Abstract
Peripheral arterial disease (PAD) is a complex, multifactorial atherosclerotic disease that primarily affects the arteries supplying the lower extremities, causing significant occlusion and reduced blood flow. Several studies have found an association between PAD and both genetic and environmental factors, which play a key role in the disease’s pathophysiology. Therefore, in this review, we describe the main genetic variants associated with plaque initiation, progression, and rupture in PAD. Furthermore, we identify different KEGG pathways involved in the pathological processes of these genes. We also describe gene expressions or transcriptomic studies, particularly in biopsies from patients with PAD. These findings could help identify the functional impact of genetic variants on the disease phenotype and, consequently, allow for the development of appropriate interventions that improve patient prognoses.
1. Introduction
Peripheral artery disease (PAD) is defined as an atherosclerotic arterial disease of the lower extremities [1,2]. It primarily affects arteries outside the aorta and the coronary arteries, predominantly those that supply the lower extremities, causing significant occlusion and reduction in blood flow [2].
The most commonly stenosed regions in the organism are vessels in the lower extremities, particularly the aortoiliac, femoropopliteal, and infrapopliteal vascular segments. PAD may manifest as intermittent claudication, defined as variable post-exertional pain that ceases with rest, or as chronic limb-threatening ischemia (CLTI) in advanced stages of the disease [3]. Notably, a significant number of individuals with PAD are either asymptomatic or have mild symptoms, often dismissing them as a normal part of aging. This perception can downplay the seriousness of PAD and contribute to its misrecognition [4,5,6].
In the evaluation of PAD, systematic lower-extremity examination is required, aiming to identify muscle or skin atrophy, loss of hair, and nail hypertrophy. Auscultation of bruits may be practical, but an exhaustive assessment of peripheral pulses is indispensable [4]. In appropriately selected patients, the ankle–brachial index (ABI) might be useful; this is a noninvasive test that calculates the ratio of systolic blood pressure (SBP) at the ankle to the SBP in the arm [4,6]. This test is used to assess vascular health, especially to detect PAD. A normal ABI value ranges from 1 to 1.4, borderline values are 0.91–0.99, and abnormal values are ≤0.9 for clinical and epidemiological purposes [4,6]. ABI is the standard method for diagnosing PAD; however, it is not reliable in patients with calcified noncompressible vessels, who tend to exhibit ABI values greater than 1.4.
This is commonly seen in individuals with diabetes and those with chronic kidney disease, who depend on the toe–brachial index (TBI) or more sophisticated imaging methods to establish an accurate diagnosis [4]. After diagnosis, additional imaging techniques (computed tomography angiography) may be necessary to properly assess the anatomical distribution of the disease and determine the most appropriate therapeutic approach [5,7].
For instance, individuals with vascular calcifications may exhibit an elevated ABI as a result of arterial stiffness, commonly referred to as medial calcification or Mönckeberg’s arteriosclerosis. This condition is prevalent among patients with diabetes mellitus or end-stage renal disease, rendering blood vessels less compressible and resulting in artificially elevated systolic pressures in the ankle compared to the arm, thus increasing the ABI above the normal range [8,9]. Recently, the Plantar Acceleration Time approach emerged as a reliable and novel technique that could serve as a valuable alternative in diabetic patients, especially when ABI and/or TBI are inconclusive or not applicable [10].
The recognition of PAD is of growing significance in light of the worldwide demographic shift toward an aging population. It is estimated that PAD affects over 230 million people globally. Studies indicate that PAD is slightly more common in younger individuals (40–60 years old) in low- and middle-income countries, but this prevalence rises sharply with age in high-income countries [6,11]. In this regard, by 2040, the elderly population is expected to increase by 22%, which will likely contribute to a significant healthcare burden due to atherosclerotic disease. Although evidence regarding sex differences in PAD is conflicting, men have traditionally been considered more prone to this disease; however, in recent years, there has been a notable rise in PAD cases among women, particularly in high-income countries.
The role of genetics for PAD is well known, as well as additional risk factors including, smoking, type 2 diabetes mellitus (DM2), hypertension, increased cholesterol levels, obesity, and older age [12].
Early efforts to study PAD genetics involved estimating heritability through family-based and twin studies, but these methods often overestimated the actual trait heritability. Subsequently, researchers turned to linkage analysis (LA), a traditional method centered on tracing disease-causing genes within families; however, this approach struggles to examine diseases such as PAD that involve polygenic interactions and environmental factors. As a result, LA has been replaced by association studies, such as candidate gene association studies (CGASs) and genome-wide association studies (GWASs) [7,13,14,15,16,17,18]. Therefore, the early detection of genetic factors associated with PAD is vital for the timely management of therapeutic approaches to reduce the growing healthcare burden. Thus, this narrative review presents a descriptive approach to identify the genetic determinants involved in the pathophysiology of PAD. We include the updated ESC 2024 PAD clinical guidelines to provide specific recommendations for appropriate disease management. We also identify the pathways involved and transcriptomics in genes that converge in PAD. Finally, we describe the relationship of genetic variants associated with PAD with environment, lifestyle, metabolic risk, and drugs.
2. Management of PAD in ESC 2024 Guidelines
The 2024 ESC Guidelines on the Management of Peripheral Arterial and Aortic Diseases (PAAD) underscore PAD as a chronic, progressive atherosclerotic condition associated with a significantly increased risk of cardiovascular events and limb-related complications. The guidelines emphasize the importance of early diagnosis, particularly through ABI screening in high-risk populations [Class I, Level B] as well as the comprehensive management of modifiable risk factors, including dyslipidemia, hypertension, DM2, and smoking [Class I, Level A]. Structured exercise therapy is also recommended as a core component of conservative treatment [Class I, Level A evidence]. Long-term follow-up within a multidisciplinary care framework is advised [Class I, Level C], and revascularization—either endovascular or surgical—should be considered in patients with critical limb-threatening ischemia (CLTI) [Class I, Level B] or non-healing ulcers. The need for revascularization and the risk of major amputation may now be stratified using validated clinical tools such as the WIfI (Wound, Ischemia, and foot Infection) classification system [Class IIa, Level C]. Future predictive models may now be stratified using validated clinical tools such as the WIfI classification system [Class IIa, Level C]. Future predictive models may incorporate genetic profiling to further refine risk stratification and personalize therapeutic decision-making in PAD management [2].
On the other hand, it is important to differentiate the genetic pattern for PAD rather than atherosclerosis. Recent studies emphasize the importance of differentiating PAD-specific genetic signatures from those driving coronary or cerebrovascular disease. Several methodological approaches may help delineate this specificity, including case-only analyses in PAD patients without CAD or stroke, conditional GWAS adjusting for shared atherosclerotic risk, and tissue-specific expression studies in peripheral arteries [19]. Transcriptomic and expression quantitative trait locus analyses suggest that certain variants influence gene expression in limb vasculature more strongly than in coronary or cerebral beds, pointing to localized mechanisms such as impaired angiogenesis and collateral formation [12]. Importantly, PAD progression is shaped by unique biomechanical and hemodynamic factors in peripheral circulation, such as shear stress, collateral vessel development, and ischemia–reperfusion dynamics, which may account for functional differences in the genetic determinants of disease expression. Thus, PAD genetics are characterized by both shared systemic pathways and modifiers with context-specific effects in the peripheral vasculature.
3. PAD Genetic Factors
PAD is not only shaped by lifestyle and aging, but also by genetic makeup. Some people are born with genetic variations that make their blood vessels more vulnerable to inflammation, cholesterol buildup, and clot formation. These inherited factors can silently increase the risk of developing PAD, especially when combined with conditions such as DM2 or high blood pressure. Understanding these genetic influences can help us detect the disease earlier and offer more personalized care to those at higher risk [20,21,22].
Recent genetic discoveries in PAD hold promise for translation into clinical practice. Several susceptibility loci, including SH2B3 and ABO, have been linked to PAD and may eventually be integrated into polygenic risk scores alongside conventional risk factors, thereby improving patient stratification and early detection [12]. Beyond risk prediction, these findings open up the possibility of precision prevention, where individuals at elevated genetic risk could benefit from earlier vascular screening (e.g., ankle–brachial index, Doppler ultrasound) and the more aggressive management of modifiable risk factors [12]. Furthermore, the identification of molecular pathways unique to PAD may provide novel therapeutic targets, paving the way for personalized interventions that differ from strategies typically applied to coronary artery disease or stroke [23]. Figure 1 represents the main associated genes in PAD.
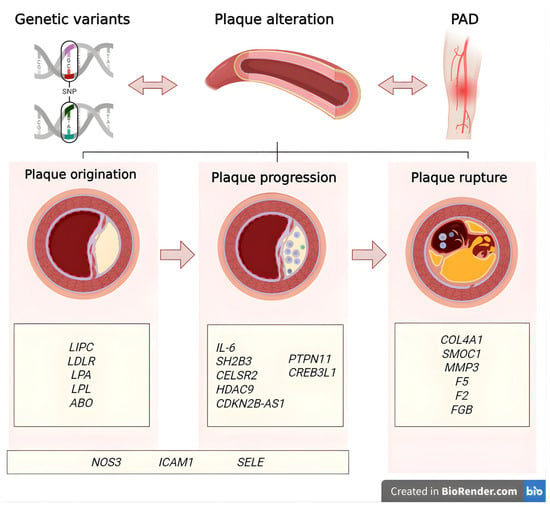
Figure 1.
