
DNA Methylation Signatures in Paired Placenta and Umbilical Cord Samples: Relationship with Maternal Pregestational Body Mass Index and Offspring Metabolic Outcomes

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population and Ethics
2.2. Mother–Newborn Assessments
2.3. Umbilical Cord and Placenta Samples
2.4. Children Assessments
2.5. Genome-Wide DNA Methylation Array
2.6. CpG Methylation by Pyrosequencing
2.7. Gene Expression by Real-Time PCR
2.8. Statistical Methods
3. Results
3.1. Maternal and Offspring Characteristics
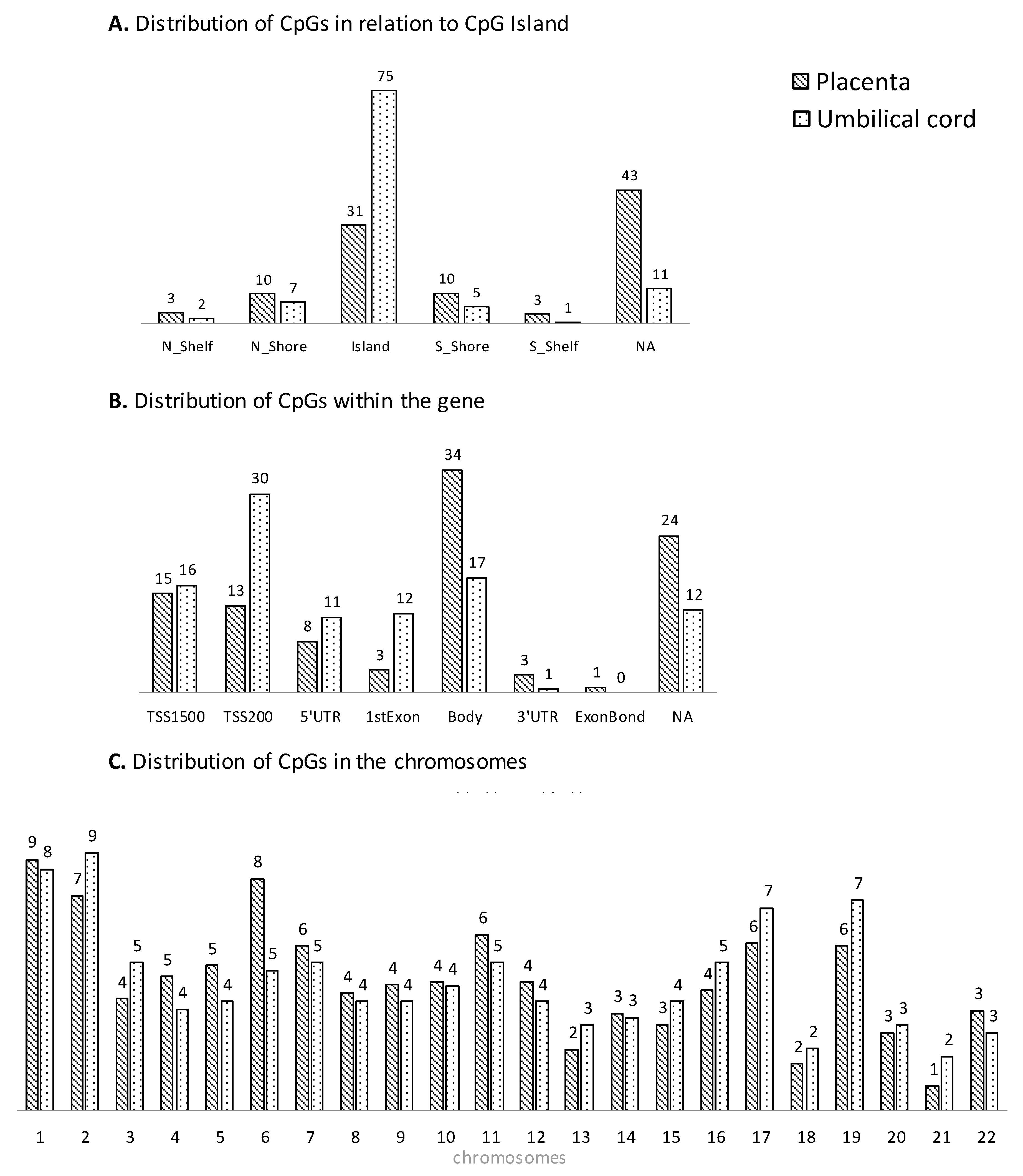
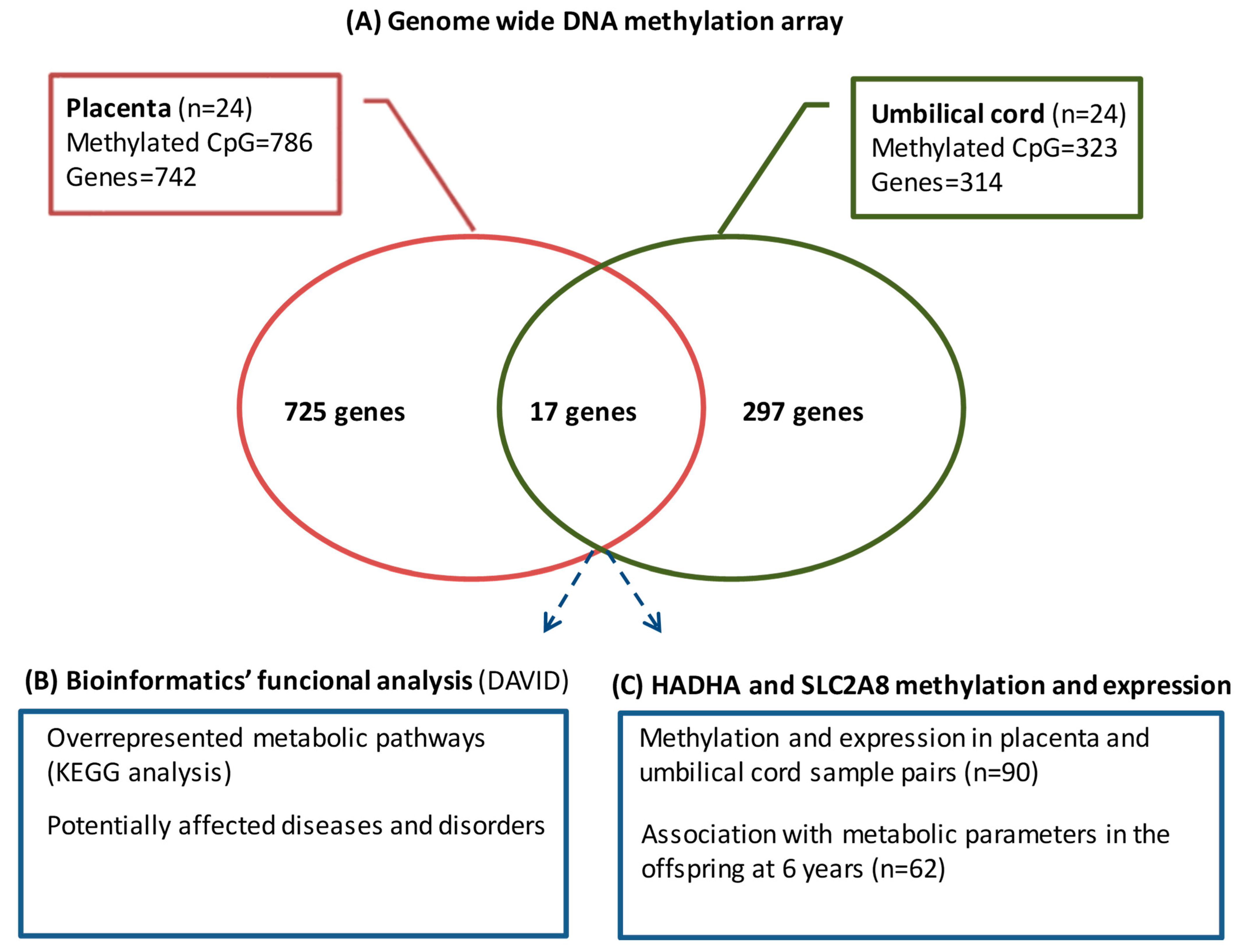
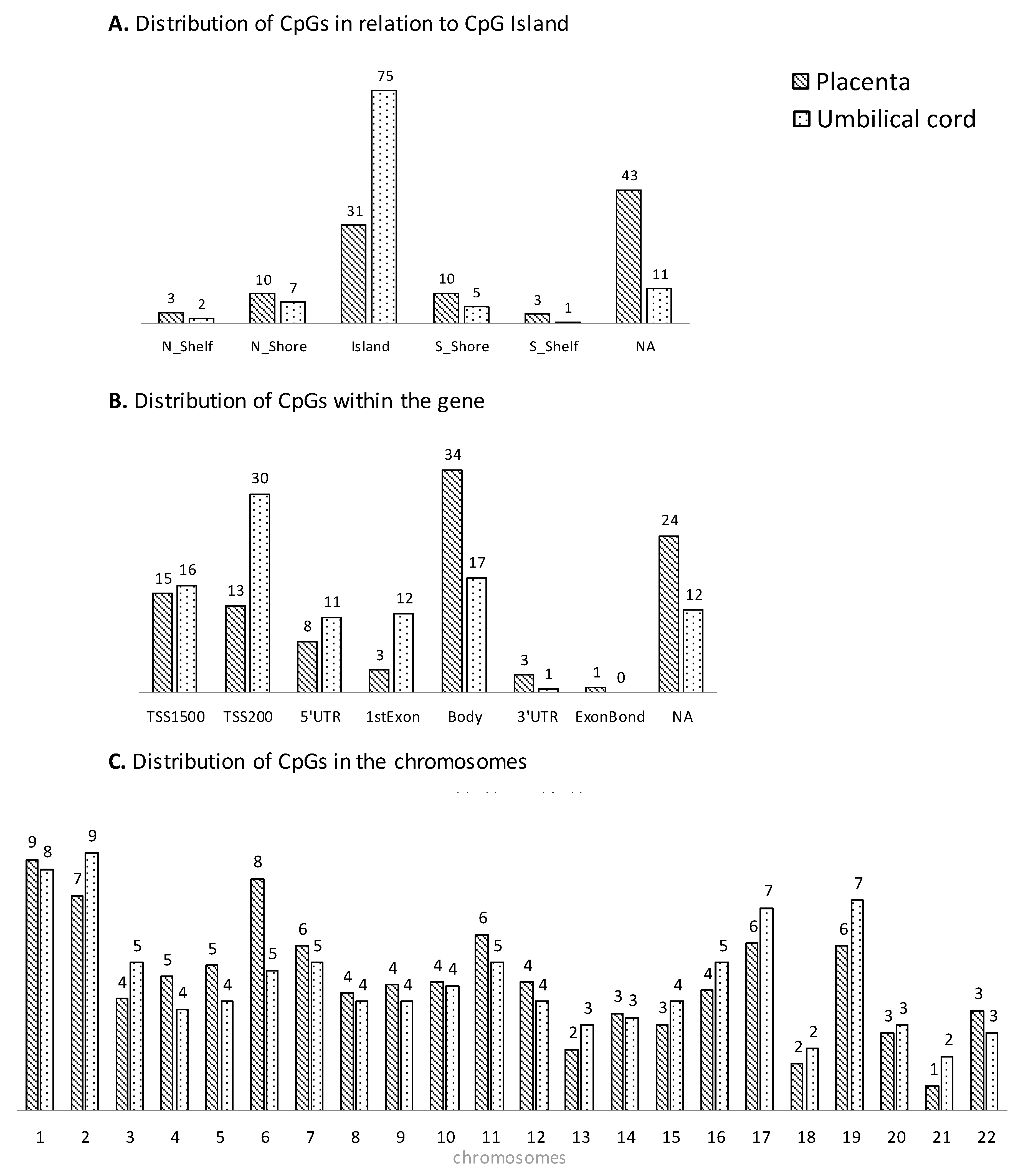
3.2. Genome-Wide DNA Methylation Array
3.3. Bioinformatics Functional Analysis
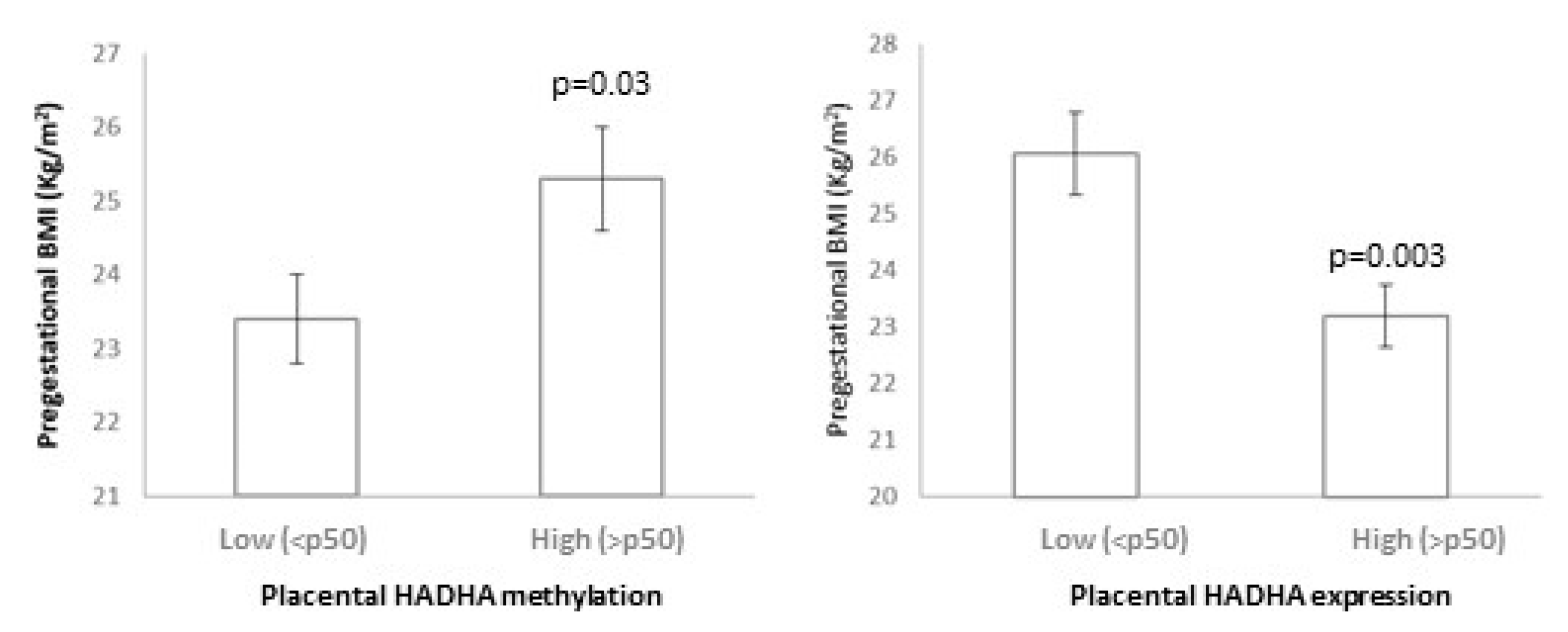
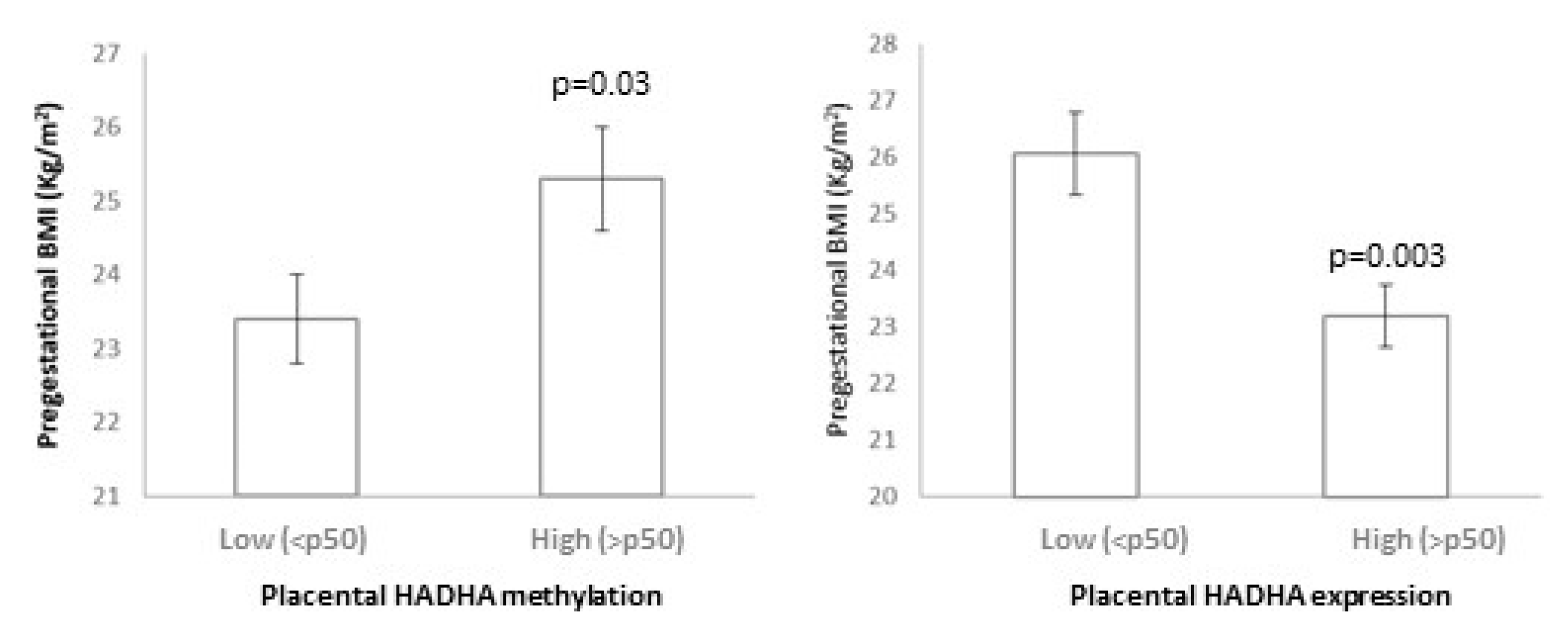
3.4. HADHA and SLC2A8 Methylation and Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, G.D.; Steer, C.; Leary, S.; Ness, A. Is there an intrauterine influence on obesity? Evidence from parent child associations in the Avon Longitudinal Study of Parents and Children (ALSPAC). Arch. Dis. Child. 2007, 92, 876–880. [Google Scholar] [CrossRef]
- Warner, M.J.; Ozanne, S.E. Mechanisms involved in the developmental programming of adulthood disease. Biochem. J. 2010, 427, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Poston, L. Maternal obesity, gestational weight gain and diet as determinants of offspring long term health. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, R.; Steegers, E.A.; Duijts, L.; Felix, J.F.; Hofman, A.; Franco, O.H.; Jaddoe, V.W. Childhood cardiometabolic outcomes of maternal obesity during pregnancy: The Generation R Study. Hypertension 2014, 63, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.; Luyten, L.J.; Dockx, Y.; Provost, E.; Madhloum, N.; De Boever, P.; Neven, K.Y.; Sassi, F.; Sleurs, H.; Vrijens, K.; et al. Association Between Maternal Prepregnancy Body Mass Index and Anthropometric Parameters, Blood Pressure, and Retinal Microvasculature in Children Age 4 to 6 Years. JAMA Netw. Open 2020, 3, e204662. