Abstract
Background/Objectives: Organ transplantation is a life-saving intervention for patients with terminal organ failure, but long-term success is hindered by graft rejection and dependence on lifelong immunosuppressants. These drugs pose risks such as opportunistic infections and malignancies. Chimeric antigen receptor (CAR) technology, originally developed for cancer immunotherapy, has been adapted to regulatory T cells (Tregs) to enhance their antigen-specific immunosuppressive function. This systematic review evaluates the preclinical development of CAR-Tregs in promoting graft tolerance and suppressing graft-versus-host disease (GvHD). Methods: A systematic review following PROSPERO guidelines (CRD420251073207) was conducted across PubMed, Scopus, and Web of Science for studies published from 2015 to 2024. After screening 105 articles, 17 studies involving CAR-Tregs in preclinical or in vivo transplant or GvHD models were included. Results: CAR-Tregs exhibited superior graft-protective properties compared to unmodified or polyclonal Tregs. HLA-A2-specific CAR-Tregs consistently improved graft survival, reduced inflammatory cytokines, and suppressed immune cell infiltration across skin, heart, and pancreatic islet transplant models. The inclusion of CD28 as a co-stimulatory domain enhanced Treg function and FOXP3 expression. However, challenges such as Treg exhaustion, tonic signaling, and reduced in vivo persistence were noted. Some studies reported synergistic effects when CAR-Tregs were combined with immunosuppressants like rapamycin or tacrolimus. Conclusions: CAR-Tregs offer a promising strategy for inducing targeted immunosuppression in allogeneic transplantation. While preclinical findings are encouraging, further work is needed to optimize CAR design, ensure in vivo stability, and establish clinical-scale manufacturing before translation to human trials.
1. Introduction
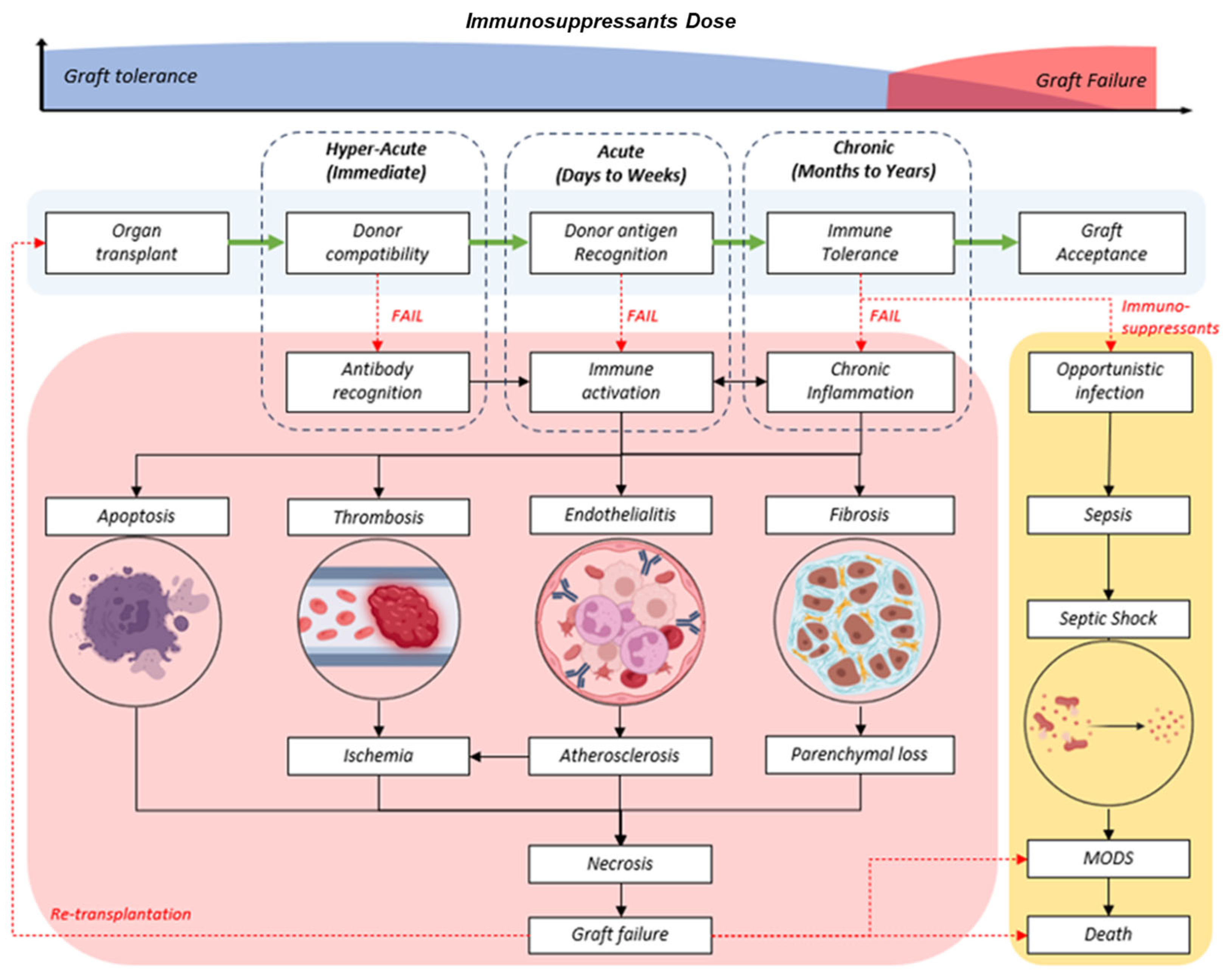
Lifelong immunosuppression is necessary to prevent graft-versus-host disease (GvHD) following allogenic graft or organ transplantations. The lymphodepletion strategy has become the standard protocol for solid organ transplants and has been shown to improve overall quality of life [1]. However, the long-term survival of the transplanted graft cannot be guaranteed. Prolonged intake of conventional immunosuppressants is associated with several complications, with opportunistic infections being high on the list. Take, for instance, the frequently reported EBV-associated malignancy of post-transplant lymphoproliferative disorders (PTLD) [2]. To achieve a successful graft assimilation, there should be an ideal balance between the persistence of allogeneic transplants and the prevention of graft rejection, as illustrated in Figure 1. This biological revision should take place during the refractory period of a depleted patient’s immunity and contact with donor tissue antigens to allow the emergence and expansion of graft-tolerant immune cells. However, the absence of the required signalling factors and poorly regulated conditions has led to chronic or delayed rejection. Premature graft failure from hyperacute or acute rejection of the graft is prevalent despite highly stringent screening protocols in the selection of compatible donors.

Figure 1.
(Top) Trend of life-long immunosuppressants in graft tolerant (blue) or increased dose during chronic, delayed, or failed transplants (red). (Bottom) Generalized flow chart of successful graft progress (light blue) and the phases of rejection (red) at different phases (dotted box) in order of duration post-transplant: hyperacute, acute, and chronic.
Regulatory T cells (Tregs) are a specialized subpopulation of CD4+ T lymphocytes that are crucial in maintaining immune homeostasis and self-tolerance. These cells express high expression of CD25 (the α-chain of the IL-2 receptor) and low expression of CD127 (IL-7 receptor α-chain). More importantly, the transcription factor FOXP3 that enables Tregs acts as the immune system’s natural suppressors [3,4]. They exert immunosuppressive functions via multiple mechanisms, including the secretion of anti-inflammatory cytokines (IL-10 and TGF-β), dendritic cell modulation, deactivation of effector T cells (Teff), and cytolysis of activated immune cells. In contrast, Teff cells (Th1, Th2, and Th17) are responsible for executing immune responses by generating pro-inflammatory cytokines (IFN-γ, TNF-α, IL-2, and IL-17), aimed at clearing infections or target mismatched antigens [5]. However, in the context of transplantation or autoimmunity, unregulated activity of Teff cells often results in graft rejection.
