The Androgen Receptor in Prostate Cancer: Effect of Structure, Ligands and Spliced Variants on Therapy

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
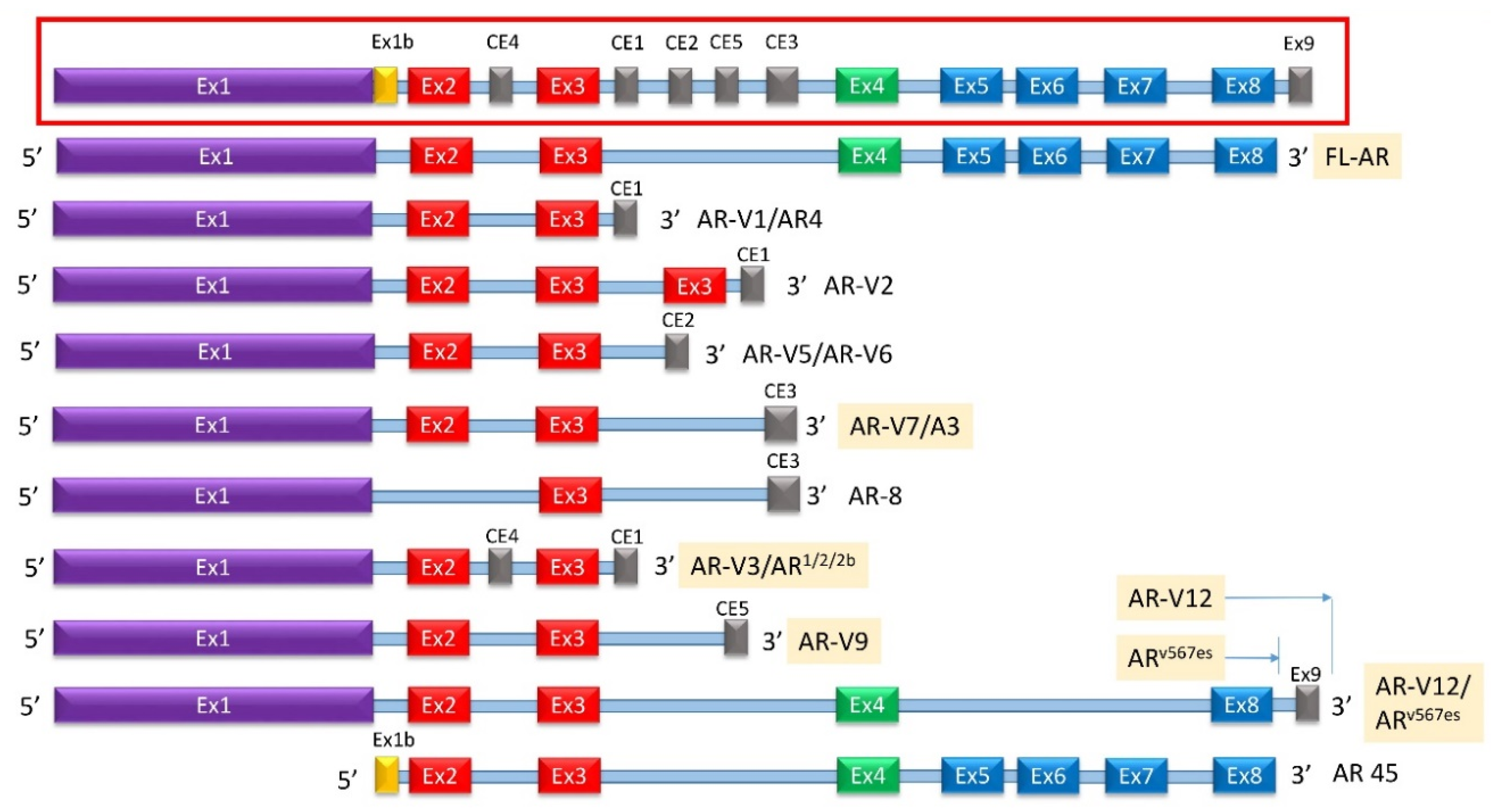{kind=link}
{kind=link}
Abstract
1. Introduction
2. Structure of the Androgen Receptor
3. The Spaciotemporal Regulation of AR Transcriptional Activity
4. AR-Driven Resistance to Current Treatments for Advanced and Metastatic CRPC
5. AR Variants in the Development of Resistance to Current AR Inhibition Strategies in CRPC
6. AR-Targeting Strategies to Overcome ADT and ASI Resistance
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, D.; Zhao, S.; Li, X.; Kirk, J.S.; Tang, D.G. Prostate Luminal Progenitor Cells in Development and Cancer. Trends Cancer 2018, 4, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Packer, J.R.; Maitland, N.J. The molecular and cellular origin of human prostate cancer. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 1238–1260. [Google Scholar] [CrossRef] [PubMed]
- Villers, A.; Steg, A.; Boccon-Gibod, L. Anatomy of the prostate: Review of the different models. Eur. Urol. 1991, 20, 261–268. [Google Scholar] [CrossRef]
- Lee, J.J.; Thomas, I.C.; Nolley, R.; Ferrari, M.; Brooks, J.D.; Leppert, J.T. Biologic differences between peripheral and transition zone prostate cancer. Prostate 2015, 75, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Carter, H.B.; Albertsen, P.C.; Barry, M.J.; Etzioni, R.; Freedland, S.J.; Greene, K.L.; Holmberg, L.; Kantoff, P.; Konety, B.R.; Murad, M.H.; et al. Early detection of prostate cancer: AUA Guideline. J. Urol. 2013, 190, 419–426. [Google Scholar] [CrossRef]
- National Cancer Institute. Available online: www.seer.cancer.gov (accessed on 15 October 2020).