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.C.; Roberts, J.; Catov, J.; Krishnamurthy, R.; Shypailo, R.; Bacha, F. Mother’s pre-pregnancy BMI is an important determinant of adverse cardiometabolic risk in childhood. Pediatr. Diabetes 2015, 16, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Sharp, G.C.; Lawlor, D.A.; Richmond, R.C.; Fraser, A.; Simpkin, A.; Suderman, M.; Shihab, H.A.; Lyttleton, O.; McArdle, W.; Ring, S.M.; et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: Findings from the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol. 2015, 44, 1288–1304. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Wahl, S.; Drong, A.; Lehne, B.; Loh, M.; Scott, W.R.; Kunze, S.; Tsai, P.-C.; Ried, J.S.; Zhang, W.; Yang, Y.; et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017, 541, 81–86. [Google Scholar] [CrossRef]
- Lima, R.S.; de Assis Silva Gomes, J.; Moreira, P.R. An overview about DNA methylation in childhood obesity: Characteristics of the studies and main findings. J. Cell. Biochem. 2020, 121, 3042–3057. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, H.; Snieder, H.; Su, S.; Munn, D.; Harshfield, G.; Maria, B.L.; Dong, Y.; Treiber, F.; Gutin, B.; et al. Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med. 2010, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Su, S.; Barnes, V.A.; De Miguel, C.; Pollock, J.; Ownby, D.; Shi, H.; Zhu, H.; Snieder, H.; Wang, X. A genome-wide methylation study on obesity: Differential variability and differential methylation. Epigenetics 2013, 8, 522–533. [Google Scholar] [CrossRef]
- García-Cardona, M.C.; Huang, F.; García-Vivas, J.M.; López-Camarillo, C.; Navarro, B.E.d.R.; Olivos, E.N.; Hong-Chong, E.; Bolaños-Jiménez, F.; Marchat, L.A. DNA methylation of leptin and adiponectin promoters in children is reduced by the combined presence of obesity and insulin resistance. Int. J. Obes. 2014, 38, 1457–1465. [Google Scholar] [CrossRef]
- Engel, S.M.; Joubert, B.R.; Wu, M.C.; Olshan, A.F.; Håberg, S.E.; Ueland, P.M.; Nystad, W.; Nilsen, R.M.; Vollset, S.E.; Peddada, S.D.; et al. Neonatal genome-wide methylation patterns in relation to birth weight in the Norwegian Mother and Child Cohort. Am. J. Epidemiol. 2014, 179, 834–842. [Google Scholar] [CrossRef]
- Michels, K.B.; Harris, H.R.; Barault, L. Birthweight, maternal weight trajectories and global DNA methylation of LINE-1 repetitive elements. PLoS ONE 2011, 6, e25254. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Hivert, M.-F.; Perron, P.; Poirier, P.; Guay, S.-P.; Brisson, D.; Bouchard, L. IGF2 DNA methylation is a modulator of newborn’s fetal growth and development. Epigenetics 2012, 7, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Lesseur, C.; Armstrong, D.A.; Paquette, A.G.; Koestler, D.C.; Padbury, J.F.; Marsit, C.J. Tissue-specific Leptin promoter DNA methylation is associated with maternal and infant perinatal factors. Mol. Cell. Endocrinol. 2013, 381, 160–167. [Google Scholar] [CrossRef]