Besides, conventional T cells (Tconv), which refer to CD4+FOXP3− T cells, are the pool of naive and memory T cells that differentiate into Teff subsets during antigen stimulation [5,6]. These cells orchestrate immune responses against foreign antigens and are central to both protective immunity and immunopathology. Compared to Tregs, Tconv cells do not express intrinsic regulatory functions and can become activated and drive graft rejection via secretion of inflammatory mediators and recruitment of Teffs. Thus, a higher Treg to Tconv ratio is considered favourable for transplant outcome and immune tolerance [7].
Hence, Tregs possess the advantage as a platform to initiate suppression of autoreactive and alloreactive T cells. Tregs are also valuable because they offer a biologically compatible means of generating immune tolerance without entirely suppressing the graft recipient’s immunity, unlike conventional treatment with immunosuppressive drugs [8]. This selective ability makes them attractive for cell-based immunotherapies for transplantation. In therapeutic models, Tregs have been shown to prolong graft survival, reduce chronic inflammation, and promote immune tolerance, highlighting their potential as an alternative to existing anti-rejection solutions [9]. However, Tregs alone often do not generate a sufficient or timely response, leading to masked symptoms and eventual graft rejection.
Over the years, there has been tremendous development of chimeric antigen receptor (CAR) technology. The foundation of CAR technology lies in engineering patient or donor immune cells, typically T cells, to recognize target cells. This technology essentially utilizes the target specificity of antibodies along with the powerful effector functions of T cells [10]. Leveraging their natural tumoricidal efficacy, CAR-modified T cells have become a promising tool to combat various cancers. Currently, CAR T cells are under investigation for haematological cancers, leukaemia, and lymphomas by targeting the CD19 antigen exclusive to B cells. Besides, research is well underway for CAR T cells to target viral antigens in infected tissues [11].
In parallel to the anti-tumour role of CAR T cells, CAR-regulatory T cells (CAR-Tregs) were recently proposed as candidates that could modulate erratic immune cells against self-tissues or transplanted organs [12]. Naturally, regulatory T cells (Tregs) contribute to donor-specific transplantation tolerance while having much fewer adverse effects than non-specific or polyclonal Treg immunosuppression [13]. Previously, the co-administration of donor Tregs with the corresponding graft has been shown to prevent transplant rejection [14]. However, the number of antigen-specific Tregs is often low or eventually diminished post-infusion. Thus, the ideal model of CAR Treg should be able to elevate their antigen-specificity function while remedying the viability issues to improve the overall retention and performance of conventional Tregs.
CAR is a modular platform, first consisting of an extracellular region, the binding domain of an antibody towards a specific antigen (e.g., HER2 or CD19); a transmembrane, co-signalling domain of a hinge and/or stalk for receptor dynamicity and flexibility (e.g., CD8 or CD28); and intracellular signalling domain for metabolic activation (e.g., CD28 and/or CD3ζ). Beyond their basic construct, the CAR can be additionally modified through ‘armour’ molecules that protect or react opposingly to inhibitory factors (e.g., enriched anti-inflammatory cytokines in tumour microenvironment); over- or under-expression of activating and inhibitory receptors; enhanced lymphocyte homing capabilities (e.g., VCAM-1) and other deficiencies that can be compensated for [15]. Granted that there are many different facets possible through CAR technology, but T cells already possess a high metabolic niche, which has led to biological “exhaustion” or senescence [16]. Ultimately, there remains much work to be completed and optimised in CAR design.
In the existing literature, there is critically limited evidence on the clinical applications of CAR Tregs. There is ongoing research on the safety of prototypes and preclinical studies using animal transplant models. Therefore, this article aims to evaluate the current available data and discuss the role of modified CAR T cells as a treatment for alloreactivity and tissue transplant tolerance.
2. Materials and Methods
A systematic review was performed with adherence to the guidelines of the International Prospective Register of Systematic Review (PROSPERO; CRD420251073207). The Search keywords were selected from medical subject headings (MESH) available from PubMed, namely, (i) receptors, chimeric antigen; (ii) organ transplantation; and (iii) graft rejection. Unconventional terms or synonyms not registered as MESH were included in the search. Database access and bibliography retrieval from Scopus, PubMed, and Web of Science (WOS) were accessible by the National University of Malaysia (UKM). Only “research articles” or “journal articles” published in the last 10 years (2015–2024) were downloaded as bibliographies containing title, keywords, and abstract. The bibliographies were labelled appropriately according to the databases, date of access, and results. The duplicates were merged using reference software: Mendeley v1.19.8 (Elsevier, Amsterdam, The Netherlands). The primary screening was performed relevant to the titles, abstract, and keywords. Thereafter, full-text screening was performed according to set inclusion and exclusion criteria as described below. Inclusion criteria: (i) CAR T, (ii) GvHD or any tissue transplant, (iii) preclinical or in vivo model, and (iv) controlled experimental studies. Exclusion criteria: (i) non-CAR T, (ii) not GvHD or any tissue transplant, (iii) non-preclinical or animal model studies, and (iv) uncontrolled experimental study.
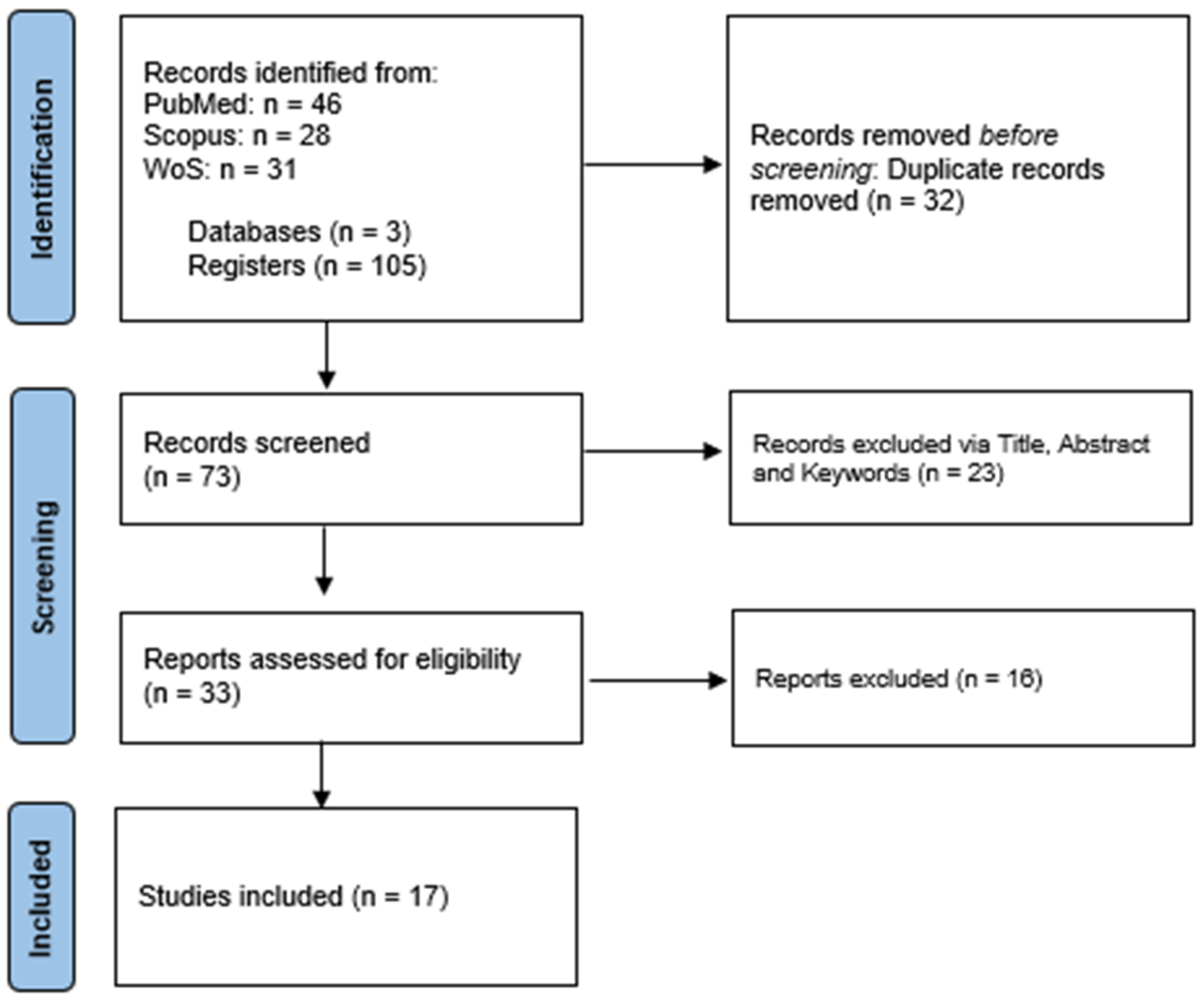
A total of 105 records were acquired from merging the three bibliographic sources: PUBMED (46), SCOPUS (28), and WOS (31). Thirty-two records were removed from merging record duplicates, yielding 73 individual records (Figure 2). The primary screening eliminated 23 records, leaving 33 eligible studies for full-text screening. Finally, seventeen accepted records were selected for data extraction and analysis. The text screening and consensus were performed by authors (A.M.L.C. and Y.L).

