Emerging Medical Treatments for Meningioma in the Molecular Era


 ,
, 
Abstract
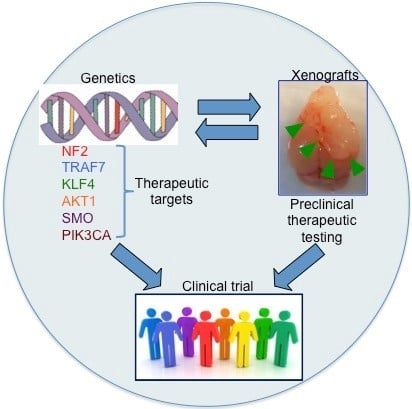
1. Introduction
2. Genetic Background
3. Clinical and Histological Features in Connection to Genotypes
4. Medical Treatment for Meningioma
4.1. Chemotherapy
4.2. Targeted Therapy
4.3. Epidermal Growth Factor Receptor (EGFR)
4.4. Platelet-Derived Growth Factor Receptor (PDGFR)
4.5. Anti-Angiogenesis
4.6. Hormonal Therapy
4.7. Interferons
4.8. Oncolytic Virus
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nigim, F.; Esaki, S.; Hood, M.; Lelic, N.; James, M.F.; Ramesh, V.; Stemmer-Rachamimov, A.; Cahill, D.P.; Brastianos, P.K.; Rabkin, S.D.; et al. A new patient-derived orthotopic malignant meningioma model treated with oncolytic herpes simplex virus. Neuro Oncol. 2016, 18, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Schrimpf, D.; Stichel, D.; Jones, D.T.W.; Hielscher, T.; Schefzyk, S.; Okonechnikov, K.; Koelsche, C.; Reuss, D.E.; Capper, D.; et al. DNA methylation-based classification and grading system for meningioma: A multicentre, retrospective analysis. Lancet Oncol. 2017, 18, 682–694. [Google Scholar] [CrossRef]
- Wen, P.Y.; Quant, E.; Drappatz, J.; Beroukhim, R.; Norden, A.D. Medical therapies for meningiomas. J. Neurooncol. 2010, 99, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Rushing, E.J.; Olsen, C.; Mena, H.; Rueda, M.E.; Lee, Y.S.; Keating, R.F.; Packer, R.J.; Santi, M. Central nervous system meningiomas in the first two decades of life: A clinicopathological analysis of 87 patients. J. Neurosurg. 2005, 103, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Ozduman, K.; Avsar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Scheithauer, B.W.; Stafford, S.L.; Lohse, C.M.; Wollan, P.C. “Malignancy” in meningiomas: A clinicopathologic study of 116 patients, with grading implications. Cancer 1999, 85, 2046–2056. [Google Scholar] [PubMed]
- Kalamarides, M.; Stemmer-Rachamimov, A.O.; Niwa-Kawakita, M.; Chareyre, F.; Taranchon, E.; Han, Z.Y.; Martinelli, C.; Lusis, E.A.; Hegedus, B.; Gutmann, D.H.; et al. Identification of a progenitor cell of origin capable of generating diverse meningioma histological subtypes. Oncogene 2011, 30, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W.; Kim, T.M.; Song, S.S.; Shrinath, N.; Park, R.; Kalamarides, M.; Park, P.J.; Black, P.M.; Carroll, R.S.; Johnson, M.D. Alternative splicing of CHEK2 and codeletion with NF2 promote chromosomal instability in meningioma. Neoplasia 2012, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 who classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Buschges, R.; Ichimura, K.; Weber, R.G.; Reifenberger, G.; Collins, V.P. Allelic gain and amplification on the long arm of chromosome 17 in anaplastic meningiomas. Brain Pathol. 2002, 12, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.X.; Banerjee, R.; Scheithauer, B.W.; Lohse, C.M.; Kleinschmidt-Demasters, B.K.; Perry, A. Chromosome 1p and 14q FISH analysis in clinicopathologic subsets of meningioma: Diagnostic and prognostic implications. J. Neuropathol. Exp. Neurol. 2001, 60, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Lomas, J.; Bello, M.J.; Arjona, D.; Gonzalez-Gomez, P.; Alonso, M.E.; de Campos, J.M.; Vaquero, J.; Ruiz-Barnes, P.; Sarasa, J.L.; Casartelli, C.; et al. Analysis of p73 gene in meningiomas with deletion at 1p. Cancer Genet. Cytogenet. 2001, 129, 88–91. [Google Scholar] [CrossRef]
- Bostrom, J.; Meyer-Puttlitz, B.; Wolter, M.; Blaschke, B.; Weber, R.G.; Lichter, P.; Ichimura, K.; Collins, V.P.; Reifenberger, G. Alterations of the tumor suppressor genes CDKN2A (p16(INK4a)), p14(ARF), CDKN2B (p15(INK4b)), and CDKN2c (p18(INK4c)) in atypical and anaplastic meningiomas. Am. J. Pathol. 2001, 159, 661–669. [Google Scholar] [CrossRef]
- Mendiola, M.; Bello, M.J.; Alonso, J.; Leone, P.E.; Vaquero, J.; Sarasa, J.L.; Kusak, M.E.; De Campos, J.M.; Pestana, A.; Rey, J.A. Search for mutations of the hRAD54 gene in sporadic meningiomas with deletion at 1p32. Mol. Carcinog. 1999, 24, 300–304. [Google Scholar] [CrossRef]
- Piaskowski, S.; Rieske, P.; Szybka, M.; Wozniak, K.; Bednarek, A.; Pluciennik, E.; Jaskolski, D.; Sikorska, B.; Liberski, P.P. GADD45A and EPB41 as tumor suppressor genes in meningioma pathogenesis. Cancer Genet. Cytogenet. 2005, 162, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Lusis, E.A.; Watson, M.A.; Chicoine, M.R.; Lyman, M.; Roerig, P.; Reifenberger, G.; Gutmann, D.H.; Perry, A. Integrative genomic analysis identifies NDRG2 as a candidate tumor suppressor gene frequently inactivated in clinically aggressive meningioma. Cancer Res. 2005, 65, 7121–7126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gejman, R.; Mahta, A.; Zhong, Y.; Rice, K.A.; Zhou, Y.; Cheunsuchon, P.; Louis, D.N.; Klibanski, A. Maternally expressed gene 3, an imprinted noncoding RNA gene, is associated with meningioma pathogenesis and progression. Cancer Res. 2010, 70, 2350–2358. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Banerjee, R.; Lohse, C.M.; Kleinschmidt-DeMasters, B.K.; Scheithauer, B.W. A role for chromosome 9p21 deletions in the malignant progression of meningiomas and the prognosis of anaplastic meningiomas. Brain Pathol. 2002, 12, 183–190. [Google Scholar] [PubMed]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2a locus, p14(ARF), participates in a regulatory feedback loop with p53 and mdm2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef] [PubMed]