- Kochmanski, J.; Goodrich, J.M.; Peterson, K.E.; Lumeng, J.C.; Dolinoy, D.C. Neonatal bloodspot DNA methylation patterns are associated with childhood weight status in the Healthy Families Project. Pediatr. Res. 2019, 85, 848–855. [Google Scholar] [CrossRef]
- Martin, C.L.; Jima, D.; Sharp, G.C.; McCullough, L.E.; Park, S.S.; Gowdy, K.M.; Skaar, D.; Cowley, M.; Maguire, R.L.; Fuemmeler, B.; et al. Maternal pre-pregnancy obesity, offspring cord blood DNA methylation, and offspring cardiometabolic health in early childhood: An epigenome-wide association study. Epigenetics 2019, 14, 325–340. [Google Scholar] [CrossRef]
- Agha, G.; Hajj, H.; Rifas-Shiman, S.L.; Just, A.C.; Hivert, M.-F.; Burris, H.H.; Lin, X.; Litonjua, A.A.; Oken, E.; DeMeo, D.L.; et al. Birth weight-for-gestational age is associated with DNA methylation at birth and in childhood. Clin. Epigenet. 2016, 8, 118. [Google Scholar] [CrossRef]
- Relton, C.L.; Groom, A.; Pourcain, B.S.; Sayers, A.E.; Swan, D.C.; Embleton, N.D.; Pearce, M.S.; Ring, S.M.; Northstone, K.; Tobias, J.H.; et al. DNA methylation patterns in cord blood DNA and body size in childhood. PLoS ONE 2012, 7, e31821. [Google Scholar] [CrossRef]
- Simpkin, A.J.; Suderman, M.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Tilling, K.; Davey Smith, G.; Relton, C.L. Longitudinal analysis of DNA methylation associated with birth weight and gestational age. Hum. Mol. Genet. 2015, 24, 3752–3763. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Mantay, M.; Brannan, C.; Griffiths, S. Placental Tissues as Biomaterials in Regenerative Medicine. Biomed. Res. Int. 2022, 2022, 6751456. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Morriseau, T.S.; Kereliuk, S.M.; Doucette, C.A.; Wicklow, B.A.; Dolinsky, V.W. Maternal obesity, diabetes during pregnancy and epigenetic mechanisms that influence the developmental origins of cardiometabolic disease in the offspring. Crit. Rev. Clin. Lab. Sci. 2018, 55, 71–101. [Google Scholar] [CrossRef]