Figure 2.
PRISMA flow diagram.
3. Results
3.1. Conventional Tregs (Tconv) Have Limited Suppression Capacity
Regulatory T cells or Tregs play a critical role in maintaining immune homeostasis and adjusting irregular immune activities. Tregs are responsible for mediating between multiple target cells, utilizing various methods including induced apoptosis, regulating cellular metabolic pathways, and promoting or inhibiting anti- and pro-inflammatory cytokines, respectively. Some of these mutually interacting cells include antigen-presenting cells (APC), dendritic cells (DC), effector T cells, macrophages, and more.
In most studies reviewed (Table 1), the groups employing naive (n-), polyclonal (poly-), non-specific (NT-), or untransduced (UT-) Tregs did cause harm in either allo- or xenograft transplant models, compared to the effector (Teff) or conventional T (Tconv) cells where deliberate cytotoxicity was simulated [14,17,18,19,20,21]. However, the overall immunosuppression capacity of these Tregs did not last very long due to diminishing signals, which eventually caused the graft to be rejected by the recipient. Although polyclonal Tregs could theoretically be considered for universal model therapy, their well-established off-target effects remain a legitimate concern for clinical and translational application. For example, Noyan et al., 2017 reported significantly increased delayed-type hypersensitivity (DTH) and the ear-swelling response by 70% (p < 0.01) in the UT-Tregs vs. CAR-treated group [17].

Table 1.
Preclinical model of induced GvHD or tissue transplant, dose of CAR T model, and in vivo analysis (see end of document).
Unbiasedly, the overall performance of Tregs was greatly enhanced post-transduction regardless of the CAR model induced. The study by Boardman et al., 2017 reported that CAR Tregs prevented immune cytokine trafficking to the graft by greatly suppressing IFN-γ and promoting IL-10 secretion in situ (p < 0.05) [18]. Similar outcomes were reported in Boroughs et al., 2019, whereby CAR Tregs prevented Teff-mediated tissue infiltration and apoptosis, prolonging the graft’s survival in the skin xenograft model (p < 0.0001) [24]. Additionally, the bioluminescence imaging by Dawson et al., 2019 observed fewer keratinocytes and involucrin destruction, prompting conserved function of the graft in the A2-CAR Treg-treated mice (p < 0.001) [25]. Beyond the commonly employed skin graft models, similar results were recreated from more biologically relevant transplants, including pancreatic islet [23,29,32], heart [28,30], and even CD19- or BCMA-CAR T cells for targeting B-cell malignancies [32].
3.2. HLA-A2 as the Basis of Immune Tolerance
In the context of organ and tissue transplantation, HLA mismatch is the primary source between the donor and the recipient that could trigger graft rejection. The HLA-A locus, especially HLA-A2, is one of the most important determinants of transplant compatibility. The HLA-A2 is a globally common Major Histocompatibility Class 1 (MHC-1) molecule found in humans [22,35]. Among the hundreds of HLA-A2 variants, the HLA-A*02:01 is the most prevalent model to study immune cytotoxic responses to foreign particle elimination [36]. Since nearly 40 to 50% of the population expresses HLA-A2, this frequent recurrence makes it an ideal target for immunological studies and the development of therapies catered to a wider range of populations.
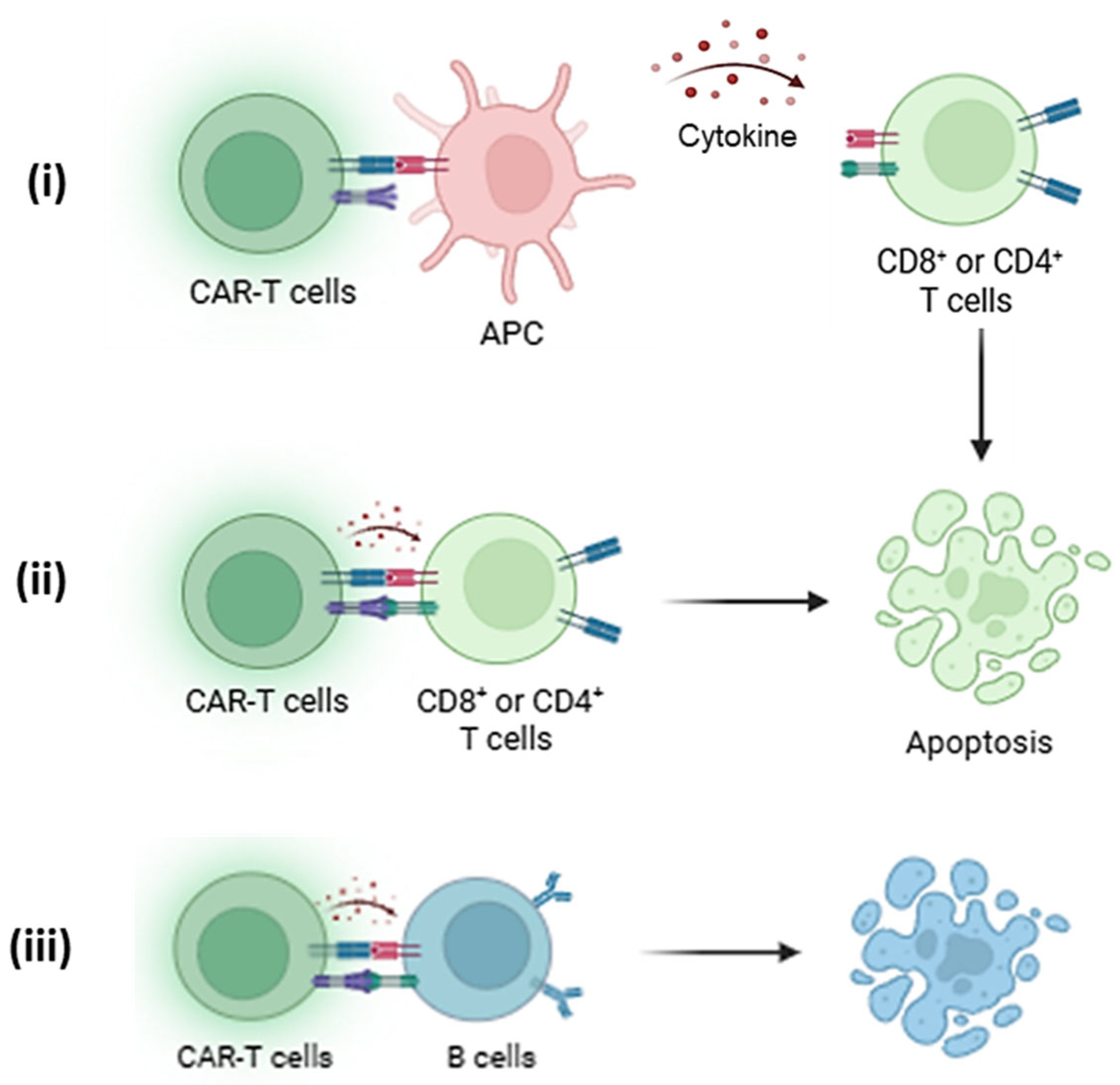
Targeting HLA-A2 specifically with CAR Tregs or other CAR-based models has allowed researchers to create immunotherapy models of tolerance where the recipient’s immune system could be retrained to accept the transplanted organ, even if it is bearing HLA-A2 that would otherwise be rejected. This specificity also helps develop the recipient’s immune system to selectively tolerate the donor’s graft without relying on generalized immune suppression strategies, as seen in Figure 3. Thereby, reducing the risk of infections and other systemic complications associated with broad immunosuppression medications.