Gene classification by role in peripheral arterial disease. Genes are categorized by their predominant roles in the pathophysiology of peripheral arterial disease: those involved in plaque origination, progression through inflammatory and vascular remodeling processes, and plaque rupture leading to thrombotic events [24]. Created in BioRender. Villamil C (2025) https://app.biorender.com/illustrations/6368c2ffb888492c0c82d0c8 (accessed on 30 May 2025).
3.1. Variants Associated with Plaque Origination in PAD
The endothelium is a layer of cells that lines the interior of blood and lymphatic vessels and is essential for maintaining cardiovascular health and overall body function. It acts as a dynamic organ that regulates blood flow, inflammation, coagulation, and immune response [25]. A healthy endothelium produces substances that relax blood vessels (vasodilators). When endothelial dysfunction occurs, this ability is compromised, impairing blood flow. Endothelial dysfunction can trigger an inflammatory response in vessel walls, contributing to plaque formation and narrowing of the arteries [5,25].
Therefore, endothelial dysfunction and inflammation are central pathways to the pathogenesis of PAD. The endothelium plays a key role in regulating inflammation; however, in PAD, these processes become dysregulated, reducing nitric oxide (NO) production, increasing oxidative stress, and triggering harmful inflammatory responses. This contributes to vascular stiffness, plaque formation, and calcification. Inflammation then accelerates atherosclerosis progression by promoting arterial damage through cellular activation and the release of pro-inflammatory cytokines [5,25]. Genetic factors that regulate both endothelial function and inflammation are critical in understanding the molecular mechanisms leading to PAD. Variations in genes involved in NO synthesis, oxidative stress management, and inflammatory pathways offer the potential to identify novel markers and targets to restore vascular health and limit irreversible complications [26].
3.1.1. NOS3
The endothelial nitric oxide synthase gene (eNOS or NOS3) is located on chromosome 7q36, which produces NO from L-arginine. NO plays a crucial role in vascular homeostasis by preventing atherogenesis and promoting vasodilation because it promotes the relaxation of blood vessels by reducing blood pressure. Moreover, it inhibits both platelet aggregation and adhesion. Genetic variants such as rs3918226 reduce eNOS activity, impairing NO production and disrupting vascular function. This polymorphism has been linked to vascular conditions such as PAD, where decreased NO bioavailability contributes to vascular dysfunction. Additionally, this variant has also been associated with low ABI values (OR = 2.86, 95% CI = 1.89–4.32, p < 0.0001), highlighting its role in PAD susceptibility [25,27,28].
3.1.2. ICAM1
The ICAM1 gene is located on chromosome 19p13.2 and encodes intercellular adhesion molecule 1 (ICAM-1), a glycoprotein with several functions that actively participates in cell adhesion, migration, and signaling, with a prominent role in the inflammatory response and the immune system. ICAM-1 may be a useful marker for detecting and evaluating inflammatory processes in the body as it mediates leukocyte adhesion during inflammation. The variant rs5498 has been associated with structural changes in ICAM-1, potentially increasing the risk of PAD by 67% among carriers [25,27,28].
3.1.3. SELE
The SELE gene, located on chromosome 1q24.3, encodes E-selectin, a cell adhesion molecule expressed on cytokine-stimulated endothelial cells. E-selectin mediates the adhesion of leukocytes to the vascular lining, facilitating their migration to sites of inflammation, and plays a key role in atherosclerosis [29]. In addition, E-selectin’s involvement in leukocyte recruitment and vascular inflammation underscores its contribution to the development and progression of PAD. A genetic variant in SELE, rs5361, has been associated with an increased risk of PAD [30].
3.1.4. LIPC
The LIPC gene, located on chromosome 15q21.3, encodes hepatic lipase (LIPC), an enzyme primarily expressed in the liver that plays a critical role in lipoprotein metabolism. LIPC hydrolyzes triglycerides and phospholipids in circulating lipoproteins such as high-density lipoprotein (HDL) and intermediate-density lipoprotein (IDL), facilitating their remodeling and clearance. This enzymatic activity is essential for maintaining cholesterol homeostasis and regulating plasma lipid levels. Genetic variants in LIPC can alter hepatic lipase activity, impacting the concentration and composition of atherogenic lipoproteins, further increasing the risk of vascular disease [31]. For instance, Ochoa et al. reported a specific variant (rs1800588) in the LIPC gene associated with elevated levels of these lipoproteins and an increased risk of PAD [30].
3.1.5. LDLR
The LDLR gene, located on 19p13.2, encodes the low-density lipoprotein receptor (LDLR), which mediates the endocytosis of LDL and regulates cholesterol levels. This gene is linked to familial hypercholesterolemia, an autosomal-dominant disorder. The dysfunction or deficiency of LDLR causes LDL to accumulate in circulation, promoting atheroma formation [5,7,32,33]. Variants in the LDLR gene have also been associated with PAD [34].
3.1.6. LPA
The LPA gene, located on chromosome 6q25.3-q26, encodes lipoprotein(a) [Lp(a)]. The pathogenicity of this protein is largely attributed to its Apo(a) subunit, where smaller isoforms promote oxidative reactions, increasing thrombotic risk and inhibiting plasmin activity [35,36]. Elevated Lp(a) serum levels are associated with an increased risk of atherosclerosis and vascular disease [37]. For instance, the variant rs118039278, associated with elevated Lp(a) concentrations, increases thrombotic risk and has been linked to PAD, highlighting its pathogenic role in vascular disease [7,38].
3.1.7. LPL
The LPL gene, located on chromosome 8p21.3, encodes lipoprotein lipase (LPL), a key enzyme responsible for the hydrolysis of triglycerides in circulating lipoproteins such as chylomicrons and VLDL. LPL functions by releasing free fatty acids used for energy production or storage, and it also facilitates the uptake of lipoprotein remnants by interacting with endothelial cell surface proteins. Variants in the LPL gene can disrupt lipid metabolism, leading to elevated plasma triglycerides and contributing to vascular dysfunction. Notably, in GWAS related to PAD, rs322 in the LPL gene has been linked to altered lipid profiles and an increased risk of vascular disease, highlighting its importance in PAD pathophysiology [7,34,35].
3.1.8. ABO
The ABO gene is located on 9q34.2 and encodes a glycosyltransferase that modifies the H antigen on the surface of red blood cells by adding specific sugar residues, thereby determining an individual’s ABO blood type (A, B, AB, or O). Beyond its hematologic role, the ABO glycosyltransferase also modifies glycoproteins and glycolipids expressed in endothelial cells, platelets, and other tissues, influencing intercellular interactions, inflammation, and thrombosis. It is suggested that variants in the ABO gene have been associated with a role in modulating lipid levels and influencing cardiovascular outcomes [39]. In this sense, ABO variant rs505922 has been tied to an increased PAD risk [7].
3.2. Variants Associated with Plaque Progression in PAD
Once plaques have formed, ongoing inflammation and changes in vascular structure promote their growth and complexity. Genetic factors that affect lipid metabolism contribute to fat accumulation inside the arterial walls, which worsens plaque features. At the same time, genes that regulate the structure and repair of the vessel walls may be altered, weakening the artery’s integrity. This can lead to the stiffening, calcification, and abnormal remodeling of the vessel, making plaques larger and more susceptible to rupture. Inflammation continues as immune cells release substances that break down the supporting tissue, further destabilizing plaques. The combined genetic influence on lipid metabolism, vessel structure, and inflammation drives this progression [19].
3.2.1. IL-6
The IL-6 gene, located on chromosome 7p15.3, encodes interleukin 6 (IL-6), a cytokine with important roles in inflammation, immune response, and B cell maturation. Moreover, IL-6 induces inflammatory responses through its receptor, IL6R, and is secreted at sites of acute and chronic inflammation. It plays a critical role in inflammatory diseases such as atherosclerosis, leading the production of other pro-inflammatory cytokines alongside IL-1 to amplify the immune response [40]. Furthermore, elevated levels of the IL-6 receptor, in either its membrane-bound form (IL6R) or soluble form (sIL6R), have been linked to a reduced risk of PAD, suggesting that genetic variants promoting these effects confer a protective profile [41].
3.2.2. SH2B3
The SH2B3 gene, located on chromosome 12q24.12, encodes the SH2B adaptor protein 3 (LNK), a key negative regulator of multiple signaling pathways involved in hematopoiesis, immune function, and vascular homeostasis by attenuating the JAK-STAT, MAPK, and PI3K pathways. When SH2B3 is functioning correctly, it helps limit excessive immune responses and vascular cell proliferation, maintaining a balanced inflammatory environment. However, polymorphisms in SH2B3 can impair this regulatory control, leading to dysregulated cytokine signaling and exaggerated inflammatory responses, promoting vascular injury, endothelial dysfunction, and smooth muscle proliferation; therefore, genetic variants have been linked to an increased susceptibility to PAD [42].
3.2.3. CELSR2
The CELSR2 gene encodes a member of the cadherin superfamily, which plays an important role in cell adhesion and receptor–ligand interactions for maintaining vascular tissue integrity. CELSR2 is particularly important in regulating the structure and function of endothelial cells lining blood vessels. Genetic variations in CELSR2 have been associated with altered levels of lipoprotein-associated phospholipase A2 (Lp-PLA2), an enzyme linked to vascular inflammation. Lp-PLA2 contributes to atherosclerosis by hydrolyzing oxidized phospholipids in LDL generating pro-inflammatory mediators that promote arterial wall inflammation and plaque instability. When CELSR2 function is altered due to polymorphisms, Lp-PLA2 activity can be dysregulated, intensifying inflammatory processes in the arterial walls and accelerating endothelial dysfunction and plaque development [30,43].