- Robinson, W.P.; Price, E.M. The human placental methylome. Cold Spring Harb. Perspect. Med. 2015, 5, a023044. [Google Scholar] [CrossRef]
- Pogozhykh, O.; Prokopyuk, V.; Figueiredo, C.; Pogozhykh, D. Placenta and Placental Derivatives in Regenerative Therapies: Experimental Studies, History, and Prospects. Stem. Cells Int. 2018, 2018, 4837930. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.C.; Van De Walle, A.; Chang, J.; Juran, C.; McFetridge, P.S. Human Perinatal-Derived Biomaterials. Adv. Healthc. Mater. 2017, 6, 1700345. [Google Scholar] [CrossRef]
- Carrascosa, A.; Fernandez, J.M.; Fernandez, C.; Ferrandez, A.; Lopez-Siguero, J.P.; Sanchez, E.; Sobradillo, B.; Yeste, D. Spanish growth studies 2008. New anthropometric standards. Endocrinol. Nutr. 2008, 55, 484–506. [Google Scholar] [CrossRef]
- Gómez-Vilarrubla, A.; Mas-Parés, B.; Carreras-Badosa, G.; Xargay-Torrent, S.; Prats-Puig, A.; Bonmatí-Santané, A.; de Zegher, F.; Ibañez, L.; López-Bermejo, A.; Bassols, J. Placental epigenetic marks related to gestational weight gain reveal potential genes associated with offspring obesity parameters. Obesity 2023, 31, 1903–1912. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Shrestha, D.; Ouidir, M.; Workalemahu, T.; Zeng, X.; Tekola-Ayele, F. Placental DNA methylation changes associated with maternal prepregnancy BMI and gestational weight gain. Int. J. Obes. 2020, 44, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Mitsuya, K.; Parker, A.N.; Liu, L.; Ruan, J.; Vissers, M.C.M.; Myatt, L. Alterations in the placental methylome with maternal obesity and evidence for metabolic regulation. PLoS ONE 2017, 12, e0186115. [Google Scholar] [CrossRef] [PubMed]
- Ghildayal, N.; Fore, R.; Lutz, S.M.; Cardenas, A.; Perron, P.; Bouchard, L.; Hivert, M.-F. Early-pregnancy maternal body mass index is associated with common DNA methylation markers in cord blood and placenta: A paired-tissue epigenome-wide association study. Epigenetics 2022, 17, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Opsahl, J.O.; Moen, G.-H.; Qvigstad, E.; Böttcher, Y.; Birkeland, K.I.; Sommer, C. Epigenetic signatures associated with maternal body mass index or gestational weight gain: A systematic review. J. Dev. Orig. Health Dis. 2021, 12, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Bozack, A.K.; Colicino, E.; Just, A.C.; Wright, R.O.; Baccarelli, A.A.; Wright, R.J.; Lee, A.G. Associations between infant sex and DNA methylation across umbilical cord blood, artery, and placenta samples. Epigenetics 2022, 17, 1080–1097. [Google Scholar] [CrossRef] [PubMed]