- Clark, V.E.; Harmanci, A.S.; Bai, H.; Youngblood, M.W.; Lee, T.I.; Baranoski, J.F.; Ercan-Sencicek, A.G.; Abraham, B.J.; Weintraub, A.S.; Hnisz, D.; et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat. Genet. 2016, 48, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Yuzawa, S.; Nishihara, H.; Tanaka, S. Genetic landscape of meningioma. Brain Tumor Pathol. 2016, 33, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Boetto, J.; Bielle, F.; Sanson, M.; Peyre, M.; Kalamarides, M. SMO mutation status defines a distinct and frequent molecular subgroup in olfactory groove meningiomas. Neuro-Oncology 2017, 19, 345–351. [Google Scholar] [PubMed]
- Strickland, M.R.; Gill, C.M.; Nayyar, N.; D’Andrea, M.R.; Thiede, C.; Juratli, T.A.; Schackert, G.; Borger, D.R.; Santagata, S.; Frosch, M.P.; et al. Targeted sequencing of SMO and AKT1 in anterior skull base meningiomas. J. Neurosurg. 2017, 127, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Bouwmeester, T.; Bauch, A.; Ruffner, H.; Angrand, P.O.; Bergamini, G.; Croughton, K.; Cruciat, C.; Eberhard, D.; Gagneur, J.; Ghidelli, S.; et al. A physical and functional map of the human TNF-alpha/NF-kappaB signal transduction pathway. Nat. Cell Biol. 2004, 6, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Zhang, S.; Qu, G.; Zhang, D.; Li, S.; Liu, S. Downregulation of ubiquitin E3 ligase TNF receptor-associated factor 7 leads to stabilization of p53 in breast cancer. Oncol. Rep. 2013, 29, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Reuss, D.E.; Piro, R.M.; Jones, D.T.; Simon, M.; Ketter, R.; Kool, M.; Becker, A.; Sahm, F.; Pusch, S.; Meyer, J.; et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol. 2013, 125, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Beaver, J.A.; Gustin, J.P.; Yi, K.H.; Rajpurohit, A.; Thomas, M.; Gilbert, S.F.; Rosen, D.M.; Ho Park, B.; Lauring, J. PIK3CA and AKT1 mutations have distinct effects on sensitivity to targeted pathway inhibitors in an isogenic luminal breast cancer model system. Clin. Cancer Res. 2013, 19, 5413–5422. [Google Scholar] [CrossRef] [PubMed]
- Carpten, J.D.; Faber, A.L.; Horn, C.; Donoho, G.P.; Briggs, S.L.; Robbins, C.M.; Hostetter, G.; Boguslawski, S.; Moses, T.Y.; Savage, S.; et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Tetreault, M.P.; Yang, Y.; Katz, J.P. Kruppel-like factors in cancer. Nat. Rev. Cancer 2013, 13, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Hisamuddin, I.M.; Nandan, M.O.; Babbin, B.A.; Lamb, N.E.; Yang, V.W. Identification of kruppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene 2004, 23, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Zammarchi, F.; Morelli, M.; Menicagli, M.; Di Cristofano, C.; Zavaglia, K.; Paolucci, A.; Campani, D.; Aretini, P.; Boggi, U.; Mosca, F.; et al. KLF4 is a novel candidate tumor suppressor gene in pancreatic ductal carcinoma. Am. J. Pathol. 2011, 178, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Goutagny, S.; Nault, J.C.; Mallet, M.; Henin, D.; Rossi, J.Z.; Kalamarides, M. High incidence of activating TERT promoter mutations in meningiomas undergoing malignant progression. Brain Pathol. 2014, 24, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Schrimpf, D.; Olar, A.; Koelsche, C.; Reuss, D.; Bissel, J.; Kratz, A.; Capper, D.; Schefzyk, S.; Hielscher, T.; et al. TERT promoter mutations and risk of recurrence in meningioma. J. Natl. Cancer Inst. 2016, 108, djv377. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, P.S.; Hosen, I.; de Verdier, P.J.; Fallah, M.; Heidenreich, B.; Ryk, C.; Wiklund, N.P.; Steineck, G.; Schadendorf, D.; Hemminki, K.; et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc. Natl. Acad. Sci. USA 2013, 110, 17426–17431. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.S.; Wang, Z.; He, X.J.; Diplas, B.H.; Yang, R.; Killela, P.J.; Meng, Q.; Ye, Z.Y.; Wang, W.; Jiang, X.T.; et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur. J. Cancer 2015, 51, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Reuss, D.E.; Kratz, A.; Sahm, F.; Capper, D.; Schrimpf, D.; Koelsche, C.; Hovestadt, V.; Bewerunge-Hudler, M.; Jones, D.T.; Schittenhelm, J.; et al. Adult IDH wild type astrocytomas biologically and clinically resolve into other tumor entities. Acta Neuropathol. 2015, 130, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, integrative genomic analysis of diffuse lower-grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [PubMed]
