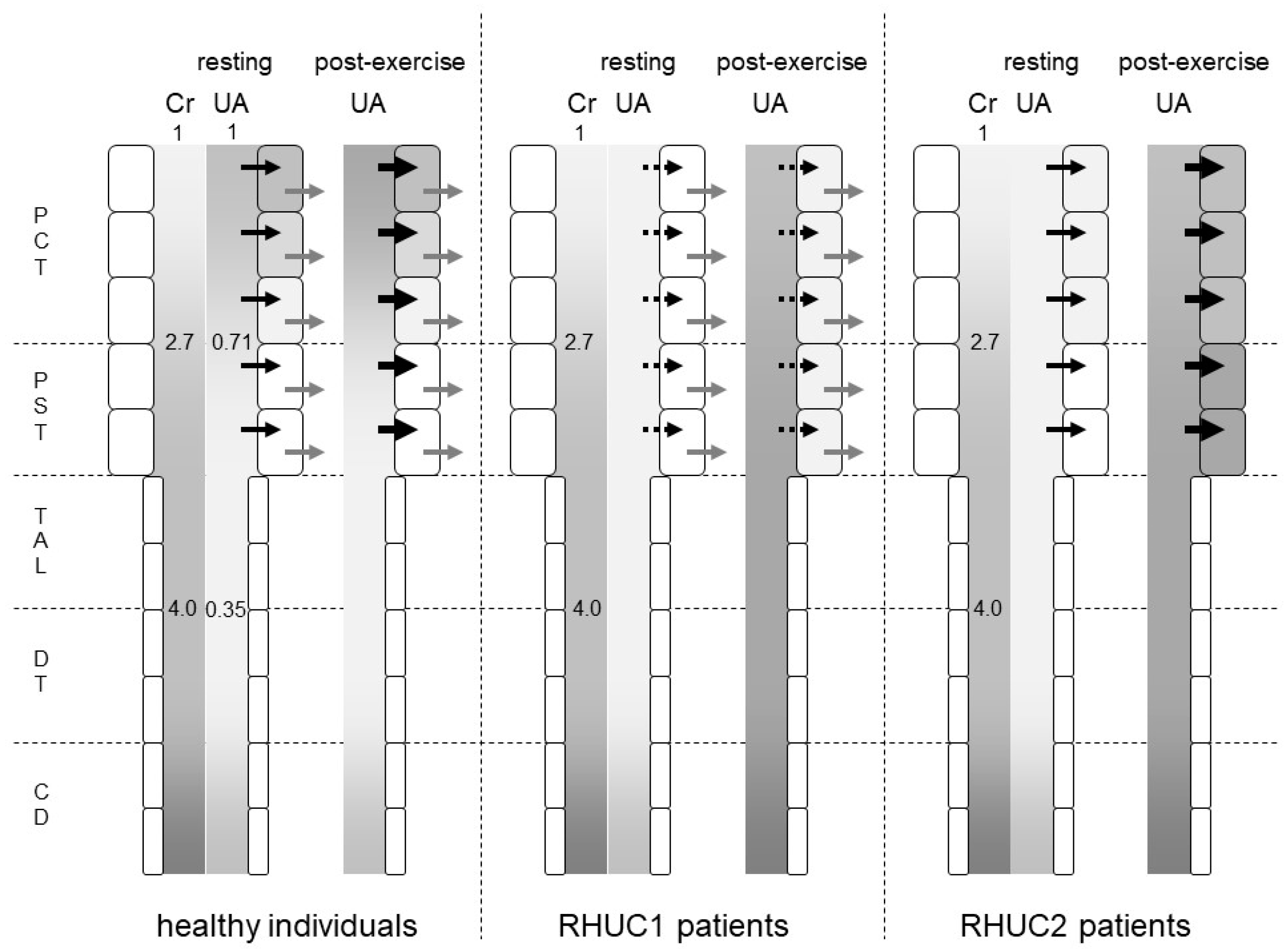
6. Post-Exercise Urate Production in Patients with Renal Hypouricemia
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Nakayama, A.; Matsuo, H.; Ohtahara, A.; Ogino, K.; Hakoda, M.; Hamada, T.; Hosoyamada, M.; Yamaguchi, S.; Hisatome, I.; Ichida, K.; et al. Clinical practice guideline for renal hypouricemia (1st edition). Hum. Cell 2019, 32, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef]
- Matsuo, H.; Chiba, T.; Nagamori, S.; Nakayama, A.; Domoto, H.; Phetdee, K.; Wiriyasermkul, P.; Kikuchi, Y.; Oda, T.; Nishiyama, J.; et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am. J. Hum. Genet. 2008, 83, 744–751. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, I.; Sakurai, Y.; Masuzaki, S.; Sugishita, N.; Shinoda, A.; Shikura, N. Exercise-induced acute renal failure in 3 patients with renal hypouricemia. Nihon Jinzo Gakkai Shi 1990, 32, 923–928. [Google Scholar]
- Erley, C.M.; Hirschberg, R.R.; Hoefer, W.; Schaefer, K. Acute renal failure due to uric acid nephropathy in a patient with renal hypouricemia. Klin. Wochenschr. 1989, 67, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, I. Acute renal failure with severe loin pain and patchy renal ischemia after anaerobic exercise in patients with or without renal hypouricemia. Nephron 2002, 91, 559–570. [Google Scholar] [CrossRef]
- Yeun, J.Y.; Hasbargen, J.A. Renal hypouricemia: Prevention of exercise-induced acute renal failure and a review of the literature. Am. J. Kidney Dis. 1995, 25, 937–946. [Google Scholar] [CrossRef]
- Ichida, K.; Hosoyamada, M.; Kimura, H.; Takeda, M.; Utsunomiya, Y.; Hosoya, T.; Endou, H. Urate transport via human PAH transporter hOAT1 and its gene structure. Kidney Int. 2003, 63, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Bakhiya, A.; Bahn, A.; Burckhardt, G.; Wolff, N. Human organic anion transporter 3 (hOAT3) can operate as an exchanger and mediate secretory urate flux. Cell Physiol. Biochem. 2003, 13, 249–256. [Google Scholar] [CrossRef]
- Hosoya, T.; Uchida, S.; Shibata, S.; Tomioka, N.; Matsumoto, K.; Hosoyamada, M. Xanthine Oxidoreductase Inhibitors Suppress the Onset of Exercise-induced AKI in High HPRT Activity Urat1-Uox Double Knockout Mice. J. Am. Soc. Nephrol. 2021, 32, ASN.2021050616. [Google Scholar] [CrossRef]
- Roch-Ramel, F.; Diezi-Chomety, F.; Roth, L.; Weiner, I.M. A micropuncture study of urate excretion by Cebus monkeys employing high performance liquid chromatography with amperometric detection of urate. Pflug. Arch. 1980, 383, 203–207. [Google Scholar] [CrossRef]