- Nomura, Y.; Lambertini, L.; Rialdi, A.; Lee, M.; Mystal, E.Y.; Grabie, M.; Manaster, I.; Huynh, N.; Finik, J.; Davey, M.; et al. Global methylation in the placenta and umbilical cord blood from pregnancies with maternal gestational diabetes, preeclampsia, and obesity. Reprod. Sci. 2014, 21, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Wang, C.; Feng, H.; Lin, L.; Liu, X.; Wei, Y.; Yang, H. Alteration in Expression and Methylation of IGF2/H19 in Placenta and Umbilical Cord Blood Are Associated with Macrosomia Exposed to Intrauterine Hyperglycemia. PLoS ONE 2016, 11, e0148399. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J.; Kakongoma, N.; Hua, W.; Xu, J.; Wang, Y.; He, S.; Gu, H.; Shi, J.; Hu, W. DNA methylation and expression profiles of placenta and umbilical cord blood reveal the characteristics of gestational diabetes mellitus patients and offspring. Clin. Epigenet. 2022, 14, 69. [Google Scholar] [CrossRef]
- Rector, R.S.; Payne, R.M.; Ibdah, J.A. Mitochondrial trifunctional protein defects: Clinical implications and therapeutic approaches. Adv. Drug Deliv. Rev. 2008, 60, 1488–1496. [Google Scholar] [CrossRef]
- Liao, C.C.; Lin, Y.L.; Kuo, C.F. Effect of high-fat diet on hepatic proteomics of hamsters. J. Agric. Food Chem. 2015, 63, 1869–1881. [Google Scholar] [CrossRef]
- Liao, C.-C.; Lin, Y.-L.; Kuo, C.-F. Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J. Clin. Investig. 2001, 107, 1403–1409. [Google Scholar]
- Ibdah, J.A.; Perlegas, P.; Zhao, Y.; Angdisen, J.; Borgerink, H.; Shadoan, M.K.; Wagner, J.D.; Matern, D.; Rinaldo, P.; Cline, J.M. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology 2005, 128, 1381–1390. [Google Scholar] [CrossRef]
- Li, L.; Lu, D.Z.; Li, Y.M.; Zhang, X.Q.; Zhou, X.X.; Jin, X. Proteomic analysis of liver mitochondria from rats with nonalcoholic steatohepatitis. World J. Gastroenterol. 2014, 20, 4778–4786. [Google Scholar] [CrossRef]
- Pepin, M.E.; Ha, C.-M.; Crossman, D.K.; Litovsky, S.H.; Varambally, S.; Barchue, J.P.; Pamboukian, S.V.; Diakos, N.A.; Drakos, S.G.; Pogwizd, S.M.; et al. Genome-wide DNA methylation encodes cardiac transcriptional reprogramming in human ischemic heart failure. Lab. Investig. 2019, 99, 371–386. [Google Scholar] [CrossRef]