- Johanns, T.M.; Fu, Y.; Kobayashi, D.K.; Mei, Y.; Dunn, I.F.; Mao, D.D.; Kim, A.H.; Dunn, G.P. High incidence of TERT mutation in brain tumor cell lines. Brain Tumor Pathol. 2016, 33, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Ahn, S.; Lee, J.I.; Bulman, M.; Plessis, D.D.; Suh, Y.L. SMARCE1 mutation screening in classification of clear cell meningiomas. Histopathology 2017, 70, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Abedalthagafi, M.; Vaubel, R.A.; Merrill, P.H.; Nayyar, N.; Gill, C.M.; Brewster, R.; Bi, W.L.; Agarwalla, P.K.; Thorner, A.R.; et al. Germline and somatic BAP1 mutations in high-grade rhabdoid meningiomas. Neuro-Oncology 2017, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, M.; Jara-Acevedo, M.; Nieto, A.B.; Caballero, A.R.; Otero, A.; Sousa, P.; Goncalves, J.; Domingues, P.H.; Orfao, A. Association between mutation of the NF2 gene and monosomy 22 in menopausal women with sporadic meningiomas. BMC Med. Genet. 2013, 14, 114. [Google Scholar] [CrossRef] [PubMed]
- Abedalthagafi, M.; Bi, W.L.; Aizer, A.A.; Merrill, P.H.; Brewster, R.; Agarwalla, P.K.; Listewnik, M.L.; Dias-Santagata, D.; Thorner, A.R.; Van Hummelen, P.; et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro-Oncology 2016, 18, 649–655. [Google Scholar] [CrossRef] [PubMed]
- McMullen, K.P.; Stieber, V.W. Meningioma: Current treatment options and future directions. Curr. Treat. Options Oncol. 2004, 5, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C.; Blumenthal, D.T. Intracranial meningiomas: Diagnosis and treatment. Expert Rev. Neurother. 2004, 4, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.D.; Drappatz, J.; Wen, P.Y. Advances in meningioma therapy. Curr. Neurol. Neurosci. Rep. 2009, 9, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Sioka, C.; Kyritsis, A.P. Chemotherapy, hormonal therapy, and immunotherapy for recurrent meningiomas. J. Neurooncol. 2009, 92, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C. Adjuvant combined modality therapy for malignant meningiomas. J. Neurosurg. 1996, 84, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Marosi, C.; Hassler, M.; Roessler, K.; Reni, M.; Sant, M.; Mazza, E.; Vecht, C. Meningioma. Crit. Rev. Oncol. Hematol. 2008, 67, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; Sade, B.; Milano, M.T.; Lee, J.H.; Toms, S.A. New prospects for management and treatment of inoperable and recurrent skull base meningiomas. J. Neurooncol. 2008, 86, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Dashti, S.R.; Sauvageau, E.; Smith, K.A.; Ashby, L.S. Nonsurgical treatment options in the management of intracranial meningiomas. Front. Biosci. 2009, 1, 494–500. [Google Scholar] [CrossRef]
- Schrell, U.M.; Rittig, M.G.; Anders, M.; Kiesewetter, F.; Marschalek, R.; Koch, U.H.; Fahlbusch, R. Hydroxyurea for treatment of unresectable and recurrent meningiomas. I. Inhibition of primary human meningioma cells in culture and in meningioma transplants by induction of the apoptotic pathway. J. Neurosurg. 1997, 86, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Schrell, U.M.; Rittig, M.G.; Anders, M.; Koch, U.H.; Marschalek, R.; Kiesewetter, F.; Fahlbusch, R. Hydroxyurea for treatment of unresectable and recurrent meningiomas. II. Decrease in the size of meningiomas in patients treated with hydroxyurea. J. Neurosurg. 1997, 86, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.P.; Gentili, F.; Macdonald, D.R.; Hariharan, S.; Cruz, C.R.; Abrey, L.E. Stabilization of disease progression by hydroxyurea in patients with recurrent or unresectable meningioma. J. Neurosurg. 2002, 97, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Newton, H.B.; Scott, S.R.; Volpi, C. Hydroxyurea chemotherapy for meningiomas: Enlarged cohort with extended follow-up. Br. J. Neurosurg. 2004, 18, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, M.A.; Ashley, D.L.; Cher, L. Treatment of high risk or recurrent meningiomas with hydroxyurea. J. Clin. Neurosci. 2002, 9, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Loven, D.; Hardoff, R.; Sever, Z.B.; Steinmetz, A.P.; Gornish, M.; Rappaport, Z.H.; Fenig, E.; Ram, Z.; Sulkes, A. Non-resectable slow-growing meningiomas treated by hydroxyurea. J. Neurooncol. 2004, 67, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Cusimano, M.D. Hydroxyurea for treatment of meningioma. J. Neurosurg. 1998, 88, 938–939. [Google Scholar] [PubMed]
- Newton, H.B. Hydroxyurea chemotherapy in the treatment of meningiomas. Neurosurg. Focus 2007, 23, E11. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Norden, A.D.; Desjardins, A.; Vredenburgh, J.J.; Herndon, J.E., 2nd; Coan, A.; Sampson, J.H.; Gururangan, S.; Peters, K.B.; McLendon, R.E.; et al. Phase ii study of Gleevec(R) plus hydroxyurea (HU) in adults with progressive or recurrent meningioma. J. Neurooncol. 2012, 106, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Ragel, B.T.; Couldwell, W.T.; Wurster, R.D.; Jensen, R.L. Chronic suppressive therapy with calcium channel antagonists for refractory meningiomas. Neurosurg. Focus 2007, 23, E10. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C.; Tsao-Wei, D.D.; Groshen, S. Temozolomide for treatment-resistant recurrent meningioma. Neurology 2004, 62, 1210–1212. [Google Scholar] [CrossRef] [PubMed]