- Xiao, J.; Zhang, X.L.; Fu, C.; Han, R.; Chen, W.; Lu, Y.; Ye, Z. Soluble uric acid increases NALP3 inflammasome and interleukin-1beta expression in human primary renal proximal tubule epithelial cells through the Toll-like receptor 4-mediated pathway. Int. J. Mol. Med. 2015, 35, 1347–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- El-Achkar, T.M.; Huang, X.; Plotkin, Z.; Sandoval, R.M.; Rhodes, G.J.; Dagher, P.C. Sepsis induces changes in the expression and distribution of Toll-like receptor 4 in the rat kidney. Am. J. Physiol. Ren. Physiol. 2006, 290, F1034–F1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Good, D.W.; George, T.; Watts, B.A., 3rd. Lipopolysaccharide directly alters renal tubule transport through distinct TLR4-dependent pathways in basolateral and apical membranes. Am. J. Physiol. Ren. Physiol. 2009, 297, F866–F874. [Google Scholar] [CrossRef] [Green Version]
- El-Achkar, T.M.; Wu, X.R.; Rauchman, M.; McCracken, R.; Kiefer, S.; Dagher, P.C. Tamm-Horsfall protein protects the kidney from ischemic injury by decreasing inflammation and altering TLR4 expression. Am. J. Physiol. Ren. Physiol. 2008, 295, F534–F544. [Google Scholar] [CrossRef]
- Watts, B.A., 3rd; George, T.; Good, D.W. Lumen LPS inhibits HCO3(-) absorption in the medullary thick ascending limb through TLR4-PI3K-Akt-mTOR-dependent inhibition of basolateral Na+/H+ exchange. Am. J. Physiol. Ren. Physiol. 2013, 305, F451–F462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.Y.; Ye, X.J.; He, X.H.; Ouyang, D.Y. The Signaling Pathways Regulating NLRP3 Inflammasome Activation. Inflammation 2021, 44, 1229–1245. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 2017, 68, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Yao, X.; Li, H.; Xue, G.; Guo, Q.; Yang, G.; An, L.; Zhang, Y.; Meng, G. Cutting Edge: TRAF6 Mediates TLR/IL-1R Signaling-Induced Nontranscriptional Priming of the NLRP3 Inflammasome. J. Immunol. 2017, 199, 1561–1566. [Google Scholar] [CrossRef] [Green Version]
- Watts, B.A., 3rd; Tamayo, E.; Sherwood, E.R.; Good, D.W. Monophosphoryl lipid A induces protection against LPS in medullary thick ascending limb through induction of Tollip and negative regulation of IRAK-1. Am. J. Physiol. Ren. Physiol. 2019, 317, F705–F719. [Google Scholar] [CrossRef] [PubMed]