- Doege, H.; Schürmann, A.; Bahrenberg, G.; Brauers, A.; Joost, H.-G. GLUT8, a novel member of the sugar transport facilitator family with glucose transport activity. J. Biol. Chem. 2000, 275, 16275–16280. [Google Scholar] [CrossRef]
- DeBosch, B.J.; Chen, Z.; Finck, B.N.; Chi, M.; Moley, K.H. Glucose transporter-8 (GLUT8) mediates glucose intolerance and dyslipidemia in high-fructose diet-fed male mice. Mol. Endocrinol. 2013, 27, 1887–1896. [Google Scholar] [CrossRef]
- Limesand, S.W.; Regnault, T.R.; Hay, W.W. Characterization of glucose transporter 8 (GLUT8) in the ovine placenta of normal and growth restricted fetuses. Placenta 2004, 25, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Janzen, C.; Lei, M.Y.; Jeong, I.S.D.; Ganguly, A.; Sullivan, P.; Paharkova, V.; Capodanno, G.; Nakamura, H.; Perry, A.; Shin, B.C.; et al. Humanin (HN) and glucose transporter 8 (GLUT8) in pregnancies complicated by intrauterine growth restriction. PLoS ONE 2018, 13, e0193583. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.; Garde, E.; Lopez-Bermejo, A.; de Zegher, F.; Ibañez, L. Differential DNA methylation profile in infants born small-for-gestational-age: Association with markers of adiposity and insulin resistance from birth to age 24 months. BMJ Open Diabetes Res. Care 2020, 8, e001402. [Google Scholar] [CrossRef] [PubMed]
- Ollikainen, M.; Smith, K.R.; Joo, E.J.-H.; Ng, H.K.; Andronikos, R.; Novakovic, B.; Aziz, N.K.A.; Carlin, J.B.; Morley, R.; Saffery, R.; et al. DNA methylation analysis of multiple tissues from newborn twins reveals both genetic and intrauterine components to variation in the human neonatal epigenome. Hum. Mol. Genet. 2010, 19, 4176–4188. [Google Scholar] [CrossRef] [PubMed]
- Titus, A.J.; Gallimore, R.M.; Salas, L.A.; Christensen, B.C. Cell-type deconvolution from DNA methylation: A review of recent applications. Hum. Mol. Genet. 2017, 26, R216–R224. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Array | Validation | p Value | |
|---|---|---|---|
| Mother (n) | 24 | 90 | |
| Age (yr) | 31 ± 1 | 31 ± 1 | Ns |
| Height (cm) | 164 ± 1 | 163 ± 1 | Ns |
| Pregestational weight (kg) | 68.5 ± 2.9 | 65.3 ± 1.4 | Ns |
| Pregestational BMI (Kg/m2) | 25.2 ± 1.0 | 24.5 ± 0.4 | Ns |
| Pregestational Obesity (%) | 31 | 33 | Ns |
| Pre-delivery weight (kg) | 84.7 ± 2.9 | 79.6 ± 1.4 | Ns |