- De Robles, P.; McIntyre, J.; Kalra, S.; Roldan, G.; Cairncross, G.; Forsyth, P.; Magliocco, T.; Hamilton, M.; Easaw, J. Methylation status of MGMT gene promoter in meningiomas. Cancer Genet. Cytogenet. 2008, 187, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C.; Tsao-Wei, D.D.; Groshen, S. Salvage chemotherapy with CPT-11 for recurrent meningioma. J. Neurooncol. 2006, 78, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Toms, S. Mitogenic signal transduction pathways in meningiomas: Novel targets for meningioma chemotherapy? J. Neuropathol. Exp. Neurol. 2005, 64, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, J.; Oskouian, R.J.; Yeoh, H.K.; Saulle, D.; Dumont, A.S. Molecular biology of unreresectable meningiomas: Implications for new treatments and review of the literature. Skull Base 2008, 18, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, M.J.; Perry, A.; Reifenberger, G. Histological classification and molecular genetics of meningiomas. Lancet Neurol. 2006, 5, 1045–1054. [Google Scholar] [CrossRef]
- Ragel, B.; Jensen, R.L. New approaches for the treatment of refractory meningiomas. Cancer Control 2003, 10, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Bostrom, J.P.; Hartmann, C. Molecular genetics of meningiomas: From basic research to potential clinical applications. Neurosurgery 2007, 60, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, M.; Tian, H.; de Sauvage, F.J. The hedgehog signaling pathway in cancer. Clin. Cancer Res. 2006, 12, 5924–5928. [Google Scholar] [CrossRef] [PubMed]
- Chari, N.S.; McDonnell, T.J. The sonic hedgehog signaling network in development and neoplasia. Adv. Anat. Pathol. 2007, 14, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Romer, J.; Curran, T. Targeting medulloblastoma: Small-molecule inhibitors of the sonic hedgehog pathway as potential cancer therapeutics. Cancer Res. 2005, 65, 4975–4978. [Google Scholar] [CrossRef] [PubMed]
- Lupi, O. Correlations between the sonic hedgehog pathway and basal cell carcinoma. Int. J. Dermatol. 2007, 46, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.L.; de Sauvage, F.J. Targeting the hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Ma, Y.; Chu, S.; Le, N.; Cao, J.; Wang, Y. Identification of key genes and pathways in meningioma by bioinformatics analysis. Oncol. Lett. 2018, 15, 8245–8252. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Jeong, E.G.; Yoo, N.J.; Lee, S.H. Mutational analysis of oncogenic AKT E17K mutation in common solid cancers and acute leukaemias. Br. J. Cancer 2008, 98, 1533–1535. [Google Scholar] [CrossRef] [PubMed]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, F.E.; Felicioni, L.; Buttitta, F.; Lamba, S.; Cardone, L.; Rodolfo, M.; Scarpa, A.; Leenstra, S.; Frattini, M.; Barbareschi, M.; et al. AKT1(E17K) in human solid tumours. Oncogene 2008, 27, 5648–5650. [Google Scholar] [CrossRef] [PubMed]
- Shoji, K.; Oda, K.; Nakagawa, S.; Hosokawa, S.; Nagae, G.; Uehara, Y.; Sone, K.; Miyamoto, Y.; Hiraike, H.; Hiraike-Wada, O.; et al. The oncogenic mutation in the pleckstrin homology domain of AKT1 in endometrial carcinomas. Br. J. Cancer 2009, 101, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Askham, J.M.; Platt, F.; Chambers, P.A.; Snowden, H.; Taylor, C.F.; Knowles, M.A. AKT1 mutations in bladder cancer: Identification of a novel oncogenic mutation that can co-operate with E17K. Oncogene 2010, 29, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Smyth, L.M.; Donoghue, M.T.A.; Westin, S.N.; Bedard, P.L.; Dean, E.J.; Bando, H.; El-Khoueiry, A.B.; Perez-Fidalgo, J.A.; Mita, A.; et al. AKT inhibition in solid tumors with AKT1 mutations. J. Clin. Oncol. 2017, 35, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Fan, H.; Nagy, T.; Wei, H.; Wang, C.; Liu, S.; Wicha, M.S.; Guan, J.L. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 2009, 69, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Watermann, D.O.; Gabriel, B.; Jager, M.; Orlowska-Volk, M.; Hasenburg, A.; zur Hausen, A.; Gitsch, G.; Stickeler, E. Specific induction of pp125 focal adhesion kinase in human breast cancer. Br. J. Cancer 2005, 93, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Xiao, G.H.; Gallagher, R.; Jablonski, S.; Jhanwar, S.C.; Testa, J.R. Re-expression of the tumor suppressor NF2/merlin inhibits invasiveness in mesothelioma cells and negatively regulates FAK. Oncogene 2006, 25, 5960–5968. [Google Scholar] [CrossRef] [PubMed]
- James, M.F.; Beauchamp, R.L.; Manchanda, N.; Kazlauskas, A.; Ramesh, V. A NHERF binding site links the betaPDGFR to the cytoskeleton and regulates cell spreading and migration. J. Cell Sci. 2004, 117, 2951–2961. [Google Scholar] [CrossRef] [PubMed]