- National Center for Health Statistics. Available online: www.cdc.gov/nchs (accessed on 15 October 2020).
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef]
- Ahmed, A.; Ali, S.; Sarkar, F.H. Advances in androgen receptor targeted therapy for prostate cancer. J. Cell. Physiol. 2014, 229, 271–276. [Google Scholar] [CrossRef]
- Tosoian, J.J.; Carter, H.B.; Lepor, A.; Loeb, S. Active surveillance for prostate cancer: Current evidence and contemporary state of practice. Nat. Rev. Urol. 2016, 13, 205–215. [Google Scholar] [CrossRef]
- Litwin, M.S.; Tan, H.J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Feng, Z.; Trock, B.J.; Humphreys, E.B.; Carducci, M.A.; Partin, A.W.; Walsh, P.C.; Eisenberger, M.A. The natural history of metastatic progression in men with prostate-specific antigen recurrence after radical prostatectomy: Long-term follow-up. BJU Int. 2012, 109, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Brand, L.J.; Dehm, S.M. Androgen Receptor Gene Rearrangements: New Perspectives on Prostate Cancer Progression. Curr. Drug Targets 2013, 14, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef]
- Rottach, A.M.; Ahrend, H.; Martin, B.; Walther, R.; Zimmermann, U.; Burchardt, M.; Stope, M.B. Cabazitaxel inhibits prostate cancer cell growth by inhibition of androgen receptor and heat shock protein expression. World J. Urol. 2019, 37, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Zieren, R.C.; Xue, W.; de Reijke, T.M.; Pienta, K.J. Metastatic prostate cancer remains incurable, why? Asian J. Urol. 2019, 6, 26–41. [Google Scholar] [CrossRef]
- Tucci, M.; Caffo, O.; Buttigliero, C.; Cavaliere, C.; D’Aniello, C.; Di Maio, M.; Kinspergher, S.; Maines, F.; Rizzo, M.; Rossetti, S.; et al. Therapeutic options for first-line metastatic castration-resistant prostate cancer: Suggestions for clinical practise in the CHAARTED and LATITUDE era. Cancer Treat. Rev. 2019, 74, 35–42. [Google Scholar] [CrossRef]
- Tucci, M.; Scagliotti, G.V.; Vignani, F. Metastatic castration-resistant prostate cancer: Time for innovation. Future Oncol. 2015, 11, 91–106. [Google Scholar] [CrossRef]
- Wang, Y.; Kreisberg, J.I.; Ghosh, P.M. Cross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr. Cancer Drug Targets 2007, 7, 591–604. [Google Scholar] [CrossRef]
- Gao, W.; Bohl, C.E.; Dalton, J.T. Chemistry and structural biology of androgen receptor. Chem. Rev. 2005, 105, 3352–3370. [Google Scholar] [CrossRef]
- Hu, R.; Isaacs, W.B.; Luo, J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate 2011, 71, 1656–1667. [Google Scholar] [CrossRef]
- Matsumoto, T.; Sakari, M.; Okada, M.; Yokoyama, A.; Takahashi, S.; Kouzmenko, A.; Kato, S. The androgen receptor in health and disease. Annu. Rev. Physiol. 2013, 75, 201–224. [Google Scholar] [CrossRef]
- Nadal, M.; Prekovic, S.; Gallastegui, N.; Helsen, C.; Abella, M.; Zielinska, K.; Gay, M.; Vilaseca, M.; Taules, M.; Houtsmuller, A.B.; et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat. Commun. 2017, 8, 14388. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, P.L.; Jivan, A.; Dollins, D.E.; Claessens, F.; Gewirth, D.T. Structural basis of androgen receptor binding to selective androgen response elements. Proc. Natl. Acad. Sci. USA 2004, 101, 4758–4763. [Google Scholar] [CrossRef]
- Claessens, F.; Denayer, S.; Van Tilborgh, N.; Kerkhofs, S.; Helsen, C.; Haelens, A. Diverse roles of androgen receptor (AR) domains in AR-mediated signaling. Nucl. Recept. Signal. 2008, 6, e008. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharm. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Kemppainen, J.A.; Wilson, E.M. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J. Biol. Chem. 2000, 275, 22986–22994. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.C.; Dehm, S.M. Constitutive activity of the androgen receptor. Adv. Pharmacol. 2014, 70, 327–366. [Google Scholar] [CrossRef]