Figure 3.
(i) HLA-A2-specific CAR-Tregs interact with antigen-presenting cells (APCs) by recognizing HLA-A2 on their surface, leading to the suppression of co-stimulatory molecule expression and promoting a tolerogenic phenotype through the release of IL-10 and CTLA-4-mediated signalling [14]. This dampens APC-mediated activation of effector T cells (Teffs), which are further suppressed by (ii) CAR-Tregs through cytokine deprivation, inhibitory cytokines, and contact-dependent mechanisms. Thus, leading to deactivation or apoptosis of Teffs [17]. (iii) Additionally, CAR-Tregs can modulate B cell activity by reducing alloantibody production within the HLA-A2+ immune environment or trigger apoptosis, contributing to broader immune tolerance [37].
Out of the 16 studies reviewed, 11 studies that selected HLA-A2 CAR Tregs successfully demonstrated excellent immune suppression without compromising the survival of the animals [14,17,18,22,25,26,29,30,31,33,34]. Compared to non-specific, control CAR Tregs such as HER2- or CD19-CAR Tregs, the HLA-A2 CAR Tregs explicitly demonstrated their antigen specificity in the presence of HLA-A2+ graft.
In other notable models to consider, Lee et al., 2022 explored the application of anti-C4d-CAR Tregs to counteract ABO-incompatible (ABOi) heart transplantation [28]. From their study, the treatment significantly prolonged graft survival (p < 0.05) compared to control CAR Tregs, validated by low inflammatory activity in sample histology and cytokine analysis (IFN-γ and TNF-α). Compared to T cell-mediated rejection patterns, antibody-mediated rejection (ABMR) remains culpable for blood-incompatible (ABOi) hyperacute or rapid transplant rejection. Therefore, strategies to overcome the challenge of ABO blood type barriers could hasten the waiting time for donor screening and broaden the therapeutic window for life-saving transplants. Till then, the current regime of strong immunosuppression enforces excess immune suppression and could likely cause secondary symptoms such as sepsis.
Even perfect ABO matching remains susceptible to the innate immunity mechanism, as HLA mismatch could still trigger NK cell-mediated rejection from the absent signal of donor MHC Class I molecules. Since autologous T cells have compromised immune function while allogeneic T cells risk interception by NK cells or cause severe GvHD, Degagné et al., 2024 explored immune cloaking methods by engineering B2M-HLA-E onto CB-011 (anti-BCMA) CAR T cells to enable safer and effective treatment for multiple myeloma [38]. The study reported significant evasion and survival from clearance by natural killer (NK) cells at nearly 60% (p < 0.05) more than the control group.
3.3. Hypo-Responsive and Metabolic Exhaustion in Tregs
Thematically, studies have frequently cited a pronounced decrease in the viability and function of Tregs post-infusion [22,31]. With CAR Tregs, a moderate degree of retention could be observed, but most of the transfused cells were lost after weeks. One of the predominant reasons for poor retention of the CAR Tregs stems from the interleukin-2 (IL-2) paradox. Principally, one of the Tregs’ immunosuppressive behaviours is to deregulate the pro-inflammatory cytokines, including interleukins (IL), interferons (IFN), and the tumor necrosis factor (TNF) family of proteins, resulting in lesser invasion of immune cells in situ. However, this becomes counterproductive as Tregs are devoid of specific cytokines, especially IL-2, which is among the essential stimuli for immune cell proliferation [39].
Of note, Rosado-Sánchez et al., 2023 reported that the engraftment of CAR Tregs could translate into the uptake of HLA-A2 into CAR Tregs via trogocytosis [31]. This exchange could encourage Ab-specific suppression and elimination of HLA-A2 availability, affecting overall Treg functionality and graft survival. To ensure the transplanted cells’ survival, the supplement of exogenous IL-2 or repeated dosing of CAR Tregs was suggested. However, pro-stimulus factors possess highly complex interactions that could affect the outcome of therapy and incite Tregs or T cell anergy.
The chronic activation of CAR Tregs in the presence of highly expressed auto- or alloantigens is an underrepresented aspect in numerous studies. Tonic signalling was described as a constant, diminutive signal in T cells under basic conditions [40]. Although no reactions typically come of it, the constant engagement of TCR emits low-level signals that cumulatively build fatigue in cells from various feedback mechanisms. Other than the immune surveillance in vivo, purposely designed cultured scenarios for ex vivo, clinical expansion of CAR T cells may also contribute to tonic signalling [41]. In a study by Lamarche et al., 2023, tonic-signalling CAR (TS-CAR) resulted in exhausted Tregs with significant metabolic, transcriptomic, and epigenetic alterations [21]. Although the in vitro assessment was determinably similar, the TS-CAR Tregs failed to operate in the GvHD animal model, resulting in morbidity and mortality rates similar to the untreated group and significantly (p < 0.01) worse than control or UT-Tregs.
Thus, ex vivo expansion strategies become critical, not only to ensure the process does not alter Treg phenotype, function, and cause exhaustion in the process of generating sufficient cell numbers for therapeutic purposes. Early methods rely on tissue culture flasks or well plates that are either bead-based (e.g., anti-CD3/CD28 beads) and/or cytokine-tailored (e.g., IL-2 with IL-15) into the growth media. Studies have shown that both methods involving prolonged, specific stimuli led to cellular exhaustion. Therefore, newer strategies such as the G-REX® (Wilson Wolf Manufacturing, St. Paul, MN, USA) platform offer a more relevant culture system and microenvironment for cells to grow optimally [42]. A key characteristic of the G-REX® system is the unique, gas-permeable membrane at its base, enabling superior nutrient flow and gas exchange. An exploration conducted in 2013 by Chakraborty et al. surmised that 1 × 109 cells could be generated from the system in 21 days using one culture vessel [43]. The expanded Tregs expressed high CD25 and CD4 (>90%) and relatively stable FOXP3 (~69%), which were assessed in an in vitro and in vivo model of GvHD. With further optimization, such as the Gotti et al., 2021 manufacturing protocol, >30 × 106 cells/cm2 could be generated in <11 days using a single unit of G-REX® compared to the 24 days of seventy-two tissue culture vessels [44]. Although the cell surface marker did not report any significant differences, the tumoricidal effect of the T cells cultured in G-REX® was superior to the flasks. This technology has marked the viability of GMP manufacturing and clinical applications for T cell-based therapies. Simultaneously, there are also other commercial alternatives, such as the protocol validated by Marín Morales et al., 2019 used to compare the efficacy of the CliniMacs Prodigy® (Miltenyi Biotec, NRW, Germany) to the G-REX® platform for Treg expansion [45]. The study was able to generate a similar yield (2 × 109 cells) with >90% subpopulation immunosuppressive phenotype (CD4+CD25highFOXP3+), with the added benefit of an automated, closed system that is more befitting of GMP requirements. Albeit, the economic factor of the culture systems must be considered to ensure low-production cost and patient accessible funds for life-saving therapy.