- Hsia, D.A.; Mitra, S.K.; Hauck, C.R.; Streblow, D.N.; Nelson, J.A.; Ilic, D.; Huang, S.; Li, E.; Nemerow, G.R.; Leng, J.; et al. Differential regulation of cell motility and invasion by FAK. J. Cell Biol. 2003, 160, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.C.; Cary, L.A.; Jamieson, J.S.; Cooper, J.A.; Turner, C.E. SRC and FAK kinases cooperate to phosphorylate paxillin kinase linker, stimulate its focal adhesion localization, and regulate cell spreading and protrusiveness. Mol. Biol. Cell 2005, 16, 4316–4328. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.B.; Lu, M.; Eilers, G.; Li, H.; Ding, J.; Meng, X.; Wu, Y.; He, Q.; Sheng, Q.; Zhou, H.M.; et al. Co-targeting of FAK and MDM2 triggers additive anti-proliferative effects in mesothelioma via a coordinated reactivation of p53. Br. J. Cancer 2016, 115, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.R.; Tancioni, I.; Ward, K.K.; Lawson, C.; Chen, X.L.; Jean, C.; Sulzmaier, F.J.; Uryu, S.; Miller, N.L.; Connolly, D.C.; et al. Analyses of merlin/NF2 connection to FAK inhibitor responsiveness in serous ovarian cancer. Gynecol. Oncol. 2014, 134, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Yesiloz, U.; Kirches, E.; Hartmann, C.; Scholz, J.; Kropf, S.; Sahm, F.; Nakamura, M.; Mawrin, C. Frequent AKT1E17K mutations in skull base meningiomas are associated with mTOR and ERK1/2 activation and reduced time to tumor recurrence. Neuro-Oncology 2017, 19, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, R.L.; James, M.F.; DeSouza, P.A.; Wagh, V.; Zhao, W.N.; Jordan, J.T.; Stemmer-Rachamimov, A.; Plotkin, S.R.; Gusella, J.F.; Haggarty, S.J.; et al. A high-throughput kinome screen reveals serum/glucocorticoid-regulated kinase 1 as a therapeutic target for NF2-deficient meningiomas. Oncotarget 2015, 6, 16981–16997. [Google Scholar] [CrossRef] [PubMed]
- James, M.F.; Han, S.; Polizzano, C.; Plotkin, S.R.; Manning, B.D.; Stemmer-Rachamimov, A.O.; Gusella, J.F.; Ramesh, V. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol. Cell. Biol. 2009, 29, 4250–4261. [Google Scholar] [CrossRef] [PubMed]
- James, M.F.; Lelke, J.M.; Maccollin, M.; Plotkin, S.R.; Stemmer-Rachamimov, A.O.; Ramesh, V.; Gusella, J.F. Modeling NF2 with human arachnoidal and meningioma cell culture systems: NF2 silencing reflects the benign character of tumor growth. Neurobiol. Dis. 2008, 29, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Pachow, D.; Andrae, N.; Kliese, N.; Angenstein, F.; Stork, O.; Wilisch-Neumann, A.; Kirches, E.; Mawrin, C. mTORC1 inhibitors suppress meningioma growth in mouse models. Clin. Cancer Res. 2013, 19, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lago, M.A.; Okada, T.; Murillo, M.M.; Socci, N.; Giancotti, F.G. Loss of the tumor suppressor gene NF2, encoding merlin, constitutively activates integrin-dependent mTORC1 signaling. Mol. Cell. Biol. 2009, 29, 4235–4249. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.; Bonne, N.X.; Vitte, J.; Chareyre, F.; Tanaka, K.; Adams, R.; Fisher, L.M.; Valeyrie-Allanore, L.; Wolkenstein, P.; Goutagny, S.; et al. Mtorc1 inhibition delays growth of neurofibromatosis type 2 schwannoma. Neuro-Oncology 2014, 16, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Karajannis, M.A.; Legault, G.; Hagiwara, M.; Giancotti, F.G.; Filatov, A.; Derman, A.; Hochman, T.; Goldberg, J.D.; Vega, E.; Wisoff, J.H.; et al. Phase II study of everolimus in children and adults with neurofibromatosis type 2 and progressive vestibular schwannomas. Neuro-Oncology 2014, 16, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Goutagny, S.; Raymond, E.; Esposito-Farese, M.; Trunet, S.; Mawrin, C.; Bernardeschi, D.; Larroque, B.; Sterkers, O.; Giovannini, M.; Kalamarides, M. Phase II study of mtorc1 inhibition by everolimus in neurofibromatosis type 2 patients with growing vestibular schwannomas. J. Neurooncol. 2015, 122, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Shih, K.C.; Chowdhary, S.; Rosenblatt, P.; Weir, A.B., 3rd; Shepard, G.C.; Williams, J.T.; Shastry, M.; Burris, H.A., 3rd; Hainsworth, J.D. A phase ii trial of bevacizumab and everolimus as treatment for patients with refractory, progressive intracranial meningioma. J. Neurooncol. 2016, 129, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Graillon, T.; Defilles, C.; Mohamed, A.; Lisbonis, C.; Germanetti, A.L.; Chinot, O.; Figarella-Branger, D.; Roche, P.H.; Adetchessi, T.; Fuentes, S.; et al. Combined treatment by octreotide and everolimus: Octreotide enhances inhibitory effect of everolimus in aggressive meningiomas. J. Neurooncol. 2015, 124, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Guo, D.; Malmer, B.; Bergenheim, A.T.; Brannstrom, T.; Hedman, H.; Henriksson, R. Epidermal growth factor receptor family (EGFR, ErbB2-4) in gliomas and meningiomas. Acta Neuropathol. 2004, 108, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; Horiba, M.; Winnier, A.R.; Arteaga, C.L. The epidermal growth factor receptor is associated with phospholipase C-gamma 1 in meningiomas. Hum. Pathol. 1994, 25, 146–153. [Google Scholar] [CrossRef]
- Hsu, D.W.; Efird, J.T.; Hedley-Whyte, E.T. MIB-1 (Ki-67) index and transforming growth factor-alpha (TGF alpha) immunoreactivity are significant prognostic predictors for meningiomas. Neuropathol. Appl. Neurobiol. 1998, 24, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.D.; Raizer, J.J.; Abrey, L.E.; Lamborn, K.R.; Lassman, A.B.; Chang, S.M.; Yung, W.K.; Gilbert, M.R.; Fine, H.A.; Mehta, M.; et al. Phase II trials of erlotinib or gefitinib in patients with recurrent meningioma. J. Neurooncol. 2010, 96, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Crombet, T.; Torres, O.; Rodriguez, V.; Menendez, A.; Stevenson, A.; Ramos, M.; Torres, F.; Figueredo, R.; Veitia, I.; Iznaga, N.; et al. Phase I clinical evaluation of a neutralizing monoclonal antibody against epidermal growth factor receptor in advanced brain tumor patients: Preliminary study. Hybridoma 2001, 20, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, G.; Asai, J.; Suzuki, R.; Fujimoto, T. Different distribution of c-myc and MIB-1 positive cells in malignant meningiomas with reference to TGFs, PDGF, and PgR expression. Brain Tumor Pathol. 2001, 18, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Xu, G.M. Expression of PDGF and its receptor as well as their relationship to proliferating activity and apoptosis of meningiomas in human meningiomas. J. Clin. Neurosci. 2001, 8 (Suppl. 1), 49–53. [Google Scholar] [CrossRef]