3.2.4. HDAC9
The HDAC9 gene is located on chromosome 7p21.1 and encodes a protein that regulates transcription and cell cycle progression through histone deacetylation, influencing immune cell function. According to Klarin et al., genetic variants are linked to PAD, where inflammation contributes to vascular dysfunction and atherosclerosis. HDAC9 may play a role in PAD by modulating inflammatory responses and smooth muscle cell proliferation, key factors in the disease’s progression [7,44].
3.2.5. CDKN2B-AS1
The cyclin-dependent kinase inhibitor 2B antisense RNA 1 (CDKN2B-AS1) gene is located on chromosome 9p21 and encodes the long antisense noncoding RNA in the INK4 Locus (ANRIL) that regulates nearby genes involved in cell cycle control and vascular integrity, such as CDKN2A and CDKN2B. ANRIL interacts with chromatin-modifying complexes to silence these genes, helping maintain cellular balance. When ANRIL is dysregulated, it promotes vascular smooth muscle proliferation, impaired senescence, and chronic vascular inflammation—all key processes in plaque formation and disease progression. Studies have reported that rs1537372 is associated with an increased risk of atherosclerosis and PAD [7,38,45].
3.2.6. PTPN11
This gene is localized on chromosome 12q24.13. PTPN11 encodes a protein that is part of the protein tyrosine phosphatase (PTP) family. These proteins regulate several cellular processes, including cell growth, differentiation, and metabolism. It is expressed in many tissues and plays a role in cell signaling, cell migration, and transcription regulation. Mutations in this gene are linked to Noonan syndrome and acute myeloid leukemia. In addition, Klarin et al. and van Zuydam et al. found an association between the variant rs11066301 in this gene and PAD [7,38].
3.2.7. CREB3L1
This gene is localized in humans on chromosome 11p11.2 and encodes a protein typically found in the endoplasmic reticulum (ER) membrane. When the ER experiences stress, this protein is cleaved, and its transcription factor domain moves to the nucleus where it activates the expression of target genes by binding to specific DNA elements. In the context of PAD, inflammation contributes to vascular injury, and the CREB3L1 gene may indirectly influence these processes by regulating stress-induced inflammatory pathways. Recently, the genetic variant rs7476 in CREB3L1 was associated with PAD [7,38].
3.3. Variants Associated with Plaque Rupture in PAD
In advanced stages, plaques can become unstable and rupture, exposing material that triggers clot formation. Genetic predisposition to increased blood clotting can raise the risk of thrombosis after rupture, leading to sudden blockages and ischemic events. Additionally, genetic factors affecting coagulation, platelet activity, and vessel wall health influence clot formation and stability. The interaction of chronic inflammation, endothelial dysfunction, and a prothrombotic tendency controlled by genetics ultimately shapes the clinical outcome in PAD patients [19].
Observational studies have reported an association between thrombotic events and worse clinical outcomes in patients with PAD. They often exhibit a prothrombotic state, which can exacerbate arterial blockages and increase the risk of complications such as limb ischemia and cardiovascular events. However, it remains unclear whether this relationship is directly causal or whether it might be influenced by reverse causation, where the severity of PAD itself leads to more thrombosis, or by confounding factors such as shared risk factors (e.g., DM2, smoking, inflammation). To clarify this, methods such as Mendelian randomization (MR) have been applied, aiming to assess causal links by using genetic variants as proxies for modifiable exposures. For instance, Klarin et al. used genetic data to investigate the causal role of coagulation factors in PAD risk but found that more research is needed to conclusively determine causality [19].
3.3.1. COL4A1
The COL4A1 gene is found on chromosome 13q34 and produces the alpha-1 chain of type IV collagen (COL4A1), an important building block of basement membranes. These membranes act like supportive scaffolds around blood vessels, helping to keep their structure strong and stable. The COL4A1 protein has special regions that allow it to form a network essential for maintaining the integrity of blood vessels. Beyond just providing support, this protein also helps remodel the walls of arteries and plays a role in controlling inflammation in the local environment [36]. Variants in the COL4A1 gene can weaken these structures, leading to vascular problems such as PAD [7].
3.3.2. SMOC1
The SMOC1 gene is located on chromosome 14q24.2 and encodes SPARC-related modular calcium-binding protein 1, a multi-domain matricellular protein essential for ocular and limb development. Loss-of-function mutations in SMOC1 result in clinical conditions such as microphthalmia with limb anomalies [46]. Beyond developmental roles, GWASs have identified SMOC1 as one of 19 genetic loci significantly associated with PAD, suggesting its potential contribution to vascular pathology. Although the specific molecular mechanisms remain to be elucidated, SMOC1’s known roles in extracellular matrix organization, the modulation of BMP/TGF-β signaling, and vascular calcification provide biological plausibility for its involvement in PAD pathogenesis [47].
3.3.3. MMP3
The MMP3 gene is located on chromosome 11q22.2-q22.3 and encodes matrix metalloproteinase-3 (MMP-3), a secreted enzyme that degrades key components of the extracellular matrix, including fibronectin, laminin, and collagen types III, IV, IX, and X. It also plays essential roles in tissue remodeling, wound repair, and the progression of atherosclerosis [48]. MMP-3 can also activate other MMPs, amplifying extracellular matrix breakdown and influencing vascular remodeling. Furthermore, elevated plasma levels of MMP-3 have been linked to an increased risk of PAD, supporting a causal role in disease progression through enhanced matrix degradation and vascular instability [44]. Genetic variants near or within MMP3, identified in large GWASs, show a statistically significant association with PAD (OR = 1.08; 95% CI: 1.05–1.11; p = 4.37 × 10−9) [19].
3.3.4. F5
The F5 gene, located on chromosome 1q24.2, encodes coagulation Factor V (FV), a critical protein in the blood coagulation cascade. When activated by thrombin, FV acts as a cofactor to accelerate the conversion of prothrombin to thrombin, which ultimately leads to fibrin clot formation. The rs6025 has been found to cause resistance to inactivation by activated protein C and is also strongly associated with an increased risk of venous thrombosis as well as arterial thrombosis, including PAD [7]. The rs6025 variant has also been linked to arterial thrombosis in multiple populations, underscoring its role in thrombophilia. Furthermore, recent studies suggest that additional variants in the F5 gene may contribute to PAD complications such as chronic limb-threatening ischemia and the risk of amputation [30,49].
3.3.5. F2
The F2 gene, located on chromosome 11p11.2, encodes prothrombin, a precursor protein that is converted into thrombin, a serine protease critical for blood clot formation and maintaining vascular integrity. Thrombin plays multiple roles, including the conversion of fibrinogen to fibrin, the activation of platelets to promote aggregation, and involvement in tissue repair and angiogenesis. The rs1799963 variant in the F2 gene has been linked to increased prothrombin levels and a heightened risk of thrombosis [50]. In PAD, carriers of the A allele show increased thrombin generation, which contributes to a prothrombotic state, raising the risk of PAD development and worsening vascular damage [49].
3.3.6. FGB
The FGB gene is located on chromosome 4q31.3 and encodes the beta chain of fibrinogen, a glycoprotein essential for blood clot formation. Thrombin cleaves fibrinogen to form fibrin, which is crucial for clotting and vascular repair. Fibrinogen also regulates cell adhesion, vasoconstriction, and immune responses. Variants in FGB can lead to an increased thrombotic risk. In PAD, some variants in this gene raise plasma fibrinogen levels, which elevate the risk of PAD, stroke, and CAD. Fowkes et al., in their assessment of 121 PAD patients from the Edinburgh Artery Study, suggested that genetic variants of this gene are linked to an increased risk of PAD through structural changes in fibrinogen or interactions with nearby genes [51]. Table 1 shows the main genes and genetic variants involved in PAD.

Table 1.
Genetic variants identified in PAD.
To highlight the relevance of the genes described in Table 1 associated with PAD, a KEGG pathway enrichment analysis was performed to represent the pathways involved in these genes’ disease processes. In this regard, at the plaque formation stage, it allowed for the integration of the arginine biosynthesis pathway, the glycosphingolipid pathway, and the fluid shear stress and atherosclerosis pathway. Genes such as NOS3, ABO, and SELE are involved in these pathways (Figure 2a).
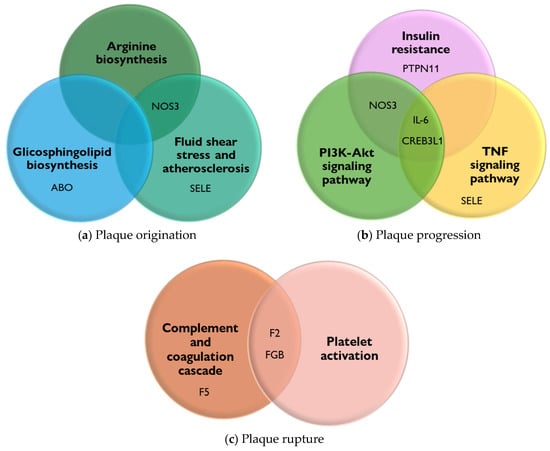
Figure 2.
PAD genes and their connection to KEGG pathways. The PAD-associated genes involved in the origin, progression, and rupture of plaque were scrutinized using ExpressAnalyst (a web-based platform) for functional enrichment analysis to KEGG pathways. The overlap of genes involved in each pathway is highlighted [55]. (a) KEGG pathways interaction of plaque origination. (b) KEGG pathways interaction of plaque progression. (c) KEGG pathways interaction of plaque rupture.
The importance of these pathways is as follows:
- (1).
- Arginine biosynthesis produces NO, a molecule that relaxes and dilates blood vessels, improving blood flow. In addition, L-arginine may improve walking ability and blood flow in these patients [56,57].