3.4. FOXP3+: A Major Consensus of Treg Function
The forkhead box P3 (FOXP3) is considered a “master regulator” of Tregs as it controls nearly all development, function, and maintenance of the Tregs. Henceforth, the FOXP3 was regarded as a lineage marker, distinctly programming precursor T cells to differentiate into Tregs and not Teffs. In the absence of dysregulated FOXP3 expression, Tregs could either fail to develop or lose their functional ability to suppress exacerbated immune responses. The reviewed studies frequently correlate the success of transducing CAR into Tregs without compromising their innate immunosuppressive qualities mostly through FOXP3+ expression. Although other factors have a role in Treg’s metabolism, the relevance of FOXP3 was unmatched by even CD25, CTLA-4, or CD69 markers.
Based on the evidence presented, the FOXP3+ expression should be maintained or increased to higher expression levels after transduction, independent of transduction efficacy or viability presented in Table 2. Lamarche et al., 2023 demonstrated that loss of FOXP3+, although created from metabolic exhaustion, still led to poor engraftment and function of the CAR Tregs [21]. In fact, Tregs inhibitory receptors—the likes of PD-1, TIM-3, LAG-3, GITR, and 4-1BB—were significantly raised (p < 0.05). Vice versa, Henschel et al., 2023 modified a CAR vector to contribute more FOXP3 expression into Tregs, finding that the CAR Tregs had continued stability and growth even under severe inflammatory, limited IL-2, and acidified conditions [34].
Table 2.
CAR design, transduction efficiency, and in vitro phenotypic analysis (see end of document).
The observed reduction in FOXP3 expression may be a cumulative effect of various stages in the CAR-Treg manufacturing process, including ex vivo expansion, post-transduction modifications, and functional assessment. Currently, there is no established consensus regarding the optimal time point for cellular characterization, whether it should be performed during upstream manufacturing or immediately prior to clinical administration. From a quality control and standardization perspective, earlier characterization is preferable. However, the temporal constraints inherent to the clinical deployment of CAR-Tregs may limit the feasibility of comprehensive phenotypic and functional validation within the available timeframe.
3.5. Strength and Compatibility of Co-Stimulatory Domain
In the overall design of a CAR (chimeric antigen receptor), setting aside the selection of the binding domain and the common intracellular signalling domain CD3ζ, there are well-documented differences between the two main co-stimulatory domains used: CD28 and 4-1BB. To further explain, the 4-1BB exerts a weaker but persistent activation signal compared to CD28, which is rapid and short-lived [46]. This has not been conveyed as the consequence of CD28, which is also complicit in the early exhaustion of Tregs by the accelerated activation through hypersensitized receptors or lowered activation threshold and increased cytokine production [47]; albeit, the moderately weaker 4-1BB could retard or hinder the timely activation of the signal transduction pathway, incapacitating Treg’s suppressive role. Boroughs et al., 2019 verified this comparison in a controlled study, determining CD28-CD3ζ was preferred over 4-1BB-CD3ζ CAR Tregs for acute graft tolerance in a mice xenograft model [24]. Likewise, Imura et al., 2020 found that CD19-CAR carrying CD28 signalling domains were superior to 4-1BB for CAR-T expansion in vitro [19].
Despite known instability under inflammatory conditions, T cell exhaustion or phenotypic drift, CD28 co-stimulation has particularly useful qualities in the context of CAR signalling. Under controlled conditions, CD28 could promote stronger and sustained activation of canonical pathways, including IL-2R-STAT5 or CD28/CTLA-4 and FOXP3 maintenance [48,49,50]. Compared to CD28, 4-1BB co-stimulation favors generalized metabolic programs and effector functions that are less directed towards Treg phenotypes [21,51]. Moreover, previous evidence suggests that the Treg-specific chromatin landscape and transcriptional circuitry could buffer or resist the pro-effector signals generated by CD28, maintaining the stability of the Treg lineage [52]. While concerns around CD28-induced Treg fragility remain valid, the benefits of enhanced potency and persistence in transplantation models certainly outweigh the risk of low immunosuppressive properties, which ultimately lead to graft rejection. Moreover, CAR-Tregs are stringently characterized prior to infusion, ensuring manufacturing consistency (uniform subpopulation) and safety via preventing the risk of effector function conversion from the presence of specific biomarkers.
3.6. Biodistribution of CAR Tregs Key to Early Immune Desensitization
Antigen-specific CAR Tregs homing to the graft early is vital to prevent the influence of polyclonal Tregs from generating an immune response. However, it has become accepted that the CAR Tregs’ circulation also plays a critical role in the success of their immunosuppressive function. The CAR Tregs’ active draining into the immunological centres is a key factor in preventing GvHD and early desensitization of immunity. For CAR Tregs to initiate immune tolerance, especially in the context of transplantation or autoimmune disorders, they must gain access to these immune hubs to modulate the activity of other immune cells. These include APCs, DC, macrophages, and Teffs that could “make or break” successful transplants. Although both expressing and non-CD19-expressing Memory B cells (Bmem) and long-lived plasma cells (LLPC) are the other major culprits of transplant failures, their complete elimination poses the risk of compromised immunity long term [53]. In the Zhang et al., 2023 study, CD19-CAR T cells targeted CD19+ Bmem and LLPC, while the APRIL-CAR T cells targeted both CD19+ and CD19− LLPC. The murine model was able to facilitate greater immune tolerance through combined CAR T (Combo-CART) vs. monotherapy, simultaneously reshaping the humoral immunity that remains functional but also tolerant to the allograft [32].
So far, none of the studies reported any challenges of CAR Tregs homing to the site of graft. Histopathology has revealed pulmonary congestion and hypercellularity of the spleen, consistent with IV administration [33]. From these studies reviewed, population or fragments of CAR Tregs were also found in the lymph nodes and bone marrow, indicative of their immune surveillance properties. Despite only a single administration of the FITC-H-2Dd-mABCAR Tregs, the accumulation of cells in lymphoid organs sustained for 3 weeks, proving long-term immunosuppressive function in the murine transplant model [23].
3.7. Complementary Action with Concomitant Therapy
Unlike CAR T cell therapy, CAR Tregs may not be making it as a first-line therapy in the foreseeable future. This is because first-line treatments must overcome numerous ethical, clinical, logistical, and risk-related hurdles. For example, long-term safety and efficacy data are still being gathered, where reports of adverse side effects are not well documented. On the other hand, first-line treatments should have proven efficacy in a significant number of patients and treatment settings. Tentatively, new therapeutic models such as CAR must be an adjunct therapy to prescribed immunosuppressants. These models will also require heavy scrutiny for underlying damage and/or dampening effects from first-line therapy.
In the latest findings, two studies showed promising results of CAR Tregs applied in tandem with immunosuppressants. Wagner et al., 2022 discovered that the haplo-identical heart transplant survived despite initial <14 days survival without treatment, the following treatment extended to >100 days for A2-CAR Tregs regardless of rapamycin treatment [30]. Supposedly, rapamycin spares Tregs but inhibits Tconv, which exceptionally contributes to its role as an immunosuppressant driver [54]. Rapamycin binds to FKBP12, which blocks the mammalian target of rapamycin complex 1 (mTORC1), leading to cell cycle arrest in T cells. On the other hand, low mTOR activity does not impede Treg activity, as it promotes their stability and enhances their suppressive function. Proics et al., 2023 also describe a CAR Treg model tolerable to tacrolimus, another common immunosuppressant for solid organ transplants [33]. Tacrolimus-FKBP complex specifically inhibits calcineurin, preventing transcription of the nuclear factor of activated T cells (NFAT) necessary for initiating T cell activation [55]. Through distinct pathways, both molecules actively suppress IL-2 production and sensitivity, indirectly retarding Treg persistence. Taken together, the success of these models suggests that patients on concomitant treatment do not need to discontinue their current treatment and may benefit from CAR Tregs simultaneously.