| Pre-delivery BMI (Kg/m2) | 31.2 ± 1.1 | 29.9 ± 0.5 | Ns |
| Gestational weight gain (Kg) | 16.1 ± 0.6 | 14.3 ± 0.5 | Ns |
| Newborn (n) | 24 | 90 | |
| Gender (% female) | 50 | 43 | Ns |
| Gestational age (wk) | 40 ± 0.1 | 39 ± 0.1 | 0.04 |
| Birth weight (Kg) | 3.4 ± 0.1 | 3.2 ± 0.1 | 0.01 |
| Birth weight SDS | 0.30 ± 0.1 | 0.01 ± 0.1 | 0.04 |
| Birth length(cm) | 50.1 ± 0.2 | 49.5 ± 0.1 | Ns |
| Birth length SDS | 0.07 ± 0.1 | −0.21 ± 0.1 | Ns |
| Offspring at 6 yr (n) | 24 | 62 | |
| Gender (% female) | 50 | 43 | Ns |
| Age (yr) | 6.2 ± 0.1 | 5.8 ± 0.1 | 0.04 |
| Weight (kg) | 23.7 ± 1.0 | 22.3 ± 0.6 | Ns |
| Weight SDS | 0.22 ± 0.2 | 0.13 ± 0.1 | Ns |
| Height (cm) | 120 ± 1 | 114 ± 1 | 0.004 |
| Height SDS | 0.46 ± 0.2 | −0.07 ± 0.1 | Ns |
| BMI (kg/m2) | 16.3 ± 0.3 | 16.8 ± 0.3 | Ns |
| BMI SDS | −0.02 ± 0.1 | 0.24 ± 0.1 | Ns |
| ∆ BW-BMI SDS | −0.30 ± 0.2 | 0.23 ± 0.1 | Ns |
| Waist (cm) | 57.1 ± 1.7 | 57.1 ± 0.9 | Ns |
| Hip (cm) | 61.1 ± 1.8 | 59.9 ± 1.0 | Ns |
| SBP (mmHg) | 96.9 ± 3.0 | 96.2 ± 1.4 | Ns |
| DBP (mmHg) | 57.1 ± 1.1 | 55.9 ± 0.9 | Ns |
| HDL-cholesterol (mg/dL) | 57.0 ± 2.7 | 54.4 ± 1.2 | Ns |
| Triglycerides (mg/dL) | 49.5 ± 2.7 | 50.8 ± 2.1 | Ns |
| Glucose (mg/dL) | 85.0 ± 1.7 | 82.5 ± 1.0 | Ns |
| Insulin (mIU/L) | 6.2 ± 0.5 | 5.5 ± 0.2 | Ns |
| HOMA-IR | 1.3 ± 0.1 | 1.1 ± 0.1 | Ns |
| Perirenal fat (cm) | 0.10 ± 0.1 | 0.11 ± 0.1 | Ns |
| FM (Kg) | 5.9 ± 0.6 | 5.7 ± 0.4 | Ns |
| FM SDS | 0.37 ± 0.3 | 0.59 ± 0.2 | Ns |
| (A) Placenta | ||||||
| IlmnID | Beta Coefficient | FDR-Adjusted p-Value | OR | Chr | Position | Gene Name |
| cg13692482 | −0.130 | 2.04 × 10−8 | 0.878 | 9 | 130168651 | SLC2A8 |
| cg13031029 | −0.124 | 3.34 × 10−6 | 0.883 | 7 | 107574783 | LAMB1 |
| cg03357803 | −0.106 | 3.76 × 10−5 | 0.899 | 15 | 28272345 | OCA2 |
| cg12006733 | −0.105 | 2.17 × 10−5 | 0.901 | 15 | 90177014 | KIF7 |
| cg12680750 | −0.104 | 7.92 × 10−7 | 0.901 | 2 | 133426182 | LYPD1 |
| cg04899492 | 0.096 | 5.82 × 10−5 | 1.101 | 6 | 24126312 | NRSN1 |
| cg05888872 | 0.098 | 1.23 × 10−7 | 1.103 | 10 | 14050479 | FRMD4A |
| cg01188578 | 0.103 | 2.48 × 10−5 | 1.108 | 2 | 26464058 | HADHA |
| cg16936887 | 0.105 | 5.28 × 10−5 | 1.111 | 18 | 52989025 | TCF4 |
| cg12877278 | 0.117 | 3.59 × 10−7 | 1.124 | 6 | 151551059 | LOC102723831 |
| (B) Umbilical Cord | ||||||
| IlmnID | Beta Coefficient | FDR-Adjusted p-Value | OR | Chr | Position | Gene Name |