- Grahammer, F.; Nesterov, V.; Ahmed, A.; Steinhardt, F.; Sandner, L.; Arnold, F.; Cordts, T.; Negrea, S.; Bertog, M.; Ruegg, M.A.; et al. mTORC2 critically regulates renal potassium handling. J. Clin. Investig. 2016, 126, 1773–1782. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Y.; Xu, Y. Pyroptosis in Kidney Disease. J. Mol. Biol. 2021, 167290. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Aksoylar, H.I.; Horng, T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin. Immunol. 2015, 27, 286–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stemkowski, P.L.; Bukhanova-Schulz, N.; Baldwin, T.; de Chaves, E.P.; Smith, P.A. Are sensory neurons exquisitely sensitive to interleukin 1beta? J. Neuroimmunol. 2021, 354, 577529. [Google Scholar] [CrossRef] [PubMed]
- Osborn, J.W.; Tyshynsky, R.; Vulchanova, L. Function of Renal Nerves in Kidney Physiology and Pathophysiology. Annu. Rev. Physiol. 2021, 83, 429–450. [Google Scholar] [CrossRef]
- Miller, A.D.; Leslie, R.A. The area postrema and vomiting. Front. Neuroendocrinol. 1994, 15, 301–320. [Google Scholar] [CrossRef]
- Binshtok, A.M.; Wang, H.; Zimmermann, K.; Amaya, F.; Vardeh, D.; Shi, L.; Brenner, G.J.; Ji, R.R.; Bean, B.P.; Woolf, C.J.; et al. Nociceptors are interleukin-1beta sensors. J. Neurosci. 2008, 28, 14062–14073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aomura, D.; Sonoda, K.; Harada, M.; Hashimoto, K.; Kamijo, Y. A Case of Acute Kidney Injury in a Patient with Renal Hypouricemia without Intense Exercise. Case Rep. Nephrol. Dial. 2020, 10, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, M.; Spriet, L.L. Skeletal muscle energy metabolism during exercise. Nat. Metab. 2020, 2, 817–828. [Google Scholar] [CrossRef]
- Hellsten-Westing, Y.; Sollevi, A.; Sjodin, B. Plasma accumulation of hypoxanthine, uric acid and creatine kinase following exhausting runs of differing durations in man. Eur. J. Appl. Physiol. Occup. Physiol. 1991, 62, 380–384. [Google Scholar] [CrossRef]
- Tullson, P.C.; Bangsbo, J.; Hellsten, Y.; Richter, E.A. IMP metabolism in human skeletal muscle after exhaustive exercise. J. Appl. Physiol. 1995, 78, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Hellsten-Westing, Y. Immunohistochemical localization of xanthine oxidase in human cardiac and skeletal muscle. Histochemistry 1993, 100, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Hellsten-Westing, Y.; Kaijser, L.; Ekblom, B.; Sjodin, B. Exchange of purines in human liver and skeletal muscle with short-term exhaustive exercise. Am. J. Physiol. 1994, 266, R81–R86. [Google Scholar] [CrossRef]