Beyond the scope of this review, a team recently validated a humanized and mutant IL-2 that selectively activates and expands endogenous Tregs in a murine model and cynomolgus monkey successfully [56]. Although both polyclonal and antigen-specific Tregs were expanded, the overall objective of an immunosuppressive microenvironment was achieved nonetheless. Thus, justifying further exploration of the mutant hIL-2. Perhaps, its effect could function synergistically with CAR Tregs to overcome viability issues and selective expansion of Tregs and not T cells in vivo. What is more, this novel cytokine could also be used as first-line therapy to modulate endogenous Tregs and simultaneously adjusted for the subsequent action of CAR Tregs if additional treatment is needed.
Owing to the fact that immune responses are primarily HLA-dependent in organ transplantation, mismatch of donor and recipient HLA alleles results in immediate graft rejection. Interestingly, the discovery of HLA-independent TCR (HIT) receptors is emerging as a potential way to bypass this issue and possibly shine light on mismatched donor transplants [57]. Since HIT receptors recognize a wider range of antigens, beyond the HLA complex, they could be designed to recognize donor-specific antigens universally expressed across different organ types. A recent study demonstrated that HIT receptors are consistently able to recognize target cells with high antigen sensitivity, despite the low abundance of target antigens [58]. Hence, combining HIT receptors with CAR-Tregs could answer how future organ transplants could be performed with less dependence on HLA compatibility.
Although CAR Treg therapy holds great promise, several obstacles continue to impede its clinical application. A key issue lies in preserving the stability and suppressive identity of Tregs following genetic modification, large-scale expansion, and in vivo administration. Under certain conditions, cells may lose FOXP3 expression and transform into a pro-inflammatory phenotype, potentially causing harmful immune responses rather than suppression [34]. The production process is also demanding, requiring compliance with stringent GMP standards to ensure cell purity, safety, and functional consistency—factors that could restrict scalability and raise manufacturing costs [59]. Additional concerns include the risk of off-target activity or unintended immune effects, particularly when donor antigens are expressed in non-target tissues. Furthermore, questions remain regarding the long-term persistence, survival, and regulatory function of CAR Tregs once infused into patients. Nevertheless, progress in areas such as synthetic biology, vector optimization, and strategies to stabilize the Treg phenotype is steadily advancing the field, bringing CAR Treg therapy closer to clinical reality as a targeted solution for preventing allograft rejection.
4. Conclusions
In summary, there is adequate evidence to support the proof-of-concept for CAR Tregs as an immunomodulatory therapy. Since it is in the relatively early stages of a working prototype, more research and experiments are needed. The design parameters of CAR Tregs are abundant but lack validation, making the interpretation subjective. The shortcomings of this review include the inability to determine the large bioprocessing and clinical translation capacity of CAR Tregs. This will be a critical factor in ensuring access to life-saving alternatives when the likelihood of success with organ transplants is low due to a high risk of allograft rejection.
Author Contributions
Conceptualization, R.S. and Y.L.; methodology, R.S. and Y.L.; software, A.M.L.C.; validation, A.M.L.C., R.S. and Y.L.; formal analysis, A.M.L.C.; investigation, A.M.L.C. and Y.L.; resources, Y.L.; data curation, A.M.L.C. and Y.L.; writing—original draft preparation, A.M.L.C.; writing—review and editing, A.M.L.C., R.S. and Y.L.; visualization, R.S. and Y.L.; supervision, R.S., and Y.L.; project administration, R.S. and Y.L.; funding acquisition, Y.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a grant provided by the University Kebangsaan Malaysia (Grant Code: GP-K020811). The granting agencies played no role in the execution of the study review and the submission of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
Author Alvin Man Lung Chan was employed by the company My CytoHealth Sdn. Bhd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ABMR | antibody-mediated rejection |
| ABO | blood group antigen system |
| APC | antigen-presenting cell |
| APRIL | a proliferation-inducing ligand |
| BCMA | B cell maturation antigen |
| CAR | chimeric antigen receptor |
| CTLA | cytotoxic T-lymphocyte-associated protein |
| DC | dendritic cell |
| DTH | delayed-type hypersensitivity |
| EBV | Epstein–Barr virus |
| FKBP | FK506-binding protein |
| GITR | glucocorticoid-induced TNFR-related protein |
| GMP | good manufacturing practice |
| GVHD | graft-versus-host disease |
| HLA | human leukocyte antigen |
| IFN | interferon |
| IL | interleukin |
| IT | intratumoral |
| IV | intravenous |
| LLPC | long-lived plasma cell |
| MHC | major histocompatibility complex |
| mTOR | mammalian target of rapamycin |
| NK | natural killer |
| NSG | NOD scid gamma (mouse strain) |
| NT | no treatment |
| PD-1 | programmed death 1 |
| PTLD | post-transplant lymphoproliferative disorder |
| TCR | T cell receptor |
| TGF-β | transforming growth factor |
| TNF-α | tumor necrosis factor alpha |
| UT | untransduced |
| VCAM | vascular cell adhesion molecule 1 |
References
- Shafiekhani, M.; Shahabinezhad, F.; Tavakoli, Z.; Tarakmeh, T.; Haem, E.; Sari, N.; Nasirabadi, S.; Dehghani, M. Quality of life Associated with Immunosuppressant Treatment Adherence in Liver Transplant Recipients: A Cross-Sectional Study. Front. Pharmacol. 2023, 14, 1051350. [Google Scholar] [CrossRef] [PubMed]
- Allen, U.D.; Preiksaitis, J.K.; AST Infectious Diseases Community of Practice. Post-Transplant Lymphoproliferative Disorders, Epstein-Barr Virus Infection, and Disease in Solid Organ Transplantation: Guidelines from the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13652. [Google Scholar] [CrossRef] [PubMed]
- Rudensky, A.Y. Regulatory T Cells and Foxp3. Immunol. Rev. 2011, 241, 260. [Google Scholar] [CrossRef] [PubMed]