- Lallous, N.; Dalal, K.; Cherkasov, A.; Rennie, P.S. Targeting alternative sites on the androgen receptor to treat castration-resistant prostate cancer. Int. J. Mol. Sci. 2013, 14, 12496–12519. [Google Scholar] [CrossRef]
- Marcelli, M. Androgen Receptor in Health and Disease; Hohl, A., Ed.; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Heemers, H.V.; Tindall, D.J. Androgen receptor (AR) coregulators: A diversity of functions converging on and regulating the AR transcriptional complex. Endocr. Rev. 2007, 28, 778–808. [Google Scholar] [CrossRef] [PubMed]
- Jenster, G.; Trapman, J.; Brinkmann, A.O. Nuclear import of the human androgen receptor. Biochem. J. 1993, 293, 761–768. [Google Scholar] [CrossRef]
- Cutress, M.L.; Whitaker, H.C.; Mills, I.G.; Stewart, M.; Neal, D.E. Structural basis for the nuclear import of the human androgen receptor. J. Cell Sci. 2008, 121, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Clinckemalie, L.; Vanderschueren, D.; Boonen, S.; Claessens, F. The hinge region in androgen receptor control. Mol. Cell. Endocrinol. 2012, 358, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dubbink, H.J.; Hersmus, R.; Pike, A.C.W.; Molier, M.; Brinkmann, A.O.; Jenster, G.; Trapman, J. Androgen Receptor Ligand-Binding Domain Interaction and Nuclear Receptor Specificity of FXXLF and LXXLL Motifs as Determined by L/F Swapping. Mol. Endocrinol. 2006, 20, 1742–1755. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jääskeläinen, J.; Deeb, A.; Schwabe, J.W.; Mongan, N.P.; Martin, H.; Hughes, I.A. Human androgen receptor gene ligand-binding-domain mutations leading to disrupted interaction between the N- and C-terminal domains. J. Mol. Endocrinol. 2006, 36, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Vis, A.N.; Schroder, F.H. Key targets of hormonal treatment of prostate cancer. Part 1: The androgen receptor and steroidogenic pathways. BJU Int. 2009, 104, 438–448. [Google Scholar] [CrossRef]
- Vis, A.N.; Schroder, F.H. Key targets of hormonal treatment of prostate cancer. Part 2: The androgen receptor and 5alpha-reductase. BJU Int. 2009, 104, 1191–1197. [Google Scholar] [CrossRef]
- Azad, A.A.; Zoubeidi, A.; Gleave, M.E.; Chi, K.N. Targeting heat shock proteins in metastatic castration-resistant prostate cancer. Nat. Rev. Urol. 2015, 12, 26–36. [Google Scholar] [CrossRef]
- Velcheti, V.; Karnik, S.; Bardot, S.F.; Prakash, O. Pathogenesis of prostate cancer: Lessons from basic research. Ochsner. J. 2008, 8, 213–218. [Google Scholar]
- Centenera, M.M.; Harris, J.M.; Tilley, W.D.; Butler, L.M. The contribution of different androgen receptor domains to receptor dimerization and signaling. Mol. Endocrinol. 2008, 22, 2373–2382. [Google Scholar] [CrossRef]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef]
- van Royen, M.E.; van Cappellen, W.A.; de Vos, C.; Houtsmuller, A.B.; Trapman, J. Stepwise androgen receptor dimerization. J. Cell Sci. 2012, 125, 1970–1979. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.; Liu, X.S.; Carroll, J.S.; Janne, O.A.; Keeton, E.K.; Chinnaiyan, A.M.; Pienta, K.J.; Brown, M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell 2007, 27, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Hörnberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikström, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE 2011, 6, e19059. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen receptor (AR) coregulators: An overview. Endocr. Rev. 2002, 23, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Sawyers, C.L. Biology of progressive, castration-resistant prostate cancer: Directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol. 2005, 23, 8253–8261. [Google Scholar] [CrossRef]