- Todo, T.; Adams, E.F.; Fahlbusch, R.; Dingermann, T.; Werner, H. Autocrine growth stimulation of human meningioma cells by platelet-derived growth factor. J. Neurosurg. 1996, 84, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Yung, W.K.; Lamborn, K.R.; Norden, A.D.; Cloughesy, T.F.; Abrey, L.E.; Fine, H.A.; Chang, S.M.; Robins, H.I.; Fink, K.; et al. Phase ii study of imatinib mesylate for recurrent meningiomas (North American Brain Tumor Consortium study 01-08). Neuro-Oncology 2009, 11, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.M.; Figg, W.D. Angiogenesis inhibitors: Current strategies and future prospects. CA Cancer J. Clin. 2010, 60, 222–243. [Google Scholar] [CrossRef] [PubMed]
- Lamszus, K. Meningioma pathology, genetics, and biology. J. Neuropathol. Exp. Neurol. 2004, 63, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, T.; Takamiya, Y.; Costello, P.C.; Mineta, T.; Menon, A.G.; Rabkin, S.D.; Martuza, R.L. Inhibition of angiogenesis and growth of human non-malignant and malignant meningiomas by TNP-470. J. Neurooncol. 1995, 23, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Lamszus, K.; Lengler, U.; Schmidt, N.O.; Stavrou, D.; Ergun, S.; Westphal, M. Vascular endothelial growth factor, hepatocyte growth factor/scatter factor, basic fibroblast growth factor, and placenta growth factor in human meningiomas and their relation to angiogenesis and malignancy. Neurosurgery 2000, 46, 938–947. [Google Scholar] [PubMed]
- Raizer, J.J.; Abrey, L.E.; Lassman, A.B.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Yung, W.K.; Gilbert, M.R.; Aldape, K.D.; Wen, P.Y.; et al. A phase I trial of erlotinib in patients with nonprogressive glioblastoma multiforme postradiation therapy, and recurrent malignant gliomas and meningiomas. Neuro-Oncology 2010, 12, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Kaley, T.J.; Wen, P.; Schiff, D.; Ligon, K.; Haidar, S.; Karimi, S.; Lassman, A.B.; Nolan, C.P.; DeAngelis, L.M.; Gavrilovic, I.; et al. Phase II trial of sunitinib for recurrent and progressive atypical and anaplastic meningioma. Neuro-Oncology 2015, 17, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Puchner, M.J.; Hans, V.H.; Harati, A.; Lohmann, F.; Glas, M.; Herrlinger, U. Bevacizumab-induced regression of anaplastic meningioma. Ann. Oncol. 2010, 21, 2445–2446. [Google Scholar] [CrossRef] [PubMed]
- Goutagny, S.; Raymond, E.; Sterkers, O.; Colombani, J.M.; Kalamarides, M. Radiographic regression of cranial meningioma in a NF2 patient treated by bevacizumab. Ann. Oncol. 2011, 22, 990–991. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.J.; Heth, J.A. Regression of a meningioma during paclitaxel and bevacizumab therapy for breast cancer. J. Clin. Neurosci. 2012, 19, 468–469. [Google Scholar] [CrossRef] [PubMed]
- Lou, E.; Sumrall, A.L.; Turner, S.; Peters, K.B.; Desjardins, A.; Vredenburgh, J.J.; McLendon, R.E.; Herndon, J.E., 2nd; McSherry, F.; Norfleet, J.; et al. Bevacizumab therapy for adults with recurrent/progressive meningioma: A retrospective series. J. Neurooncol. 2012, 109, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Nayak, L.; Iwamoto, F.M.; Rudnick, J.D.; Norden, A.D.; Lee, E.Q.; Drappatz, J.; Omuro, A.; Kaley, T.J. Atypical and anaplastic meningiomas treated with bevacizumab. J. Neurooncol. 2012, 109, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Quant, E.C.; Wen, P.Y. Response assessment in neuro-oncology. Curr. Oncol. Rep. 2011, 13, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Grimm, S.A.; Rademaker, A.; Chandler, J.P.; Muro, K.; Helenowski, I.; Rice, L.; McCarthy, K.; Johnston, S.K.; Mrugala, M.M.; et al. A phase II trial of PTK787/ZK 222584 in recurrent or progressive radiation and surgery refractory meningiomas. J. Neurooncol. 2014, 117, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Klaeboe, L.; Lonn, S.; Scheie, D.; Auvinen, A.; Christensen, H.C.; Feychting, M.; Johansen, C.; Salminen, T.; Tynes, T. Incidence of intracranial meningiomas in Denmark, Finland, Norway and Sweden, 1968–1997. Int. J. Cancer 2005, 117, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Wigertz, A.; Lonn, S.; Hall, P.; Auvinen, A.; Christensen, H.C.; Johansen, C.; Klaeboe, L.; Salminen, T.; Schoemaker, M.J.; Swerdlow, A.J.; et al. Reproductive factors and risk of meningioma and glioma. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2663–2670. [Google Scholar] [CrossRef] [PubMed]