- Rueda, C.M.; Jackson, C.M.; Chougnet, C.A. Regulatory T-Cell-Mediated Suppression of Conventional T-Cells and Dendritic Cells by Different CAMP Intracellular Pathways. Front. Immunol. 2016, 7, 195682. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and Memory T-Cell Differentiation: Implications for Vaccine Development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Mercadante, E.R.; Lorenz, U.M. Breaking Free of Control: How Conventional T Cells Overcome Regulatory T Cell Suppression. Front. Immunol. 2016, 7, 196858. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhuri, R.; Eil, R.L.; Restifo, N.P. The Interplay of Effector and Regulatory T Cells in Cancer. Curr. Opin. Immunol. 2015, 33, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Lysandrou, M.; Kefala, D.; Vinnakota, J.M.; Savvopoulos, N.; Zeiser, R.; Spyridonidis, A. Regulatory T Cell Therapy for Graft-versus-Host Disease. Bone Marrow Transplant. 2025, 60, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Im, K.I.; Song, Y.; Kim, N.; Nam, Y.S.; Jeon, Y.W.; Cho, S.G. Third-Party Regulatory T Cells Prevent Murine Acute Graft-versus-Host Disease. Korean J. Intern. Med. 2017, 33, 980. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Arany, Z.; Baur, J.A.; Epstein, J.A.; June, C.H. CAR T Therapy beyond Cancer: The Evolution of a Living Drug. Nature 2023, 619, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine Release Syndrome and Associated Neurotoxicity in Cancer Immunotherapy. Nat. Rev. Immunol. 2021, 22, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.; Hennessy, C.; Hester, J.; Issa, F. Chimeric Antigen Receptors and Regulatory T Cells: The Potential for HLA-Specific Immunosuppression in Transplantation. Engineering 2022, 10, 30–43. [Google Scholar] [CrossRef]
- Cassano, A.; Chong, A.S.; Alegre, M.-L. Tregs in Transplantation Tolerance: Role and Therapeutic Potential. Front. Transplant. 2023, 2, 1217065. [Google Scholar] [CrossRef] [PubMed]
- Sicard, A.; Lamarche, C.; Speck, M.; Wong, M.; Rosado-Sánchez, I.; Blois, M.; Glaichenhaus, N.; Mojibian, M.; Levings, M.K. Donor-Specific Chimeric Antigen Receptor Tregs Limit Rejection in Naive but Not Sensitized Allograft Recipients. Am. J. Transplant. 2020, 20, 1562–1573. [Google Scholar] [CrossRef] [PubMed]
- Safarzadeh Kozani, P.; Safarzadeh Kozani, P.; Rahbarizadeh, F.; Khoshtinat Nikkhoi, S. Strategies for Dodging the Obstacles in CAR T Cell Therapy. Front. Oncol. 2021, 11, 627549. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T Cell Exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Noyan, F.; Zimmermann, K.; Hardtke-Wolenski, M.; Knoefel, A.; Schulde, E.; Geffers, R.; Hust, M.; Huehn, J.; Galla, M.; Morgan, M.; et al. Prevention of Allograft Rejection by Use of Regulatory T Cells With an MHC-Specific Chimeric Antigen Receptor. Am. J. Transplant. 2017, 17, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Boardman, D.A.; Philippeos, C.; Fruhwirth, G.O.; Ibrahim, M.A.A.; Hannen, R.F.; Cooper, D.; Marelli-Berg, F.M.; Watt, F.M.; Lechler, R.I.; Maher, J.; et al. Expression of a Chimeric Antigen Receptor Specific for Donor HLA Class I Enhances the Potency of Human Regulatory T Cells in Preventing Human Skin Transplant Rejection. Am. J. Transplant. 2017, 17, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Imura, Y.; Ando, M.; Kondo, T.; Ito, M.; Yoshimura, A. CD19-Targeted CAR Regulatory T Cells Suppress B Cell Pathology without GvHD. JCI Insight 2020, 5, e136185. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.M.E.; Migneco, G.; Scott, G.B.; Down, J.; King, S.; Askar, B.; Jennings, V.; Oyajobi, B.; Scott, K.; West, E.; et al. Reovirus-Induced Cell-Mediated Immunity for the Treatment of Multiple Myeloma within the Resistant Bone Marrow Niche. J. Immunother. Cancer 2021, 9, e001803. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, C.; Ward-Hartstonge, K.; Mi, T.; Lin, D.T.S.; Huang, Q.; Brown, A.; Edwards, K.; Novakovsky, G.E.; Qi, C.N.; Kobor, M.S.; et al. Tonic-Signaling Chimeric Antigen Receptors Drive Human Regulatory T Cell Exhaustion. Proc. Natl. Acad. Sci. USA 2023, 120, e2219086120. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, K.G.; Hoeppli, R.E.; Huang, Q.; Gillies, J.; Luciani, D.S.; Orban, P.C.; Broady, R.; Levings, M.K. Alloantigen-Specific Regulatory T Cells Generated with a Chimeric Antigen Receptor. J. Clin. Investig. 2016, 126, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Pierini, A.; Iliopoulou, B.P.; Peiris, H.; Pérez-Cruz, M.; Baker, J.; Hsu, K.; Gu, X.; Zheng, P.-P.; Erkers, T.; Tang, S.-W.; et al. T Cells Expressing Chimeric Antigen Receptor Promote Immune Tolerance. JCI Insight 2017, 2, e92865. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, A.C.; Larson, R.C.; Choi, B.D.; Bouffard, A.A.; Riley, L.S.; Schiferle, E.; Kulkarni, A.S.; Cetrulo, C.L.; Ting, D.; Blazar, B.R.; et al. Chimeric Antigen Receptor Costimulation Domains Modulate Human Regulatory T Cell Function. JCI Insight 2019, 4, e126194. [Google Scholar] [CrossRef] [PubMed]
- Dawson, N.A.J.; Lamarche, C.; Hoeppli, R.E.; Bergqvist, P.; Fung, V.C.W.; McIver, E.; Huang, Q.; Gillies, J.; Speck, M.; Orban, P.C.; et al. Systematic Testing and Specificity Mapping of Alloantigen-Specific Chimeric Antigen Receptors in Regulatory T Cells. JCI Insight 2019, 4, e123672. [Google Scholar] [CrossRef] [PubMed]
- Bézie, S.; Charreau, B.; Vimond, N.; Lasselin, J.; Gérard, N.; Nerrière-Daguin, V.; Bellier-Waast, F.; Duteille, F.; Anegon, I.; Guillonneau, C. Human CD81 Tregs Expressing a MHC-Specific CAR Display Enhanced Suppression of Human Skin Rejection and GVHD in NSG Mice. Blood Adv. 2019, 3, 3522–3538. [Google Scholar] [CrossRef] [PubMed]
- Dawson, N.A.J.; Rosado-Sánchez, I.; Novakovsky, G.E.; Fung, V.C.W.; Huang, Q.; McIver, E.; Sun, G.; Gillies, J.; Speck, M.; Orban, P.C.; et al. Functional Effects of Chimeric Antigen Receptor Co-Receptor Signaling Domains in Human Regulatory T Cells. Sci. Transl. Med. 2020, 12, eaaz3866. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-K.; Han, J.; Piao, H.; Shin, N.; Jang, J.Y.; Yan, J.-J.; Kim, H.; Chung, J.; Yang, J. Anti-C4d Chimeric Antigen Receptor Regulatory T Cells Suppressed Allograft Rejection in ABO-Incompatible Heart Transplantation. Genes Dis. 2022, 9, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Muller, Y.D.; Ferreira, L.M.R.; Ronin, E.; Ho, P.; Nguyen, V.; Faleo, G.; Zhou, Y.; Lee, K.; Leung, K.L.; Skartsis, N.; et al. Precision Engineering of an Anti-HLA-A2 Chimeric Antigen Receptor in Regulatory T Cells for Transplant Immune Tolerance. Front. Immunol. 2021, 12, 686439. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.C.; Ronin, E.; Ho, P.; Peng, Y.; Tang, Q. Anti-HLA-A2-CAR Tregs Prolong Vascularized Mouse Heterotopic Heart Allograft Survival. Am. J. Transplant. 2022, 22, 2237–2245. [Google Scholar] [CrossRef] [PubMed]
- Rosado-Sánchez, I.; Haque, M.; Salim, K.; Speck, M.; Fung, V.C.W.; Boardman, D.A.; Mojibian, M.; Raimondi, G.; Levings, M.K. Tregs Integrate Native and CAR-Mediated Costimulatory Signals for Control of Allograft Rejection. JCI Insight 2023, 8, e167215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Markmann, C.; Yu, M.; Agarwal, D.; Rostami, S.; Wang, W.; Liu, C.; Zhao, H.; Ochoa, T.; Parvathaneni, K.; et al. Immunotherapy Targeting B Cells and Long-Lived Plasma Cells Effectively Eliminates Pre-Existing Donor-Specific Allo-Antibodies. Cell Reports Med. 2023, 4, 101336. [Google Scholar] [CrossRef] [PubMed]