- Bedolla, R.G.; Wang, Y.; Asuncion, A.; Chamie, K.; Siddiqui, S.; Mudryj, M.M.; Prihoda, T.J.; Siddiqui, J.; Chinnaiyan, A.M.; Mehra, R.; et al. Nuclear versus cytoplasmic localization of filamin A in prostate cancer: Immunohistochemical correlation with metastases. Clin. Cancer Res. 2009, 15, 788–796. [Google Scholar] [CrossRef]
- Mooso, B.A.; Vinall, R.L.; Tepper, C.G.; Savoy, R.M.; Cheung, J.P.; Singh, S.; Siddiqui, S.; Wang, Y.; Bedolla, R.G.; Martinez, A.; et al. Enhancing the effectiveness of androgen deprivation in prostate cancer by inducing Filamin A nuclear localization. Endocr. Relat. Cancer 2012, 19, 759–777. [Google Scholar] [CrossRef]
- Savoy, R.M.; Chen, L.; Siddiqui, S.; Melgoza, F.U.; Durbin-Johnson, B.; Drake, C.; Jathal, M.K.; Bose, S.; Steele, T.M.; Mooso, B.A.; et al. Transcription of Nrdp1 by the androgen receptor is regulated by nuclear filamin A in prostate cancer. Endocr. Relat. Cancer 2015, 22, 369–386. [Google Scholar] [CrossRef]
- Savoy, R.M.; Ghosh, P.M. The dual role of filamin A in cancer: Can’t live with (too much of) it, can’t live without it. Endocr. Relat. Cancer 2013, 20, R341–R356. [Google Scholar] [CrossRef]
- Wang, Y.; Kreisberg, J.I.; Bedolla, R.G.; Mikhailova, M.; deVere White, R.W.; Ghosh, P.M. A 90 kDa fragment of filamin A promotes Casodex-induced growth inhibition in Casodex-resistant androgen receptor positive C4-2 prostate cancer cells. Oncogene 2007, 26, 6061–6070. [Google Scholar] [CrossRef]
- He, B.; Kemppainen, J.A.; Voegel, J.J.; Gronemeyer, H.; Wilson, E.M. Activation function 2 in the human androgen receptor ligand binding domain mediates interdomain communication with the NH(2)-terminal domain. J. Biol. Chem. 1999, 274, 37219–37225. [Google Scholar] [CrossRef]
- Klocker, H.; Culig, Z.; Eder, I.E.; Nessler-Menardi, C.; Hobisch, A.; Putz, T.; Bartsch, G.; Peterziel, H.; Cato, A.C. Mechanism of androgen receptor activation and possible implications for chemoprevention trials. Eur. Urol. 1999, 35, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Foley, C.; Mitsiades, N. Moving Beyond the Androgen Receptor (AR): Targeting AR-Interacting Proteins to Treat Prostate Cancer. Horm. Cancer 2016, 7, 84–103. [Google Scholar] [CrossRef] [PubMed]
- Obinata, D.; Takayama, K.; Takahashi, S.; Inoue, S. Crosstalk of the Androgen Receptor with Transcriptional Collaborators: Potential Therapeutic Targets for Castration-Resistant Prostate Cancer. Cancers 2017, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Gioeli, D.; Paschal, B.M. Post-translational modification of the androgen receptor. Mol. Cell. Endocrinol. 2012, 352, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, P.A. Diagnosis of adenocarcinoma in prostate needle biopsy tissue. J. Clin. Pathol. 2007, 60, 35–42. [Google Scholar] [CrossRef]
- Nelson, W.G. Commentary on Huggins and Hodges: “Studies on Prostatic Cancer”. Cancer Res. 2016, 76, 186–187. [Google Scholar] [CrossRef]
- Perlmutter, M.A.; Lepor, H. Androgen deprivation therapy in the treatment of advanced prostate cancer. Rev. Urol. 2007, 9 (Suppl. 1), S3–S8. [Google Scholar]
- van Loenen, A.C.D.; Huirne, J.A.F.; Schats, R.; Hompes, P.G.A.; Lambalk, C.B. GnRH agonists, antagonists, and assisted conception. Semin. Reprod. Med. 2002, 20, 349–364. [Google Scholar] [CrossRef]
- Fontana, D.; Mari, M.; Martinelli, A.; Boccafoschi, C.; Magno, C.; Turriziani, M.; Maymone, S.S.; Cunico, S.C.; Zanollo, A.; Montagna, G.; et al. 3-month formulation of goserelin acetate (‘Zoladex’ 10.8-mg depot) in advanced prostate cancer: Results from an Italian, open, multicenter trial. Urol. Int. 2003, 70, 316–320. [Google Scholar] [CrossRef]
- Shore, N.D.; Guerrero, S.; Sanahuja, R.M.; Gambús, G.; Parente, A. A New Sustained-release, 3-Month Leuprolide Acetate Formulation Achieves and Maintains Castrate Concentrations of Testosterone in Patients with Prostate Cancer. Clin. Ther. 2019, 41, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Pieczonka, C.M.; Twardowski, P.; Renzulli, J., 2nd; Hafron, J.; Boldt-Houle, D.M.; Atkinson, S.; Eggener, S. Effectiveness of Subcutaneously Administered Leuprolide Acetate to Achieve Low Nadir Testosterone in Prostate Cancer Patients. Rev. Urol. 2018, 20, 63–68. [Google Scholar] [CrossRef]