- (2).
- The biosynthesis of glycosphingolipids and their metabolites, when altered, can lead to their accumulation in tissues. This accumulation has been linked to the development and progression of PAD. It is suggested that alterations in sphingolipid metabolism contribute to the cellular and tissue damage that occurs during the atherosclerotic process [58].
- (3).
- Fluid shear stress and atherosclerosis, due to the force exerted by the constant flow of blood on the walls of blood vessels, play an important role in atherogenesis by altering the integrity of the endothelium, increasing its permeability, and allowing the entry of lipoproteins and inflammatory cells, which initiates the process of plaque formation and thrombi in the arteries and causes PAD [59].
Moreover, the pathways related to the stage of plaque progression include the PI3K-Akt signaling pathway, TNF signaling pathway, and insulin resistance pathway. Genes such as NOS3, IL-6, CREB3L1, PTPN11, and SELE are involved in these pathways (Figure 2b). The relevance of these pathways is as follows:
- (1).
- The PI3K-Akt signaling pathway is involved in regulating inflammation and oxidative stress, both of which are key factors in the pathogenesis of PAD [60].
- (2).
- The TNF signaling pathway promotes oxidative stress and decreases the bioavailability of NO, a crucial vasodilator, contributing to endothelial dysfunction. Furthermore, the TNF signaling pathway impacts vascular remodeling processes, leading to structural changes in the arteries of patients [61].
- (3).
- The insulin resistance pathway causes vascular damage through endothelial dysfunction via the inhibition of NO production. In addition, insulin resistance can activate pro-inflammatory molecular pathways, such as the MAP kinase (MAPK) pathway, which contribute to the disease [62].
Finally, the pathways related to the plaque rupture stage allow for the integration of complement activation, coagulation cascade, and platelet activation, which are involved in exacerbating the inflammatory process, which, together with endothelial dysfunction promotes thrombosis. Genes such as F5, F2, and FGB are involved in these pathways (Figure 2c) [63,64].
In parallel, we describe gene expression or transcriptomic studies that are crucial for the study of PAD. These studies are relevant as they demonstrate how changes in transcriptional activity could contribute to disease and reveal information not available at the genomic level. It is important to note that studies of this nature at the tissue level are scarce in PAD.
In this sense, transcriptomic and proteomic analyses performed in humans on gastrocnemius muscle biopsies from patients with PAD and control participants without PAD showed several enriched pathways, including those related to hypoxia, such as phosphatase and tensin homolog (PTEN), phosphoinositide 3-kinase (PI3K), and mitogen-activated protein kinase (MAPK) signaling [65].
Furthermore, WNT, Hedgehog, and Notch are among the key signaling pathways involved in the repair of damage caused by chronic hypoxia or ischemia/reperfusion. In this regard, a prolonged hypoxic environment in muscle is associated with induced mitochondrial damage, reduced ATP production, and the stimulation of inflammatory responses, including NF-κB activation [65]. It has also been shown that samples from patients with PAD are characterized by increased inflammation and decreased glycolysis compared to control participants without PAD [66].
On the other hand, transcriptomic studies in human gastrocnemius muscle biopsies showed a global overexpression of genes involved in stress response, autophagy, hypoxia, and muscle atrophy, as well as deficiencies in angiogenic protein signaling, response to misfolded proteins, and nerve repair, which could contribute to poor limb function in PAD [66,67]. Each of these mechanisms has potential for future research to develop effective interventions for the treatment of PAD.
4. Discussion
This review article involved recent contributions to PAD research from a genetics perspective.
All organisms are susceptible to variations in the DNA sequence, leading to genetic diversity. Genetic variants occur in different regions of genes, indicating that their product (a protein) will undergo structural or functional changes. Some diseases arise from mutations in a single gene, known as monogenic disorders, which follow Mendelian inheritance patterns. However, several diseases are polygenic, meaning that they are influenced by multiple genes, and they do not strictly adhere to inheritance. In fact, when both genetic and environmental factors contribute to the development of a disease, they fall under a different category, known as multifactorial or complex diseases [16,17]. For instance, PAD is clearly influenced by genetic variations and traditional metabolic risk factors. Table 2 presents genetic variants in different studies associated with PAD and their relationships with environmental and lifestyle factors, metabolic risks, and drugs.
Table 2.
Genetic variants replicated in multiple studies associated with PAD and their relationship with environmental and lifestyle factors, metabolic risks, and drugs.
Given the relevance of GWAS in PAD, Matsukura et al. identified three novel PAD susceptibility loci at IPO5/RAP2A, EDNRA, and HDAC9 in a Japanese population. In particular, the IPO5/RAP2A locus revealed that rs9584669 conferred the risk of PAD. In addition, this variant showed reduced expression levels of IPO5 when validated through functional studies. This finding represented the first genetic risk evidence for PAD. Furthermore, few GWASs have identified the pathophysiological mechanisms associated with lipid metabolism, highlighting the genetic complexity of the disease [68].
In this regard, genetic association studies such CGASs and GWASs have identified variants associated with vascular diseases including PAD, small artery stroke, and atherosclerosis. Although this approach can reveal potential correlations, further analysis is necessary to validate their significance [14,15]. It is important to acknowledge that genetic heterogeneity is a well-recognized reason for the failure to replicate genetic association findings, including sample size, heterogeneity in the diagnostic criteria/definition of PAD, study power, study design, and the standardization of techniques and determinations. Table 3 depicts the potential causes of the differences observed in various studies.
Table 3.
Factors that influence the failure to replicate findings in genetic association studies.
Nevertheless, recent genetic discoveries in PAD hold promise for translation into clinical practice. Several susceptibility loci, including SH2B3 and ABO, have been linked to PAD and may eventually be integrated into polygenic risk scores alongside conventional risk factors, thereby improving patient stratification and early detection [12]. Beyond risk prediction, these findings open up the possibility of precision prevention, where individuals at elevated genetic risk could benefit from earlier vascular screening (e.g., ankle–brachial index, Doppler ultrasound) and the better management of modifiable risk factors [12]. Furthermore, the identification of molecular pathways unique to PAD may provide novel therapeutic targets, paving the way for personalized interventions that differ from strategies typically applied to coronary artery disease or stroke [23].
5. Perspectives
The integration of genetic insights into PAD management has important clinical implications, particularly for risk stratification and personalized prevention. A proposed clinical pathway begins with the identification of at-risk individuals, combining family history and conventional factors with polygenic risk scores when available [104]. In those at elevated genetic risk, early non-invasive vascular screening (ankle–brachial index, duplex ultrasound) may be warranted, followed by intensified preventive strategies, including optimized lipid, blood pressure, and glycemic control, as well as structured smoking cessation. Genetic insights may also guide patient selection for novel therapies or clinical trials targeting PAD-specific molecular mechanisms. This stepwise integration of genetic risk into vascular practice exemplifies the potential of precision medicine to improve outcomes in PAD [12] (Figure 3).
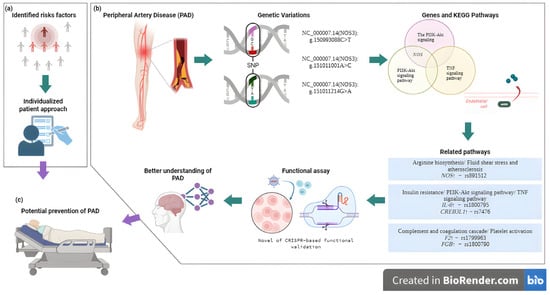
Figure 3.
Overview of the integrative approach connecting polymorphisms, functional assays, and molecular pathways in PAD, providing insights into disease mechanisms and guiding potential preventive and therapeutic approaches. (a) Medical assessment and identification of individual risk factors for PAD. (b) Integration of genetic variation analysis and functional assays to elucidate key genes and pathways involved in PAD pathogenesis. (c) A future application of findings to personalized strategies for PAD prevention [105]. Created in BioRender. Villamil C (2025). https://app.biorender.com/illustrations/6904eab05a145cdd59b382d8 (accessed on 30 May 2025).
Cardiovascular risk modeling focused on PAD prevention as well as pharmacogenomics applied to this context [106]. Monogenic risk models allow for early interventions in patients with genetic variants to prevent adverse outcomes, personalize therapy for comorbidities, and tailor the pathology to the specific mutation [107,108].
Furthermore, additional experimental strategies may be integrated in the research of gene functions by activating or silencing them. Therefore, gene editing in PAD is emerging as a promising but still developing and underexplored new field of research. Gene editing is being investigated through a gene therapy and genetic modification approach. This has made it an effective project for precisely modifying DNA. However, it is still an experimental therapy under development that could allow for the correction of genetic mutations or the silencing of harmful genes in the future, thus restoring vascular function in this group of patients. Although, this technology fits more for functional assay profiles; for example, recent studies have employed induced pluripotent stem cells as a model, utilizing CRISPR-Cas9 technology to introduce genetic variants into cell clones. This strategy might enable the functional validation of specific variants and the elucidation of their role as mechanisms in various complex diseases such as PAD [109,110,111,112].
6. Conclusions
Given its complexity, the future of PAD management lies partly in translational research. The study of genetic variants is crucial in this field, as they serve as a bridge between genetic knowledge and clinical applications. These genetic variants influence susceptibility to disease, response to treatment, and drug efficacy. They also act as markers that allow us to identify individuals at higher risk of developing complex diseases. By knowing an individual’s genetic risk, it is possible to implement prevention and early detection strategies to mitigate the impact of the disease. Despite advances in research, translation into clinical practice still presents challenges in terms of validation and accessibility. Overall, genetic analysis offers transformative opportunities for understanding and managing PAD. Continued efforts to conduct diverse large-scale studies and validate genetic tools in clinical settings will be crucial to fully realize the potential of genetic insights in PAD care in the era of molecular cardiology.