- Proics, E.; David, M.; Mojibian, M.; Speck, M.; Lounnas-Mourey, N.; Govehovitch, A.; Baghdadi, W.; Desnouveaux, J.; Bastian, H.; Freschi, L.; et al. Preclinical Assessment of Antigen-Specific Chimeric Antigen Receptor Regulatory T Cells for Use in Solid Organ Transplantation. Gene Ther. 2023, 30, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Henschel, P.; Landwehr-Kenzel, S.; Engels, N.; Schienke, A.; Kremer, J.; Riet, T.; Redel, N.; Iordanidis, K.; Saetzler, V.; John, K.; et al. Supraphysiological FOXP3 Expression in Human CAR-Tregs Results in Improved Stability, Efficacy, and Safety of CAR-Treg Products for Clinical Application. J. Autoimmun. 2023, 138, 103057. [Google Scholar] [CrossRef] [PubMed]
- Arrieta-Bolaños, E.; Hernández-Zaragoza, D.I.; Barquera, R. An HLA Map of the World: A Comparison of HLA Frequencies in 200 Worldwide Populations Reveals Diverse Patterns for Class I and Class II. Front. Genet. 2023, 14, 866407. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.Y.; Liu, J.; Ren, E.C. Structural and Functional Distinctiveness of HLA-A2 Allelic Variants. Immunol. Res. 2012, 53, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Durgam, S.S.; Rosado-Sánchez, I.; Yin, D.; Speck, M.; Mojibian, M.; Sayin, I.; Hynes, G.E.; Alegre, M.L.; Levings, M.K.; Chong, A.S. CAR Treg Synergy with Anti-CD154 Promotes Infectious Tolerance and Dictates Allogeneic Heart Transplant Acceptance. JCI Insight 2025, 10, e188624. [Google Scholar] [CrossRef] [PubMed]
- Degagné, É.; Donohoue, P.D.; Roy, S.; Scherer, J.; Fowler, T.W.; Davis, R.T.; Reyes, G.A.; Kwong, G.; Stanaway, M.; Vicena, V.L.; et al. High-Specificity CRISPR-Mediated Genome Engineering in Anti-BCMA Allogeneic CAR T Cells Suppresses Allograft Rejection in Preclinical Models. Cancer Immunol. Res. 2024, 12, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.; Berdugo, Y.A.; Tree, T. IL-2-Based Approaches to Treg Enhancement. Clin. Exp. Immunol. 2023, 211, 149. [Google Scholar] [CrossRef] [PubMed]
- Myers, D.R.; Zikherman, J.; Roose, J.P. Tonic Signals: Why Do Lymphocytes Bother? Trends Immunol. 2017, 38, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Mo, F.; McKenna, M.K. Impact of Manufacturing Procedures on CAR T Cell Functionality. Front. Immunol. 2022, 13, 876339. [Google Scholar] [CrossRef] [PubMed]
- Rothe, M.; Suerth, J.; Zychlinski, D.; Meyer, J.; Brendel, C.; Grez, M.; Cavazzana-Calvo, M.; Schambach, A.; Baum, C.; Modlich, U. 487. Large-Scale Expansion of Functional Regulatory T Cells Using a Gas-Permeable Rapid Expansion Cultureware (G-Rex). Mol. Ther. 2012, 20, S189. [Google Scholar] [CrossRef]
- Chakraborty, R.; Mahendravada, A.; Perna, S.K.; Rooney, C.M.; Heslop, H.E.; Vera, J.F.; Savoldo, B.; Dotti, G. Robust and Cost Effective Expansion of Human Regulatory T Cells Highly Functional in a Xenograft Model of Graft-versus-Host Disease. Haematologica 2013, 98, 533. [Google Scholar] [CrossRef] [PubMed]
- Gotti, E.; Tettamanti, S.; Zaninelli, S.; Cuofano, C.; Cattaneo, I.; Rotiroti, M.C.; Cribioli, S.; Alzani, R.; Rambaldi, A.; Introna, M.; et al. Optimization of Therapeutic T Cell Expansion in G-Rex Device and Applicability to Large-Scale Production for Clinical Use. Cytotherapy 2022, 24, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Marín Morales, J.M.; Münch, N.; Peter, K.; Freund, D.; Oelschlägel, U.; Hölig, K.; Böhm, T.; Flach, A.C.; Keßler, J.; Bonifacio, E.; et al. Automated Clinical Grade Expansion of Regulatory T Cells in a Fully Closed System. Front. Immunol. 2019, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic Analysis of Chimeric Antigen Receptor Signaling Reveals Kinetic and Quantitative Differences that Affect Cell Function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef] [PubMed]
- Chinen, T.; Kannan, A.K.; Levine, A.G.; Fan, X.; Klein, U.; Zheng, Y.; Gasteiger, G.; Feng, Y.; Fontenot, J.D.; Rudensky, A.Y. An Essential Role for the IL-2 Receptor in Treg Cell Function. Nat. Immunol. 2016, 17, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Gardner, D.; Jeffery, L.E.; Sansom, D.M. Understanding the CD28/CTLA-4 (CD152) Pathway and Its Implications for Costimulatory Blockade. Am. J. Transplant. 2014, 14, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Golovina, T.N.; Mikheeva, T.; Suhoski, M.M.; Aqui, N.A.; Tai, V.C.; Shan, X.; Liu, R.; Balcarcel, R.R.; Fisher, N.; Levine, B.L.; et al. CD28 Costimulation Is Essential for Human T Regulatory Expansion and Function. J. Immunol. 2008, 181, 2855. [Google Scholar] [CrossRef] [PubMed]
- Lamarthée, B.; Marchal, A.; Charbonnier, S.; Blein, T.; Leon, J.; Martin, E.; Rabaux, L.; Vogt, K.; Titeux, M.; Delville, M.; et al. Transient MTOR Inhibition Rescues 4-1BB CAR-Tregs from Tonic Signal-Induced Dysfunction. Nat. Commun. 2021, 12, 6446. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, E.; Omori, H.; Tabata, Y.; Akieda, Y.; Watanabe, S.; Ogawa, S.; Abe, R. CD28 Co-Stimulation Is Dispensable for the Steady State Homeostasis of Intestinal Regulatory T Cells. Int. Immunol. 2018, 30, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Lightman, S.M.; Utley, A.; Lee, K.P. Survival of Long-Lived Plasma Cells (LLPC): Piecing Together the Puzzle. Front. Immunol. 2019, 10, 965. [Google Scholar] [CrossRef] [PubMed]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted Role of MTOR (Mammalian Target of Rapamycin) Signaling Pathway in Human Health and Disease. Signal Transduct. Target. Ther. 2023, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Vafadari, R.; Kraaijeveld, R.; Weimar, W.; Baan, C.C. Tacrolimus Inhibits NF-KB Activation in Peripheral Human T Cells. PLoS ONE 2013, 8, 60784. [Google Scholar] [CrossRef] [PubMed]
- Efe, O.; Gassen, R.B.; Morena, L.; Ganchiku, Y.; Jurdi, A.A.; Lape, I.T.; Ventura-Aguiar, P.; LeGuern, C.; Madsen, J.C.; Shriver, Z.; et al. A Humanized IL-2 Mutein Expands Tregs and Prolongs Transplant Survival in Preclinical Models. J. Clin. Investig. 2024, 134, e173107. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, S.K.; Daccache, A.; Azzi, J.R. Chimeric Antigen Receptor Treg Therapy in Transplantation. Trends Immunol. 2024, 45, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Mansilla-Soto, J.; Eyquem, J.; Haubner, S.; Hamieh, M.; Feucht, J.; Paillon, N.; Zucchetti, A.E.; Li, Z.; Sjöstrand, M.; Lindenbergh, P.L.; et al. HLA-Independent T Cell Receptors for Targeting Tumors with Low Antigen Density. Nat. Med. 2022, 28, 345. [Google Scholar] [CrossRef] [PubMed]
- Gee, A.P. GMP CAR-T Cell Production. Best Pract. Res. Clin. Haematol. 2018, 31, 126–134. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).