- Masiello, D.; Cheng, S.; Bubley, G.J.; Lu, M.L.; Balk, S.P. Bicalutamide Functions as an Androgen Receptor Antagonist by Assembly of a Transcriptionally Inactive Receptor. J. Biol. Chem. 2002, 277, 26321–26326. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, M.C.; Astapova, I.; Hollenberg, A.N.; Balk, S.P. Activity of Androgen Receptor Antagonist Bicalutamide in Prostate Cancer Cells Is Independent of NCoR and SMRT Corepressors. Cancer Res. 2007, 67, 8388–8395. [Google Scholar] [CrossRef] [PubMed]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006, 66, 2815–2825. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.A.; Guns, E.S.; Lubik, A.A.; Adomat, H.H.; Hendy, S.C.; Wood, C.A.; Ettinger, S.L.; Gleave, M.E.; Nelson, C.C. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008, 68, 6407–6415. [Google Scholar] [CrossRef]
- Fukami, M.; Homma, K.; Hasegawa, T.; Ogata, T. Backdoor pathway for dihydrotestosterone biosynthesis: Implications for normal and abnormal human sex development. Dev. Dyn. 2013, 242, 320–329. [Google Scholar] [CrossRef]
- Buchanan, G.; Greenberg, N.M.; Scher, H.I.; Harris, J.M.; Marshall, V.R.; Tilley, W.D. Collocation of androgen receptor gene mutations in prostate cancer. Clin. Cancer Res. 2001, 7, 1273–1281. [Google Scholar]
- Sonpavde, G.; Attard, G.; Bellmunt, J.; Mason, M.D.; Malavaud, B.; Tombal, B.; Sternberg, C.N. The Role of Abiraterone Acetate in the Management of Prostate Cancer: A Critical Analysis of the Literature. Eur. Urol. 2011, 60, 270–278. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; Fizazi, K.; Saad, F.; Mulders, P.F.; Sternberg, C.N.; Miller, K.; Logothetis, C.J.; Shore, N.D.; Small, E.J.; et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015, 16, 152–160. [Google Scholar] [CrossRef]
- Chi, K.N.; Protheroe, A.; Rodriguez-Antolin, A.; Facchini, G.; Suttman, H.; Matsubara, N.; Ye, Z.; Keam, B.; Damiao, R.; Li, T.; et al. Patient-reported outcomes following abiraterone acetate plus prednisone added to androgen deprivation therapy in patients with newly diagnosed metastatic castration-naive prostate cancer (LATITUDE): An international, randomised phase 3 trial. Lancet Oncol. 2018, 19, 194–206. [Google Scholar] [CrossRef]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; Marck, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Evans, C.P.; Kim, C.S.; Kimura, G.; et al. Enzalutamide in Men with Chemotherapy-naive Metastatic Castration-resistant Prostate Cancer: Extended Analysis of the Phase 3 PREVAIL Study. Eur. Urol. 2017, 71, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.T.; Oh, W.K.; Liaw, B.C. Profile of apalutamide in the treatment of metastatic castration-resistant prostate cancer: Evidence to date. Onco Targets Ther. 2018, 11, 2141–2147. [Google Scholar] [CrossRef]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef]
- Eisermann, K.; Wang, D.; Jing, Y.; Pascal, L.E.; Wang, Z. Androgen receptor gene mutation, rearrangement, polymorphism. Transl. Androl. Urol. 2013, 2, 137–147. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Nadiminty, N.; Gao, A.C. Mechanisms of persistent activation of the androgen receptor in CRPC: Recent advances and future perspectives. World J. Urol. 2012, 30, 287–295. [Google Scholar] [CrossRef]
- Lu, J.; Lonergan, P.E.; Nacusi, L.P.; Wang, L.; Schmidt, L.J.; Sun, Z.; Van der Steen, T.; Boorjian, S.A.; Kosari, F.; Vasmatzis, G.; et al. The cistrome and gene signature of androgen receptor splice variants in castration resistant prostate cancer cells. J. Urol. 2015, 193, 690–698. [Google Scholar] [CrossRef]