Author Contributions
Conceptualization, M.F.-G., J.M.R.-P., N.P.-H. and L.E.N.-J.; investigation, A.T.-M., J.R.G.-A., L.E.N.-J., M.F.-G., J.M.R.-P., N.P.-H. and V.M.B.-C.; writing—original draft preparation, M.F.-G., L.E.N.-J., J.M.R.-P., N.P.-H., C.V.-C. and V.M.B.-C.; writing—review and editing, A.T.-M., J.R.G.-A., J.M.R.-P., N.P.-H., V.M.B.-C. and C.V.-C.; funding acquisition, M.F.-G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
Open Access funding for this article was supported by Instituto Nacional de Cardiología Ignacio Chávez. Clara Villamil-Castañeda is a doctoral student from the Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM) and has received a CONAHCYT fellowship 1347164. The Graphical Abstract is created in BioRender. Villamil C (2025). https://app.biorender.com/illustrations/6904e76eccc9b84cd34e927f?slideId=2b7e3e45-52c5-4282-ae17-ccc3e0907b7c (accessed 30 May 2025).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Treat-Jacobson, D.; McDermott, M.M.; Bronas, U.G.; Campia, U.; Collins, T.C.; Criqui, M.H.; Gardner, A.W.; Hiatt, W.R.; Regensteiner, J.G.; Rich, K.; et al. Optimal Exercise Programs for Patients with Peripheral Artery Disease: A Scientific Statement from the American Heart Association. Circulation 2019, 139, e10–e33. [Google Scholar] [CrossRef]
- Mazzolai, L.; Teixido-Tura, G.; Lanzi, S.; Boc, V.; Bossone, E.; Brodmann, M.; Bura-Rivière, A.; De Backer, J.; Deglise, S.; Della Corte, A.; et al. 2024 ESC Guidelines for the Management of Peripheral Arterial and Aortic Diseases. Eur. Heart J. 2024, 45, 3538–3700. [Google Scholar] [CrossRef]
- Conte, M.S.; Bradbury, A.W.; Kolh, P.; White, J.V.; Dick, F.; Fitridge, R.; Mills, J.L.; Ricco, J.-B.; Suresh, K.R.; Murad, M.H.; et al. Global Vascular Guidelines on the Management of Chronic Limb-Threatening Ischemia. J. Vasc. Surg. 2019, 69, 3S–125S.e40. [Google Scholar] [CrossRef]
- Campia, U.; Gerhard-Herman, M.; Piazza, G.; Goldhaber, S.Z. Peripheral Artery Disease: Past, Present, and Future. Am. J. Med. 2019, 132, 1133–1141. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primer 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Gornik, H.L.; Aronow, H.D.; Goodney, P.P.; Arya, S.; Brewster, L.P.; Byrd, L.; Chandra, V.; Drachman, D.E.; Eaves, J.M.; Ehrman, J.K.; et al. 2024 ACC/AHA/AACVPR/APMA/ABC/SCAI/SVM/SVN/SVS/SIR/VESS Guideline for the Management of Lower Extremity Peripheral Artery Disease: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2024, 149, e1313–e1410. [Google Scholar] [CrossRef] [PubMed]
- Klarin, D.; Tsao, P.S.; Damrauer, S.M. Genetic Determinants of Peripheral Artery Disease. Circ. Res. 2021, 128, 1805–1817. [Google Scholar] [CrossRef]
- Erzinger, F.L.; Polimanti, A.C.; Pinto, D.M.; Murta, G.; Cury, M.V.; da Silva, R.B.; Biagioni, R.B.; Belckzac, S.Q.; Joviliano, E.E.; de Araujo, W.J.B.; et al. Brazilian Society of Angiology and Vascular Surgery Guidelines on Peripheral Artery Disease. J. Vasc. Bras. 2024, 23, e20230059. [Google Scholar] [CrossRef]
- Bonaca, M.P.; Hamburg, N.M.; Creager, M.A. Contemporary Medical Management of Peripheral Artery Disease. Circ. Res. 2021, 128, 1868–1884. [Google Scholar] [CrossRef] [PubMed]
- Sommerset, J.; Karmy-Jones, R.; Dally, M.; Feliciano, B.; Vea, Y.; Teso, D. Plantar Acceleration Time: A Novel Technique to Evaluate Arterial Flow to the Foot. Ann. Vasc. Surg. 2019, 60, 308–314. [Google Scholar] [CrossRef]
- Horváth, L.; Németh, N.; Fehér, G.; Kívés, Z.; Endrei, D.; Boncz, I. Epidemiology of Peripheral Artery Disease: Narrative Review. Life 2022, 12, 1041. [Google Scholar] [CrossRef]
- Kullo, I.J.; Leeper, N.J. The Genetic Basis of Peripheral Arterial Disease: Current Knowledge, Challenges, and Future Directions. Circ. Res. 2015, 116, 1551–1560. [Google Scholar] [CrossRef]
- Trudsø, L.C.; Andersen, J.D.; Jacobsen, S.B.; Christiansen, S.L.; Congost-Teixidor, C.; Kampmann, M.-L.; Morling, N. A Comparative Study of Single Nucleotide Variant Detection Performance Using Three Massively Parallel Sequencing Methods. PLoS ONE 2020, 15, e0239850. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and Limitations of Genome-Wide Association Studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; de Vries, J.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-Wide Association Studies. Nat. Rev. Methods Primer 2021, 1, 59. [Google Scholar] [CrossRef]
- Zschocke, J.; Byers, P.H.; Wilkie, A.O.M. Mendelian Inheritance Revisited: Dominance and Recessiveness in Medical Genetics. Nat. Rev. Genet. 2023, 24, 442–463. [Google Scholar] [CrossRef]
- Tomaiuolo, R. Genetics and Genomics of Reproductive Medicine. Genes 2021, 12, 1612. [Google Scholar] [CrossRef] [PubMed]
- David, S. A Current Guide to Candidate Gene Association Studies. Trends Genet. TIG 2021, 37, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Klarin, D.; Lynch, J.; Aragam, K.; Chaffin, M.; Assimes, T.L.; Huang, J.; Lee, K.M.; Shao, Q.; Huffman, J.E.; Natarajan, P.; et al. Genome-Wide Association Study of Peripheral Artery Disease in the Million Veteran Program. Nat. Med. 2019, 25, 1274–1279. [Google Scholar] [CrossRef]
- Birney, E. The International Human Genome Project. Hum. Mol. Genet. 2021, 30, R161–R163. [Google Scholar] [CrossRef]
- Belsare, S.; Levy-Sakin, M.; Mostovoy, Y.; Durinck, S.; Chaudhuri, S.; Xiao, M.; Peterson, A.S.; Kwok, P.-Y.; Seshagiri, S.; Wall, J.D. Evaluating the Quality of the 1000 Genomes Project Data. BMC Genom. 2019, 20, 620. [Google Scholar] [CrossRef] [PubMed]
- Byrska-Bishop, M.; Evani, U.S.; Zhao, X.; Basile, A.O.; Abel, H.J.; Regier, A.A.; Corvelo, A.; Clarke, W.E.; Musunuri, R.; Nagulapalli, K.; et al. High-Coverage Whole-Genome Sequencing of the Expanded 1000 Genomes Project Cohort Including 602 Trios. Cell 2022, 185, 3426–3440.e19. [Google Scholar] [CrossRef] [PubMed]
- Leeper, N.J.; Kullo, I.J.; Cooke, J.P. Genetics of Peripheral Artery Disease. Circulation 2012, 125, 3220–3228. [Google Scholar] [CrossRef]
- Villamil, C. Created in BioRender. 2025. Available online: https://app.biorender.com/illustrations/6368c2ffb888492c0c82d0c8 (accessed on 30 May 2025).