- Hellsten, Y.; Nyberg, M.; Jensen, L.G.; Mortensen, S.P. Vasodilator interactions in skeletal muscle blood flow regulation. J. Physiol. 2012, 590, 6297–6305. [Google Scholar] [CrossRef]
- Edwards, G.; Feletou, M.; Weston, A.H. Endothelium-derived hyperpolarising factors and associated pathways: A synopsis. Pflug. Arch. 2010, 459, 863–879. [Google Scholar] [CrossRef]
- Mikami, T.; Kita, K.; Tomita, S.; Qu, G.J.; Tasaki, Y.; Ito, A. Is allantoin in serum and urine a useful indicator of exercise-induced oxidative stress in humans? Free Radic. Res. 2000, 32, 235–244. [Google Scholar] [CrossRef]
- Hellsten, Y.; Tullson, P.C.; Richter, E.A.; Bangsbo, J. Oxidation of urate in human skeletal muscle during exercise. Free Radic. Biol. Med. 1997, 22, 169–174. [Google Scholar] [CrossRef]
- Sugihara, S.; Hisatome, I.; Kuwabara, M.; Niwa, K.; Maharani, N.; Kato, M.; Ogino, K.; Hamada, T.; Ninomiya, H.; Higashi, Y.; et al. Depletion of Uric Acid Due to SLC22A12 (URAT1) Loss-of-Function Mutation Causes Endothelial Dysfunction in Hypouricemia. Circ. J. 2015, 79, 1125–1132. [Google Scholar] [CrossRef] [Green Version]
- De Becker, B.; Coremans, C.; Chaumont, M.; Delporte, C.; Van Antwerpen, P.; Franck, T.; Rousseau, A.; Zouaoui Boudjeltia, K.; Cullus, P.; van de Borne, P. Severe Hypouricemia Impairs Endothelium-Dependent Vasodilatation and Reduces Blood Pressure in Healthy Young Men: A Randomized, Placebo-Controlled, and Crossover Study. J. Am. Heart Assoc. 2019, 8, e013130. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosoyamada, M. Hypothetical Mechanism of Exercise-Induced Acute Kidney Injury Associated with Renal Hypouricemia. Biomedicines 2021, 9, 1847. https://doi.org/10.3390/biomedicines9121847
Hosoyamada M. Hypothetical Mechanism of Exercise-Induced Acute Kidney Injury Associated with Renal Hypouricemia. Biomedicines. 2021; 9(12):1847. https://doi.org/10.3390/biomedicines9121847
Chicago/Turabian StyleHosoyamada, Makoto. 2021. "Hypothetical Mechanism of Exercise-Induced Acute Kidney Injury Associated with Renal Hypouricemia" Biomedicines 9, no. 12: 1847. https://doi.org/10.3390/biomedicines9121847
APA StyleHosoyamada, M. (2021). Hypothetical Mechanism of Exercise-Induced Acute Kidney Injury Associated with Renal Hypouricemia. Biomedicines, 9(12), 1847. https://doi.org/10.3390/biomedicines9121847