- Zhan, Y.; Zhang, G.; Wang, X.; Qi, Y.; Bai, S.; Li, D.; Ma, T.; Sartor, O.; Flemington, E.K.; Zhang, H.; et al. Interplay between Cytoplasmic and Nuclear Androgen Receptor Splice Variants Mediates Castration Resistance. Mol. Cancer Res. 2017, 15, 59–68. [Google Scholar] [CrossRef]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.T.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Mostaghel, E.A.; Marck, B.T.; Plymate, S.R.; Vessella, R.L.; Balk, S.; Matsumoto, A.M.; Nelson, P.S.; Montgomery, R.B. Resistance to CYP17A1 Inhibition with Abiraterone in Castration-Resistant Prostate Cancer: Induction of Steroidogenesis and Androgen Receptor Splice Variants. Clin. Cancer Res. 2011, 17, 5913–5925. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhan, Y.; Qi, Y.; Cao, B.; Bai, S.; Xu, W.; Gambhir, S.S.; Lee, P.; Sartor, O.; Flemington, E.K.; et al. Androgen Receptor Splice Variants Dimerize to Transactivate Target Genes. Cancer Res. 2015, 75, 3663–3671. [Google Scholar] [CrossRef]
- Liu, X.; Ledet, E.; Li, D.; Dotiwala, A.; Steinberger, A.; Feibus, A.; Li, J.; Qi, Y.; Silberstein, J.; Lee, B.; et al. A Whole Blood Assay for AR-V7 and AR(v567es) in Patients with Prostate Cancer. J. Urol. 2016, 196, 1758–1763. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Antonarakis, E.S.; Gjyrezi, A.; Galletti, G.; Kim, S.; Worroll, D.; Stewart, J.; Zaher, A.; Szatrowski, T.P.; Ballman, K.V.; et al. Expression of AR-V7 and ARv(567es) in Circulating Tumor Cells Correlates with Outcomes to Taxane Therapy in Men with Metastatic Prostate Cancer Treated in TAXYNERGY. Clin. Cancer Res. 2019, 25, 1880–1888. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, E.; Bergh, A.; Wikstrom, P. Clinical relevance of androgen receptor alterations in prostate cancer. Endocr. Connect. 2017, 6, R146–R161. [Google Scholar] [CrossRef]
- Kallio, H.M.L.; Hieta, R.; Latonen, L.; Brofeldt, A.; Annala, M.; Kivinummi, K.; Tammela, T.L.; Nykter, M.; Isaacs, W.B.; Lilja, H.G.; et al. Constitutively active androgen receptor splice variants AR-V3, AR-V7 and AR-V9 are co-expressed in castration-resistant prostate cancer metastases. Br. J. Cancer 2018, 119, 347–356. [Google Scholar] [CrossRef]
- Ware, K.E.; Garcia-Blanco, M.A.; Armstrong, A.J.; Dehm, S.M. Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocr. Relat. Cancer 2014, 21, T87–T103. [Google Scholar] [CrossRef]
- Lu, C.; Luo, J. Decoding the androgen receptor splice variants. Transl. Androl. Urol. 2013, 2, 178–186. [Google Scholar] [CrossRef]
- Wadosky, K.M.; Koochekpour, S. Androgen receptor splice variants and prostate cancer: From bench to bedside. Oncotarget 2017, 8, 18550–18576. [Google Scholar] [CrossRef]
- Guo, Z.; Qiu, Y. A New Trick of an Old Molecule: Androgen Receptor Splice Variants Taking the Stage?! Int. J. Biol. Sci. 2011, 7, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Yang, X.; Sun, F.; Jiang, R.; Linn, D.E.; Chen, H.; Chen, H.; Kong, X.; Melamed, J.; Tepper, C.G.; et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009, 69, 2305–2313. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef]
- Xia, N.; Cui, J.; Zhu, M.; Xing, R.; Lu, Y. Androgen receptor variant 12 promotes migration and invasion by regulating MYLK in gastric cancer. J. Pathol. 2019, 248, 304–315. [Google Scholar] [CrossRef]
- Azoitei, A.; Merseburger, A.S.; Godau, B.; Hoda, M.R.; Schmid, E.; Cronauer, M.V. C-terminally truncated constitutively active androgen receptor variants and their biologic and clinical significance in castration-resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2017, 166, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Sprenger, C.; Sun, S.; Epilepsia, K.S.; Haugk, K.; Zhang, X.; Coleman, I.; Nelson, P.S.; Plymate, S. AR variant ARv567es induces carcinogenesis in a novel transgenic mouse model of prostate cancer. Neoplasia 2013, 15, 1009–1017. [Google Scholar] [CrossRef]