- Adams, J.A.; Uryash, A.; Lopez, J.R.; Sackner, M.A. The Endothelium as a Therapeutic Target in Diabetes: A Narrative Review and Perspective. Front. Physiol. 2021, 12, 638491. [Google Scholar] [CrossRef]
- Bartoli-Leonard, F.; Zimmer, J.; Sonawane, A.R.; Perez, K.; Turner, M.E.; Kuraoka, S.; Pham, T.; Li, F.; Aikawa, M.; Singh, S.; et al. NLRP3 Inflammasome Activation in Peripheral Arterial Disease. J. Am. Heart Assoc. 2023, 12, e026945. [Google Scholar] [CrossRef]
- Aimo, A.; Botto, N.; Vittorini, S.; Emdin, M. Polymorphisms in the eNOS Gene and the Risk of Coronary Artery Disease: Making the Case for Genome-Wide Association Studies. Eur. J. Prev. Cardiol. 2019, 26, 157–159. [Google Scholar] [CrossRef]
- Trapé, Á.A.; Rodrigues, J.A.L.; Ferezin, L.P.; Ferrari, G.D.; Lizzi, E.A.d.S.; de Moraes, V.N.; da Silva, R.F.; Zago, A.S.; Brazo-Sayavera, J.; Bueno Júnior, C.R. NOS3 Polymorphisms Can Influence the Effect of Multicomponent Training on Blood Pressure, Nitrite Concentration and Physical Fitness in Prehypertensive and Hypertensive Older Adult Women. Front. Physiol. 2021, 12, 566023. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, S.; Zhu, Z.; Gatt, A.; Liu, J. E-Selectin in Vascular Pathophysiology. Front. Immunol. 2024, 15, 1401399. [Google Scholar] [CrossRef]
- Ochoa Chaar, C.I.; Kim, T.; Alameddine, D.; DeWan, A.; Guzman, R.; Dardik, A.; Grossetta Nardini, H.K.; Wallach, J.D.; Kullo, I.; Murray, M. Systematic Review and Meta-Analysis of the Genetics of Peripheral Arterial Disease. JVS-Vasc. Sci. 2024, 5, 100133. [Google Scholar] [CrossRef]
- Guerra-García, M.T.; Moreno-Macías, H.; Ochoa-Guzmán, A.; Ordoñez-Sánchez, M.L.; Rodríguez-Guillen, R.; Vázquez-Cárdenas, P.; Ortíz-Ortega, V.M.; Peimbert-Torres, M.; Aguilar-Salinas, C.A.; Tusié-Luna, M.T. The -514C>T Polymorphism in the LIPC Gene Modifies Type 2 Diabetes Risk through Modulation of HDL-Cholesterol Levels in Mexicans. J. Endocrinol. Investig. 2021, 44, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Braun, V.; Hantke, K. Lipoproteins: Structure, Function, Biosynthesis. Subcell. Biochem. 2019, 92, 39–77. [Google Scholar] [CrossRef] [PubMed]
- Porntadavity, S.; Jeenduang, N. Structure-Function Relationships of LDL Receptor Missense Mutations Using Homology Modeling. Protein J. 2019, 38, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Poredoš, P.; Šabovič, M.; Božič Mijovski, M.; Nikolajević, J.; Antignani, P.L.; Paraskevas, K.I.; Mikhailidis, D.P.; Blinc, A. Inflammatory and Prothrombotic Biomarkers, DNA Polymorphisms, MicroRNAs and Personalized Medicine for Patients with Peripheral Arterial Disease. Int. J. Mol. Sci. 2022, 23, 12054. [Google Scholar] [CrossRef] [PubMed]
- Lampsas, S.; Xenou, M.; Oikonomou, E.; Pantelidis, P.; Lysandrou, A.; Sarantos, S.; Goliopoulou, A.; Kalogeras, K.; Tsigkou, V.; Kalpis, A.; et al. Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment. Molecules 2023, 28, 969. [Google Scholar] [CrossRef]
- Li, Q.; Wang, C.; Li, W.; Zhang, Z.; Wang, S.; Wupuer, A.; Hu, X.; Wumaier, K.; Zhu, Y.; Li, H.; et al. A Novel Mutation in COL4A1 Gene in a Chinese Family with Pontine Autosomal Dominant Microangiopathy and Leukoencephalopathy. Transl. Stroke Res. 2022, 13, 238–244. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Thorgeirsson, G.; Sulem, P.; Helgadottir, A.; Gylfason, A.; Saemundsdottir, J.; Bjornsson, E.; Norddahl, G.L.; Jonasdottir, A.; Jonasdottir, A.; et al. Lipoprotein(a) Concentration and Risks of Cardiovascular Disease and Diabetes. J. Am. Coll. Cardiol. 2019, 74, 2982–2994. [Google Scholar] [CrossRef]
- van Zuydam, N.R.; Stiby, A.; Abdalla, M.; Austin, E.; Dahlström, E.H.; McLachlan, S.; Vlachopoulou, E.; Ahlqvist, E.; Di Liao, C.; Sandholm, N.; et al. Genome-Wide Association Study of Peripheral Artery Disease. Circ. Genom. Precis. Med. 2021, 14, e002862. [Google Scholar] [CrossRef]
- Ni, X.; Bai, C.; Nie, C.; Qi, L.; Liu, Y.; Yuan, H.; Zhu, X.; Sun, L.; Zhou, Q.; Li, Y.; et al. Identification and Replication of Novel Genetic Variants of ABO Gene to Reduce the Incidence of Diseases and Promote Longevity by Modulating Lipid Homeostasis. Aging 2021, 13, 24655–24674. [Google Scholar] [CrossRef]
- Abubakar, M.; Rasool, H.F.; Javed, I.; Raza, S.; Abang, L.; Hashim, M.M.A.; Saleem, Z.; Abdullah, R.M.; Faraz, M.A.; Hassan, K.M.; et al. Comparative Roles of IL-1, IL-6, IL-10, IL-17, IL-18, 1L-22, IL-33, and IL-37 in Various Cardiovascular Diseases with Potential Insights for Targeted Immunotherapy. Cureus 2023, 15, e42494. [Google Scholar] [CrossRef]
- Levin, M.G.; Klarin, D.; Georgakis, M.K.; Lynch, J.; Liao, K.P.; Voight, B.F.; O’Donnell, C.J.; Chang, K.-M.; Assimes, T.L.; Tsao, P. S et al. A Missense Variant in the IL-6 Receptor and Protection from Peripheral Artery Disease. Circ. Res. 2021, 129, 968–970. [Google Scholar] [CrossRef]
- Alexander, M.R.; Hank, S.; Dale, B.L.; Himmel, L.; Zhong, X.; Smart, C.D.; Fehrenbach, D.J.; Chen, Y.; Prabakaran, N.; Tirado, B.; et al. A Single Nucleotide Polymorphism in SH2B3/LNK Promotes Hypertension Development and Renal Damage. Circ. Res. 2022, 131, 731–747, Erratum in Circ Res. 2023, 132, e95. [Google Scholar] [CrossRef]
- Wu, C.; Zhou, T.; Zhou, Y.; Han, W. Association of Serum Lipoprotein-Associated Phospholipase A2 and A379V Gene Polymorphisms with Carotid Plaques. Genet. Test. Mol. Biomark. 2020, 24, 131–137. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, B.; Huang, S.; Chen, Y.; Yan, J. Causal Association between Circulating Inflammatory Proteins and Peripheral Artery Disease: A Bidirectional Two-Sample Mendelian Randomization Study. Front. Immunol. 2024, 15, 1432041. [Google Scholar] [CrossRef]
- Ali, M.W.; Patro, C.P.K.; Devall, M.; Dampier, C.H.; Plummer, S.J.; Kuscu, C.; Adli, M.; Lai, R.K.; Casey, G. A Functional Variant on 9p21.3 Related to Glioma Risk Affects Enhancer Activity and Modulates Expression of CDKN2B-AS1. Hum. Mutat. 2021, 42, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Reveles, V.; Owen, N.; Ching Chan, B.H.; Toms, M.; Schoenebeck, J.J.; Moosajee, M.; Rainger, J.; Genomics England Research Consortium. Identification of Novel Coloboma Candidate Genes through Conserved Gene Expression Analyses across Four Vertebrate Species. Biomolecules 2023, 13, 293. [Google Scholar] [CrossRef]
- Wang, Y.; Gu, J.; Du, A.; Zhang, S.; Deng, M.; Zhao, R.; Lu, Y.; Ji, Y.; Shao, Y.; Sun, W.; et al. SPARC-Related Modular Calcium Binding 1 Regulates Aortic Valve Calcification by Disrupting BMPR-II/p-P38 Signalling. Cardiovasc. Res. 2022, 118, 913–928. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Xu, M.; Lu, F.; He, Y. Development of Matrix Metalloproteinases-Mediated Extracellular Matrix Remodeling in Regenerative Medicine: A Mini Review. Tissue Eng. Regen. Med. 2023, 20, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Butnariu, L.I.; Gorduza, E.V.; Florea, L.; Țarcă, E.; Moisă, Ș.M.; Tradafir, L.M.; Cojocaru, E.; Luca, A.-C.; Stătescu, L.; Bădescu, M.C. The Genetic Architecture of the Etiology of Lower Extremity Peripheral Artery Disease: Current Knowledge and Future Challenges in the Era of Genomic Medicine. Int. J. Mol. Sci. 2022, 23, 10481. [Google Scholar] [CrossRef]
- Ryu, J.; Rämö, J.T.; Jurgens, S.J.; Niiranen, T.; Sanna-Cherchi, S.; Bauer, K.A.; Haj, A.; Choi, S.H.; Palotie, A.; Daly, M.; et al. Thrombosis Risk in Single- and Double-Heterozygous Carriers of Factor V Leiden and Prothrombin G20210A in FinnGen and the UK Biobank. Blood 2024, 143, 2425–2432. [Google Scholar] [CrossRef]
- Fowkes, F.G.; Connor, J.M.; Smith, F.B.; Wood, J.; Donnan, P.T.; Lowe, G.D. Fibrinogen Genotype and Risk of Peripheral Atherosclerosis. Lancet 1992, 339, 693–696. [Google Scholar] [CrossRef]