- Schoenberg, B.S.; Christine, B.W.; Whisnant, J.P. Nervous system neoplasms and primary malignancies of other sites. The unique association between meningiomas and breast cancer. Neurology 1975, 25, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Blitshteyn, S.; Crook, J.E.; Jaeckle, K.A. Is there an association between meningioma and hormone replacement therapy? J. Clin. Oncol. 2008, 26, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Ter Wengel, P.V.; Martin, E.; Gooren, L.; Den Heijer, M.; Peerdeman, S.M. Meningiomas in three male-to-female transgender subjects using oestrogens/progestogens and review of the literature. Andrologia 2016, 48, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Sanson, M.; Cornu, P. Biology of meningiomas. Acta Neurochir. 2000, 142, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.W.; Efird, J.T.; Hedley-Whyte, E.T. Progesterone and estrogen receptors in meningiomas: Prognostic considerations. J. Neurosurg. 1997, 86, 113–120. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, I.E. The biology of meningiomas. J. Neurooncol. 1996, 29, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Wolfsberger, S.; Doostkam, S.; Boecher-Schwarz, H.G.; Roessler, K.; van Trotsenburg, M.; Hainfellner, J.A.; Knosp, E. Progesterone-receptor index in meningiomas: Correlation with clinico-pathological parameters and review of the literature. Neurosurg. Rev. 2004, 27, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.W.; Crowley, J.; Eyre, H.J.; Stafford, B.; Jaeckle, K.A.; Townsend, J.J. A phase II evaluation of tamoxifen in unresectable or refractory meningiomas: A southwest oncology group study. J. Neurooncol. 1993, 15, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Markwalder, T.M.; Seiler, R.W.; Zava, D.T. Antiestrogenic therapy of meningiomas—A pilot study. Surg. Neurol. 1985, 24, 245–249. [Google Scholar] [CrossRef]
- Grunberg, S.M.; Weiss, M.H.; Spitz, I.M.; Ahmadi, J.; Sadun, A.; Russell, C.A.; Lucci, L.; Stevenson, L.L. Treatment of unresectable meningiomas with the antiprogesterone agent mifepristone. J. Neurosurg. 1991, 74, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Lamberts, S.W.; Tanghe, H.L.; Avezaat, C.J.; Braakman, R.; Wijngaarde, R.; Koper, J.W.; de Jong, H. Mifepristone (RU 486) treatment of meningiomas. J. Neurol. Neurosurg. Psychiatry 1992, 55, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Rankin, C.; Grunberg, S.; Sherrod, A.E.; Ahmadi, J.; Townsend, J.J.; Feun, L.G.; Fredericks, R.K.; Russell, C.A.; Kabbinavar, F.F.; et al. Double-blind phase iii randomized trial of the antiprogestin agent mifepristone in the treatment of unresectable meningioma: SWOG S9005. J. Clin. Oncol. 2015, 33, 4093–4098. [Google Scholar] [CrossRef] [PubMed]
- Grunberg, S.M.; Weiss, M.H.; Russell, C.A.; Spitz, I.M.; Ahmadi, J.; Sadun, A.; Sitruk-Ware, R. Long-term administration of mifepristone (RU486): Clinical tolerance during extended treatment of meningioma. Cancer Investig. 2006, 24, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Barbieri, F.; Thellung, S.; Pirani, P.; Corsaro, A.; Villa, V.; Dadati, P.; Dorcaratto, A.; Lapertosa, G.; Ravetti, J.L.; et al. Expression of somatostatin receptor mRNA in human meningiomas and their implication in in vitro antiproliferative activity. J. Neurooncol. 2004, 66, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Cavalla, P.; Schiffer, D. Neuroendocrine tumors in the brain. Ann. Oncol. 2001, 12 (Suppl. 2), S131–S134. [Google Scholar] [CrossRef]
- Chamberlain, M.C.; Glantz, M.J.; Fadul, C.E. Recurrent meningioma: Salvage therapy with long-acting somatostatin analogue. Neurology 2007, 69, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.; Lewis, I.; Briner, U.; Meno-Tetang, G.; Weckbecker, G. SOM230: A novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur. J. Endocrinol. 2002, 146, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; Kimmel, D.W.; Burch, P.A.; Cascino, T.L.; Giannini, C.; Wu, W.; Buckner, J.C. Phase II study of subcutaneous octreotide in adults with recurrent or progressive meningioma and meningeal hemangiopericytoma. Neuro-Oncology 2011, 13, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Rammo, R.; Rock, A.; Transou, A.; Raghunathan, A.; Rock, J. Anaplastic meningioma: Octreotide therapy for a case of recurrent and progressive intracranial disease. J. Neurosurg. 2016, 124, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Koper, J.W.; Zwarthoff, E.C.; Hagemeijer, A.; Braakman, R.; Avezaat, C.J.; Bergstrom, M.; Lamberts, S.W. Inhibition of the growth of cultured human meningioma cells by recombinant interferon-alpha. Eur. J. Cancer 1991, 27, 416–419. [Google Scholar] [CrossRef]
- Kaba, S.E.; DeMonte, F.; Bruner, J.M.; Kyritsis, A.P.; Jaeckle, K.A.; Levin, V.; Yung, W.K. The treatment of recurrent unresectable and malignant meningiomas with interferon alpha-2B. Neurosurgery 1997, 40, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Muhr, C.; Gudjonsson, O.; Lilja, A.; Hartman, M.; Zhang, Z.J.; Langstrom, B. Meningioma treated with interferon-alpha, evaluated with [(11)C]-L-methionine positron emission tomography. Clin. Cancer Res. 2001, 7, 2269–2276. [Google Scholar] [PubMed]