- Cao, B.; Qi, Y.; Zhang, G.; Xu, D.; Zhan, Y.; Alvarez, X.; Guo, Z.; Fu, X.; Plymate, S.R.; Sartor, O.; et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget 2014, 5, 1646–1656. [Google Scholar] [CrossRef]
- Ahrens-Fath, I.; Politz, O.; Geserick, C.; Haendler, B. Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J. 2005, 272, 74–84. [Google Scholar] [CrossRef]
- Weiss, B.; Faus, H.; Haendler, B. Phylogenetic conservation of the androgen receptor AR45 variant form in placental mammals. Gene 2007, 399, 105–111. [Google Scholar] [CrossRef]
- Ma, D.; Gao, P.; Qian, L.; Wang, Q.; Cai, C.; Jiang, S.; Xiao, G.; Cui, W. Over-Expression of Porcine Myostatin Missense Mutant Leads to A Gender Difference in Skeletal Muscle Growth between Transgenic Male and Female Mice. Int. J. Mol. Sci. 2015, 16, 20020–20032. [Google Scholar] [CrossRef] [PubMed]
- Meakin, A.S.; Saif, Z.; Tuck, A.R.; Clifton, V.L. Human placental androgen receptor variants: Potential regulators of male fetal growth. Placenta 2019, 80, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Guo, Z.; Sun, F.; Li, W.; Alfano, A.; Shimelis, H.; Chen, M.; Brodie, A.M.; Chen, H.; Xiao, Z.; et al. Novel membrane-associated androgen receptor splice variant potentiates proliferative and survival responses in prostate cancer cells. J. Biol. Chem. 2011, 286, 36152–36160. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Lim, S.D.; Kwon, G.Y. mRNA expressions of androgen receptor and its variants in matched hormone-sensitive and castration-resistant prostate cancer. Scand. J. Urol. 2019, 53, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.K.; Watt, K.; Tam, T.; Yang, Y.C.; Banuelos, C.A.; et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.C.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Chandhasin, C.; Osbourne, E.; Luo, J.; Sadar, M.D.; Perabo, F. Targeting the N-Terminal Domain of the Androgen Receptor: A New Approach for the Treatment of Advanced Prostate Cancer. Oncologist 2016, 21, 1427–1435. [Google Scholar] [CrossRef]
- Le Moigne, R.; Zhou, H.-J.; Obst, J.K.; Banuelos, C.A.; Jian, K.; Williams, D.; Virsik, P.; Andersen, R.J.; Sadar, M.; Perabo, F.; et al. Lessons learned from the metastatic castration-resistant prostate cancer phase I trial of EPI-506, a first-generation androgen receptor N-terminal domain inhibitor. JCO 2019, 37, 257. [Google Scholar] [CrossRef]
- Moigne, R.L.; Banuelos, C.A.; Mawji, N.R.; Tam, T.; Wang, J.; Jian, K.; Andersen, R.J.; Cesano, A.; Sadar, M.D.; Zhou, H.-J.; et al. Abstract B117: Treatment of castrated resistant prostate cancer with EPI-7386, a second generation N-terminal domain androgen receptor inhibitor. Mol. Cancer 2019, 18, B117. [Google Scholar] [CrossRef]
- Meimetis, L.G.; Williams, D.E.; Mawji, N.R.; Banuelos, C.A.; Lal, A.A.; Park, J.J.; Tien, A.H.; Fernandez, J.G.; de Voogd, N.J.; Sadar, M.D.; et al. Niphatenones, Glycerol Ethers from the Sponge Niphates digitalis Block Androgen Receptor Transcriptional Activity in Prostate Cancer Cells: Structure Elucidation, Synthesis, and Biological Activity. J. Med. Chem. 2012, 55, 503–514. [Google Scholar] [CrossRef]
- Sadar, M.D.; Williams, D.E.; Mawji, N.R.; Patrick, B.O.; Wikanta, T.; Chasanah, E.; Irianto, H.E.; Soest, R.V.; Andersen, R.J. Sintokamides A to E, chlorinated peptides from the sponge Dysidea sp. that inhibit transactivation of the N-terminus of the androgen receptor in prostate cancer cells. Org. Lett. 2008, 10, 4947–4950. [Google Scholar] [CrossRef] [PubMed]
- Sadar, M.D. Small molecule inhibitors targeting the “achilles’ heel” of androgen receptor activity. Cancer Res. 2011, 71, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Nickols, N.G.; Dervan, P.B. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc. Natl. Acad. Sci. USA 2007, 104, 10418–10423. [Google Scholar] [CrossRef] [PubMed]