- Lasek-Bal, A.; Kula, D.; Urbanek, T.; Puz, P.; Szymszal, J.; Jarzab, M.; Halczok, M.; Cyplinska, R.; Bal, W.; Łabuz-Roszak, B.; et al. The Association of SNPs Located in the CDKN2B-AS1 and LPA Genes with Carotid Artery Stenosis and Atherogenic Stroke. Front. Neurol. 2019, 10, 1170. [Google Scholar] [CrossRef]
- Pavlatos, N.; Kalra, D.K. The Role of Lipoprotein(a) in Peripheral Artery Disease. Biomedicines 2024, 12, 1229. [Google Scholar] [CrossRef] [PubMed]
- Kullo, I.J.; Shameer, K.; Jouni, H.; Lesnick, T.G.; Pathak, J.; Chute, C.G.; de Andrade, M. The ATXN2-SH2B3 Locus Is Associated with Peripheral Arterial Disease: An Electronic Medical Record-Based Genome-Wide Association Study. Front. Genet. 2014, 5, 166. [Google Scholar] [CrossRef]
- Ewald, J.; Zhou, G.; Lu, Y.; Xia, J. Using ExpressAnalyst for Comprehensive Gene Expression Analysis in Model and Non-Model Organisms. Curr. Protoc. 2023, 3, e922. [Google Scholar] [CrossRef]
- Kurhaluk, N.; Tkaczenko, H. L-Arginine and Nitric Oxide in Vascular Regulation-Experimental Findings in the Context of Blood Donation. Nutrients 2025, 17, 665. [Google Scholar] [CrossRef]
- Iannuzzi, A.; Iannuzzo, G.; Sapio, C.; Pauciullo, P.; Iorio, D.; Spampinato, N.; Mancini, M.; Rubba, P. L-Arginine Improves Post-Ischemic Vasodilation in Coronary Heart Disease Patients Taking Vasodilating Drugs. J. Cardiovasc. Pharmacol. Ther. 2001, 6, 121–127. [Google Scholar] [CrossRef]
- Gomez-Larrauri, A.; Larrea-Sebal, A.; Martín, C.; Gomez-Muñoz, A. The Critical Roles of Bioactive Sphingolipids in In-flammation. J. Biol. Chem. 2025, 301, 110475. [Google Scholar] [CrossRef]
- Gaffori, O.; de Wied, D. Further Evidence for a Dissociation of Peripheral and Central Effects of Vasopressin. Psychoneuroen-Docrinology 1985, 10, 439–444. [Google Scholar] [CrossRef]
- Linton, M.F.; Moslehi, J.J.; Babaev, V.R. Akt Signaling in Macrophage Polarization, Survival, and Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 2703. [Google Scholar] [CrossRef] [PubMed]
- Lamb, F.S.; Choi, H.; Miller, M.R.; Stark, R.J. TNFα and Reactive Oxygen Signaling in Vascular Smooth Muscle Cells in Hypertension and Atherosclerosis. Am. J. Hypertens. 2020, 33, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Adeva-Andany, M.M.; Ameneiros-Rodríguez, E.; Fernández-Fernández, C.; Domínguez-Montero, A.; Funcasta-Calderón, R. Insulin Resistance Is Associated with Subclinical Vascular Disease in Humans. World J. Diabetes 2019, 10, 63–77. [Google Scholar] [CrossRef]
- Miceli, G.; Basso, M.G.; Rizzo, G.; Pintus, C.; Tuttolomondo, A. The Role of the Coagulation System in Peripheral Arterial Disease: Interactions with the Arterial Wall and Its Vascular Microenvironment and Implications for Rational Therapies. Int. J. Mol. Sci. 2022, 23, 14914. [Google Scholar] [CrossRef] [PubMed]
- Lebas, H.; Yahiaoui, K.; Martos, R.; Boulaftali, Y. Platelets Are at the Nexus of Vascular Diseases. Front. Cardiovasc. Med. 2019, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.; Oropeza, B.P.; Huang, N.F. Multiomics Analyses of Peripheral Artery Disease Muscle Biopsies. Circ. Res. 2023, 132, 1444–1446. [Google Scholar] [CrossRef]
- Ferrucci, L.; Candia, J.; Ubaida-Mohien, C.; Lyashkov, A.; Banskota, N.; Leeuwenburgh, C.; Wohlgemuth, S.; Guralnik, J.M.; Kaileh, M.; Zhang, D.; et al. Transcriptomic and Proteomic of Gastrocnemius Muscle in Peripheral Artery Disease. Circ. Res. 2023, 132, 1428–1443. [Google Scholar] [CrossRef]
- Pass, C.G.; Palzkill, V.; Tan, J.; Kim, K.; Thome, T.; Yang, Q.; Fazzone, B.; Robinson, S.T.; O’Malley, K.A.; Yue, F.; et al. Single-Nuclei RNA-Sequencing of the Gastrocnemius Muscle in Peripheral Artery Disease. Circ. Res. 2023, 133, 791–809. [Google Scholar] [CrossRef]
- Matsukura, M.; Ozaki, K.; Takahashi, A.; Onouchi, Y.; Morizono, T.; Komai, H.; Shigematsu, H.; Kudo, T.; Inoue, Y.; Kimura, H.; et al. Genome-Wide Association Study of Peripheral Arterial Disease in a Japanese Population. PLoS ONE 2015, 10, e0139262. [Google Scholar] [CrossRef]
- Zhabin, S.; Lazarenko, V.; Azarova, I.; Klyosova, E.; Bashkatov, D.; Kononov, S.; Dolgintsev, M.; Churnosov, M.; Solodilova, M.; Polonikov, A. The Promoter Polymorphism Rs3918226 of the Endothelial Nitric Oxide Synthase Gene as a Novel Susceptibility Marker for Peripheral Artery Disease. Ann. Vasc. Surg. 2024, 108, 557–563. [Google Scholar] [CrossRef]
- Kardia, S.L.; Greene, M.T.; Boerwinkle, E.; Turner, S.T.; Kullo, I.J. Investigating the Complex Genetic Architecture of Ankle-Brachial Index, a Measure of Peripheral Arterial Disease, in Non-Hispanic Whites. BMC Med. Genom. 2008, 1, 16. [Google Scholar] [CrossRef]
- Casas, J.P.; Cavalleri, G.L.; Bautista, L.E.; Smeeth, L.; Humphries, S.E.; Hingorani, A.D. Endothelial Nitric Oxide Synthase Gene Polymorphisms and Cardiovascular Disease: A HuGE Review. Am. J. Epidemiol. 2006, 164, 921–935. [Google Scholar] [CrossRef]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Zintzaras, E.; Zdoukopoulos, N. A Field Synopsis and Meta-Analysis of Genetic Association Studies in Peripheral Arterial Disease: The CUMAGAS-PAD Database. Am. J. Epidemiol. 2009, 170, 1–11. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in Atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Barresi, V.; Signorelli, S.S.; Musso, N.; Anzaldi, M.; Fiore, V.; Alberghina, M.; Condorelli, D.F. ICAM-1 and SRD5A1 Gene Polymorphisms in Symptomatic Peripheral Artery Disease. Vasc. Med. 2014, 19, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Shaker, O.; Zahra, A.; Sayed, A.; Refaat, A.; El-Khaiat, Z.; Hegazy, G.; El-Hindawi, K.; Ay-El Deen, M. Role of ICAM-1 and E-Selectin Gene Polymorphisms in Pathogenesis of PAOD in Egyptian Patients. Vasc. Health Risk Manag. 2010, 6, 9–15. [Google Scholar] [CrossRef]
- Flex, A.; Gaetani, E.; Angelini, F.; Sabusco, A.; Chillà, C.; Straface, G.; Biscetti, F.; Pola, P.; Castellot, J.J.; Pola, R. Pro-Inflammatory Genetic Profiles in Subjects with Peripheral Arterial Occlusive Disease and Critical Limb Ischemia. J. Intern. Med. 2007, 262, 124–130. [Google Scholar] [CrossRef]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, Clinical and Population Relevance of 95 Loci for Blood Lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef]
- Verdier, C.; Ruidavets, J.-B.; Genoux, A.; Combes, G.; Bongard, V.; Taraszkiewicz, D.; Galinier, M.; Elbaz, M.; Ferrières, J.; Martinez, L.O.; et al. Common P2y13 Polymorphisms Are Associated with Plasma Inhibitory Factor 1 and Lipoprotein(a) Concentrations, Heart Rate and Body Fat Mass: The GENES Study. Arch. Cardiovasc. Dis. 2019, 112, 124–134. [Google Scholar] [CrossRef]
- Valdivielso, P.; Ariza, M.J.; de la Vega-Román, C.; González-Alegre, T.; Rioja, J.; Ulzurrun, E.; González-Santos, P. Association of the -250G/A Promoter Polymorphism of the Hepatic Lipase Gene with the Risk of Peripheral Arterial Disease in Type 2 Diabetic Patients. J. Diabetes Complicat. 2008, 22, 273–277. [Google Scholar] [CrossRef]
- Hopewell, J.C.; Clarke, R.; Parish, S.; Armitage, J.; Lathrop, M.; Hager, J.; Collins, R.; Heart Protection Study Collaborative Group. Lipoprotein(a) Genetic Variants Associated with Coronary and Peripheral Vascular Disease but Not with Stroke Risk in the Heart Protection Study. Circ. Cardiovasc. Genet. 2011, 4, 68–73. [Google Scholar] [CrossRef]
- Laschkolnig, A.; Kollerits, B.; Lamina, C.; Meisinger, C.; Rantner, B.; Stadler, M.; Peters, A.; Koenig, W.; Stöckl, A.; Dähnhardt, D.; et al. Lipoprotein (a) Concentrations, Apolipoprotein (a) Phenotypes, and Peripheral Arterial Disease in Three Independent Cohorts. Cardiovasc. Res. 2014, 103, 28–36. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in Atherosclerotic Cardiovascular Disease and Aortic Stenosis: A European Atherosclerosis Society Consensus Statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Erqou, S.; Thompson, A.; Di Angelantonio, E.; Saleheen, D.; Kaptoge, S.; Marcovina, S.; Danesh, J. Apolipoprotein(a) Isoforms and the Risk of Vascular Disease: Systematic Review of 40 Studies Involving 58,000 Participants. J. Am. Coll. Cardiol. 2010, 55, 2160–2167. [Google Scholar] [CrossRef]