- Chamberlain, M.C.; Glantz, M.J. Interferon-alpha for recurrent world health organization grade 1 intracranial meningiomas. Cancer 2008, 113, 2146–2151. [Google Scholar] [CrossRef] [PubMed]
- Peyre, M.; Stemmer-Rachamimov, A.; Clermont-Taranchon, E.; Quentin, S.; El-Taraya, N.; Walczak, C.; Volk, A.; Niwa-Kawakita, M.; Karboul, N.; Giovannini, M.; et al. Meningioma progression in mice triggered by NF2 and Cdkn2ab inactivation. Oncogene 2013, 32, 4264–4272. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Wakimoto, H. Oncolytic herpes simplex virus-based strategies: Toward a breakthrough in glioblastoma therapy. Front. Microbiol. 2014, 5, 303. [Google Scholar] [CrossRef] [PubMed]
- Grill, J.; Lamfers, M.L.; van Beusechem, V.W.; van der Valk, P.; Huisman, A.; Sminia, P.; Alemany, R.; Curiel, D.T.; Vandertop, W.P.; Gerritsen, W.R.; et al. Oncolytic virotherapy of meningiomas in vitro with replication-competent adenovirus. Neurosurgery 2005, 56, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Coen, D.M.; Malick, A.; Mineta, T.; Martuza, R.L. Expanded spectrum of viral therapy in the treatment of nervous system tumors. J. Neurosurg. 1992, 77, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, T.; Manz, H.J.; Rabkin, S.D.; Martuza, R.L. Treatment of human malignant meningiomas by G207, a replication-competent multimutated herpes simplex virus 1. Cancer Res. 1995, 55, 4752–4756. [Google Scholar] [PubMed]
- Liu, T.C.; Zhang, T.; Fukuhara, H.; Kuroda, T.; Todo, T.; Canron, X.; Bikfalvi, A.; Martuza, R.L.; Kurtz, A.; Rabkin, S.D. Dominant-negative fibroblast growth factor receptor expression enhances antitumoral potency of oncolytic herpes simplex virus in neural tumors. Clin. Cancer Res. 2006, 12, 6791–6799. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, S.; Messerli, S.M.; Stemmer-Rachamimov, A.O.; Liu, T.C.; Rabkin, S.; Martuza, R.; Breakefield, X.O. Treatment of implantable NF2 schwannoma tumor models with oncolytic herpes simplex virus G47delta. Cancer Gene Ther. 2007, 14, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Messerli, S.M.; Prabhakar, S.; Tang, Y.; Mahmood, U.; Giovannini, M.; Weissleder, R.; Bronson, R.; Martuza, R.; Rabkin, S.; Breakefield, X.O. Treatment of schwannomas with an oncolytic recombinant herpes simplex virus in murine models of neurofibromatosis type 2. Hum. Gene Ther. 2006, 17, 20–30. [Google Scholar] [CrossRef] [PubMed]

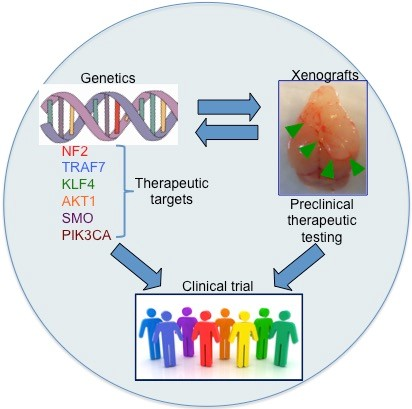{kind=link}
| Gene | Mutation Type | Frequency (%) | Histopathological Subtype | Tumor Anatomical Location |
|---|---|---|---|---|
| NF2 | Chromosome loss Var. mutations | 40–60 | Fibroblastic, transitional, atypical and anaplastic | Convexity and skull base |
| TRAF7 | Var. mutations (WD40 domains) | 12–25 | Secretory, meningothelial and atypical | Skull base |
| KLF4 | K409Q | 9–12 | Secretory | Skull base |
| AKT1 | E17K | 7–9 | Meningothelial, transitional and atypical | Skull base |
| TERT promoter | C228T, C250T | 6 | Anaplastic and atypical (secondary) | Convexity and skull base |
| POLR2A | Q403K, L438_H439del | 6 | Meningothelial | Skull base (Tuberculum sellae) |
| SMO | L412F, W535L | 1–5 | Meningothelial and atypical | Anterior skull base |
| PIK3CA | H1047R most frequent | 3–4 | Meningothelial and transitional | Skull base |
| SMARCE1 | Var. mutations | 3–4 | Clear cell | Spine and posterior fossa |
| BAP1 | Var. mutations | Rare | Rhabdoid | Convexity and skull base |
| Targeted Pathway | Agent | Phase | Dates of the Study | Trial Identifier |
|---|---|---|---|---|
| SMO | Vismodegib | II | August 2015–present | NCT02523014 |
| FAK | GSK2256098 | II | August 2015–present | NCT02523014 |
| mTORC1/2 | AZD2014 | II | August 2016–present | NCT02831257 |
| mTOR | Everolimus | I | June 2013–present | NCT01880749 |
| Somatostatin + radionucleotide | 90-YDOTA tyr3-Octreotide | II | September 2017–present | NCT03273712 |
| PD-1 | Nivolumab | II | March 2016–present | NCT02648997 |
| PDL-1 | Pembrolizumab | II | November 2017–present | NCT03279692 |
| PDL-1 + proton radiation | Avelumab | II | January 2018–present | NCT03267836 |
| MEK1/2 | Selumetinib | II | March 2017–present | NCT03095248 |
| Histone Deacetylase | AR-42 | I | September 2017–present | NCT02282917 |
| CDK4/6 | Ribociclib | I | October 2016–present | NCT02933736 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nigim, F.; Wakimoto, H.; Kasper, E.M.; Ackermans, L.; Temel, Y. Emerging Medical Treatments for Meningioma in the Molecular Era. Biomedicines 2018, 6, 86. https://doi.org/10.3390/biomedicines6030086
Nigim F, Wakimoto H, Kasper EM, Ackermans L, Temel Y. Emerging Medical Treatments for Meningioma in the Molecular Era. Biomedicines. 2018; 6(3):86. https://doi.org/10.3390/biomedicines6030086
Chicago/Turabian StyleNigim, Fares, Hiroaki Wakimoto, Ekkehard M. Kasper, Linda Ackermans, and Yasin Temel. 2018. "Emerging Medical Treatments for Meningioma in the Molecular Era" Biomedicines 6, no. 3: 86. https://doi.org/10.3390/biomedicines6030086
APA StyleNigim, F., Wakimoto, H., Kasper, E. M., Ackermans, L., & Temel, Y. (2018). Emerging Medical Treatments for Meningioma in the Molecular Era. Biomedicines, 6(3), 86. https://doi.org/10.3390/biomedicines6030086