- Dalal, K.; Roshan-Moniri, M.; Sharma, A.; Li, H.; Ban, F.; Hessein, M.; Hsing, M.; Singh, K.; LeBlanc, E.; Dehm, S.; et al. Selectively Targeting the DNA-binding Domain of the Androgen Receptor as a Prospective Therapy for Prostate Cancer. J. Biol. Chem. 2014, 289, 26417–26429, Erratum in 2017, 292, 4359. [Google Scholar] [CrossRef]
- Nazareth, L.V.; Weigel, N.L. Activation of the human androgen receptor through a protein kinase A signaling pathway. J. Biol. Chem. 1996, 271, 19900–19907. [Google Scholar] [CrossRef]
- Esumi, H.; Lu, J.; Kurashima, Y.; Hanaoka, T. Antitumor activity of pyrvinium pamoate, 6-(dimethylamino)-2-[2-(2,5-dimethyl-1-phenyl-1H-pyrrol-3-yl)ethenyl]-1-methyl-quinolinium pamoate salt, showing preferential cytotoxicity during glucose starvation. Cancer Sci. 2004, 95, 685–690. [Google Scholar] [CrossRef]
- Jones, J.O.; Bolton, E.C.; Huang, Y.; Feau, C.; Guy, R.K.; Yamamoto, K.R.; Hann, B.; Diamond, M.I. Non-competitive androgen receptor inhibition in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 7233–7238. [Google Scholar] [CrossRef]
- Lim, M.; Otto-Duessel, M.; He, M.; Su, L.; Nguyen, D.; Chin, E.; Alliston, T.; Jones, J.O. Ligand-independent and tissue-selective androgen receptor inhibition by pyrvinium. ACS Chem. Biol. 2014, 9, 692–702. [Google Scholar] [CrossRef]
- Pal, S.K.; Tew, B.Y.; Lim, M.; Stankavich, B.; He, M.; Pufall, M.; Hu, W.; Chen, Y.; Jones, J.O. Mechanistic Investigation of the Androgen Receptor DNA-Binding Domain Inhibitor Pyrvinium. ACS Omega 2019, 4, 2472–2481. [Google Scholar] [CrossRef]
- Liu, C.; Lou, W.; Zhu, Y.; Nadiminty, N.; Schwartz, C.T.; Evans, C.P.; Gao, A.C. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 3198–3210. [Google Scholar] [CrossRef]
- Liu, C.; Lou, W.; Armstrong, C.; Zhu, Y.; Evans, C.P.; Gao, A.C. Niclosamide suppresses cell migration and invasion in enzalutamide resistant prostate cancer cells via Stat3-AR axis inhibition. Prostate 2015, 75, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Armstrong, C.; Zhu, Y.; Lou, W.; Gao, A.C. Niclosamide enhances abiraterone treatment via inhibition of androgen receptor variants in castration resistant prostate cancer. Oncotarget 2016, 7, 32210–32220. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Armstrong, C.M.; Lou, W.; Lombard, A.P.; Cucchiara, V.; Gu, X.; Yang, J.C.; Nadiminty, N.; Pan, C.X.; Evans, C.P.; et al. Niclosamide and Bicalutamide Combination Treatment Overcomes Enzalutamide- and Bicalutamide-Resistant Prostate Cancer. Mol. Cancer 2017, 16, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.T.; Haugk, K.; McKiernan, J.S.; Gulati, R.; Cheng, H.H.; Maes, J.L.; Dumpit, R.F.; Nelson, P.S.; Montgomery, B.; McCune, J.S.; et al. A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PLoS ONE 2018, 13, e0198389. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messner, E.A.; Steele, T.M.; Tsamouri, M.M.; Hejazi, N.; Gao, A.C.; Mudryj, M.; Ghosh, P.M. The Androgen Receptor in Prostate Cancer: Effect of Structure, Ligands and Spliced Variants on Therapy. Biomedicines 2020, 8, 422. https://doi.org/10.3390/biomedicines8100422
Messner EA, Steele TM, Tsamouri MM, Hejazi N, Gao AC, Mudryj M, Ghosh PM. The Androgen Receptor in Prostate Cancer: Effect of Structure, Ligands and Spliced Variants on Therapy. Biomedicines. 2020; 8(10):422. https://doi.org/10.3390/biomedicines8100422
Chicago/Turabian StyleMessner, Elisabeth A., Thomas M. Steele, Maria Malvina Tsamouri, Nazila Hejazi, Allen C. Gao, Maria Mudryj, and Paramita M. Ghosh. 2020. "The Androgen Receptor in Prostate Cancer: Effect of Structure, Ligands and Spliced Variants on Therapy" Biomedicines 8, no. 10: 422. https://doi.org/10.3390/biomedicines8100422
APA StyleMessner, E. A., Steele, T. M., Tsamouri, M. M., Hejazi, N., Gao, A. C., Mudryj, M., & Ghosh, P. M. (2020). The Androgen Receptor in Prostate Cancer: Effect of Structure, Ligands and Spliced Variants on Therapy. Biomedicines, 8(10), 422. https://doi.org/10.3390/biomedicines8100422