- Zhabin, S.N.; Lazarenko, V.A.; Azarova, I.E.; Klyosova, E.Y.; Bykanova, M.A.; Churnosov, M.I.; Solodilova, M.A.; Polonikov, A.V. Lipid-Associated GWAS Loci as Important Markers of the Risk, Severity, and Clinical Course of Peripheral Artery Disease. Expert Rev. Mol. Diagn. 2024, 24, 1033–1044. [Google Scholar] [CrossRef]
- Danielsson, P.; Truedsson, L.; Eriksson, K.F.; Norgren, L. Inflammatory Markers and IL-6 Polymorphism in Peripheral Arterial Disease with and without Diabetes Mellitus. Vasc. Med. 2005, 10, 191–198. [Google Scholar] [CrossRef]
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-Reactive Protein and Other Markers of Inflammation in the Prediction of Cardiovascular Disease in Women. N. Engl. J. Med. 2000, 342, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Libra, M.; Signorelli, S.S.; Bevelacqua, Y.; Navolanic, P.M.; Bevelacqua, V.; Polesel, J.; Talamini, R.; Stivala, F.; Mazzarino, M.C.; Malaponte, G. Analysis of G(-174)C IL-6 Polymorphism and Plasma Concentrations of Inflammatory Markers in Patients with Type 2 Diabetes and Peripheral Arterial Disease. J. Clin. Pathol. 2006, 59, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Tragante, V.; Doevendans, P.A.F.M.; Nathoe, H.M.; van der Graaf, Y.; Spiering, W.; Algra, A.; de Borst, G.J.; de Bakker, P.I.W.; Asselbergs, F.W.; SMART Study Group. The Impact of Susceptibility Loci for Coronary Artery Disease on Other Vascular Domains and Recurrence Risk. Eur. Heart J. 2013, 34, 2896–2904. [Google Scholar] [CrossRef]
- Cluett, C.; McDermott, M.M.; Guralnik, J.; Ferrucci, L.; Bandinelli, S.; Miljkovic, I.; Zmuda, J.M.; Li, R.; Tranah, G.; Harris, T.; et al. The 9p21 Myocardial Infarction Risk Allele Increases Risk of Peripheral Artery Disease in Older People. Circ. Cardiovasc. Genet. 2009, 2, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Willeit, J.; Kiechl, S.; Oberhollenzer, F.; Rungger, G.; Egger, G.; Bonora, E.; Mitterer, M.; Muggeo, M. Distinct Risk Profiles of Early and Advanced Atherosclerosis: Prospective Results from the Bruneck Study. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 529–537. [Google Scholar] [CrossRef]
- Bérard, A.M.; Bedel, A.; Le Trequesser, R.; Freyburger, G.; Nurden, A.; Colomer, S.; Guérin, V.; Vergnes, M.-C.; Becker, F.; Camelot, G.; et al. Novel Risk Factors for Premature Peripheral Arterial Occlusive Disease in Non-Diabetic Patients: A Case-Control Study. PLoS ONE 2013, 8, e37882. [Google Scholar] [CrossRef] [PubMed]
- Reny, J.L.; Alhenc-Gelas, M.; Fontana, P.; Bissery, A.; Julia, P.L.; Fiessinger, J.N.; Aiach, M.; Emmerich, J. The Factor II G20210A Gene Polymorphism, but Not Factor V Arg506Gln, Is Associated with Peripheral Arterial Disease: Results of a Case-Control Study. J. Thromb. Haemost. JTH 2004, 2, 1334–1340. [Google Scholar] [CrossRef]
- Sofi, F.; Lari, B.; Rogolino, A.; Marcucci, R.; Pratesi, G.; Dorigo, W.; Pratesi, C.; Gensini, G.F.; Abbate, R.; Prisco, D. Thrombophilic Risk Factors for Symptomatic Peripheral Arterial Disease. J. Vasc. Surg. 2005, 41, 255–260. [Google Scholar] [CrossRef][Green Version]
- Vazquez, F.; Rodger, M.; Carrier, M.; Le Gal, G.; Reny, J.-L.; Sofi, F.; Mueller, T.; Nagpal, S.; Jetty, P.; Gandara, E. Prothrombin G20210A Mutation and Lower Extremity Peripheral Arterial Disease: A Systematic Review and Meta-Analysis. Eur. J. Vasc. Endovasc. Surg. Off. J. Eur. Soc. Vasc. Surg. 2015, 50, 232–240. [Google Scholar] [CrossRef][Green Version]
- Murakami, T. Atherosclerosis and Arteriosclerosis. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2023, 46, 1810–1811. [Google Scholar] [CrossRef]
- Lee, A.J.; Fowkes, F.G.; Lowe, G.D.; Connor, J.M.; Rumley, A. Fibrinogen, Factor VII and PAI-1 Genotypes and the Risk of Coronary and Peripheral Atherosclerosis: Edinburgh Artery Study. Thromb. Haemost. 1999, 81, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.B.; Connor, J.M.; Lee, A.J.; Cooke, A.; Lowe, G.D.O.; Rumley, A.; Fowkes, F.G. Relationship of the Platelet Glycoprotein PlA and Fibrinogen T/G+1689 Polymorphisms with Peripheral Arterial Disease and Ischaemic Heart Disease. Thromb. Res. 2003, 112, 209–216. [Google Scholar] [CrossRef]
- Aday, A.W.; Duncan, M.S.; Patterson, O.V.; DuVall, S.L.; Alba, P.R.; Alcorn, C.W.; Tindle, H.A.; Creager, M.A.; Bonaca, M.P.; Damrauer, S.M.; et al. Association of Sex and Race with Incident Peripheral Artery Disease Among Veterans with Normal Ankle-Brachial Indices. JAMA Netw. Open 2022, 5, e2240188. [Google Scholar] [CrossRef]
- Barnes, J.A.; Eid, M.A.; Creager, M.A.; Goodney, P.P. Epidemiology and Risk of Amputation in Patients with Diabetes Mellitus and Peripheral Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Nead, K.T.; Olin, J.W.; Cooke, J.P.; Leeper, N.J. Clinical and Socioeconomic Factors Associated with Unrecognized Peripheral Artery Disease. Vasc. Med. 2014, 19, 289–296. [Google Scholar] [CrossRef]
- Rebuffet, C.; Gillois, P.; Joly, M.; Satger, B.; Seinturier, C.; Pernod, G. Evaluation of Socio-Economic Insecurity in Peripheral Artery Disease Patients. J. Med. Vasc. 2021, 46, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Giannoli, J.-M.; Vassault, A.; Carobene, A.; Liaudet, A.P.; Blasutig, I.M.; Dabla, P.K.; Lin, J.; Thomas, A.; Tesser Poloni, J.A.; Meng, Q.H.; et al. Ensuring Internal Quality Control Practices in Medical Laboratories: IFCC Recommendations for Practical Applications Based on ISO 15189:2022. Clin. Chim. Acta Int. J. Clin. Chem. 2025, 571, 120240. [Google Scholar] [CrossRef]
- Wassel, C.L.; Loomba, R.; Ix, J.H.; Allison, M.A.; Denenberg, J.O.; Criqui, M.H. Family History of Peripheral Artery Disease Is Associated with Prevalence and Severity of Peripheral Artery Disease: The San Diego Population Study (SDPS). J. Am. Coll. Cardiol. 2011, 58, 1386–1392. [Google Scholar] [CrossRef]
- Villamil, C. Created in BioRender. 2025. Available online: https://app.biorender.com/illustrations/6904eab05a145cdd59b382d8 (accessed on 30 May 2025).
- Paynter, N.P.; Ridker, P.M.; Chasman, D.I. Are Genetic Tests for Atherosclerosis Ready for Routine Clinical Use? Circ. Res. 2016, 118, 607–619. [Google Scholar] [CrossRef]
- Mitsis, A.; Khattab, E.; Kyriakou, M.; Sokratous, S.; Sakellaropoulos, S.G.; Tzikas, S.; Kadogou, N.P.E.; Kassimis, G. Genomic and Precision Medicine Approaches in Atherosclerotic Cardiovascular Disease: From Risk Prediction to Therapy-A Review. Biomedicines 2025, 13, 1723. [Google Scholar] [CrossRef]
- Morales, A.; Goehringer, J.; Sanoudou, D. Evolving Cardiovascular Genetic Counseling Needs in the Era of Precision Medicine. Front. Cardiovasc. Med. 2023, 10, 1161029. [Google Scholar] [CrossRef] [PubMed]
- Khachigian, L.M.; Varcoe, R.L.; Suoranta, T.; Laham-Karam, N.; Ylä-Herttuala, S. Gene Therapeutic Strategies for Peripheral Artery Disease and New Opportunities Provided by Adeno-Associated Virus Vectors. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 836–851. [Google Scholar] [CrossRef]
- Li, J.-M.; Huang, J.; Liao, Y.; Hu, T.; Wang, C.-L.; Zhang, W.-Z.; Huang, C.-W. Gene and RNA Editing: Revolutionary Approaches to Treating Diseases. MedComm 2025, 6, e70389. [Google Scholar] [CrossRef]
- Forster, R.; Liew, A.; Bhattacharya, V.; Shaw, J.; Stansby, G. Gene Therapy for Peripheral Arterial Disease. Cochrane Database Syst. Rev. 2018, 10, CD012058. [Google Scholar] [CrossRef]
- Wahbeh, M.H.; Boyd, R.J.; Yovo, C.; Rike, B.; McCallion, A.S.; Avramopoulos, D. A Functional Schizophrenia-Associated Genetic Variant near the TSNARE1 and ADGRB1 Genes. HGG Adv. 2024, 5, 100303. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).