| cg13692482 | −0.133 | 2.35 × 10−6 | 0.875 | 9 | 130168651 | SLC2A8 |
| cg03751055 | −0.084 | 7.46 × 10−6 | 0.919 | 10 | 131380455 | MGMT |
| cg23143502 | −0.063 | 3.64 × 10−7 | 0.939 | 4 | 675936 | MFSD7 |
| cg25783997 | −0.058 | 7.21 × 10−6 | 0.944 | 2 | 185462928 | ZNF804A |
| cg16396228 | −0.050 | 3.06 × 10−6 | 0.952 | 17 | 46985589 | UBE2Z |
| cg03876184 | 0.074 | 1.31 × 10−5 | 1.077 | 2 | 27886755 | SUPT7L |
| cg04740665 | 0.080 | 8.39 × 10−6 | 1.083 | 1 | 205560997 | MFSD4 |
| cg16513685 | 0.081 | 6.31 × 10−7 | 1.084 | 9 | 33738831 | LINC01251 |
| cg26028074 | 0.118 | 1.39 × 10−6 | 1.125 | 17 | 32201533 | ASIC2 |
| cg01188578 | 0.179 | 2.16 × 10−6 | 1.196 | 2 | 26464058 | HADHA |
| Placenta | Genes | p Value |
| Hsa04512: ECM–receptor interaction | 3 | 0.016 |
| Hsa00062: Fatty acid elongation | 4 | 0.018 |
| Hsa03013: RNA transport | 7 | 0.031 |
| Hsa04933: AGE-RAGE signaling pathway in diabetic complications | 4 | 0.036 |
| Hsa05412: Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 3 | 0.045 |
| Umbilical Cord | Genes | p Value |
| Hsa04650: Natural killer cell-mediated cytotoxicity | 3 | 0.007 |
| Hsa04621: NOD-like receptor signaling pathway | 7 | 0.010 |
| Hsa03013: RNA transport | 4 | 0.047 |
| Placenta | Genes | p Value |
| Tobacco User Disorder | 139 | 0.0078 |
| Height | 17 | 0.014 |
| Eosinophils | 8 | 0.016 |
| Hemoglobin A, Glycosylated | 14 | 0.016 |
| Coronary Disease | 17 | 0.02 |
| Attention Deficit and Disruptive Behavior Disorders | 4 | 0.021 |
| Bone Mineral Density | 25 | 0.024 |
| Occipital Lobe | 4 | 0.029 |
| Type 2 Diabetes | 100 | 0.032 |
| Intuition | 3 | 0.034 |
| Pancreatic Neoplasms | 6 | 0.049 |
| Breath Tests | 10 | 0.05 |
| Chronic Renal Failure | 45 | 0.05 |
| Umbilical Cord | Genes | p Value |
| Type 2 Diabetes | 57 | 0.00009 |
| Cleft Lip | 14 | 0.0092 |
| Obesity | 13 | 0.013 |
| Autism | 12 | 0.025 |
| Hypertension | 19 | 0.028 |
| Respiratory Function Tests | 9 | 0.042 |
| Leukemia | 8 | 0.05 |
| (A) Placenta | ||||||
| HADHA Methylation | HADHA Expression | |||||
| Low Levels (<p50) | High Levels (>p50) | p Value | Low Levels (<p50) | High Levels (>p50) | p Value | |
| Weight SDS | 0.57 ± 0.25 | −0.10 ± 0.15 | 0.02 | −0.19 ± 0.17 | 0.41 ± 0.22 | 0.03 |
| Height SDS | 0.03 ± 0.25 | −0.14 ± 0.19 | Ns | −0.31 ± 0.21 | 0.13 ± 0.19 | Ns |
| BMI SDS | 0.58 ± 0.24 | −0.01 ± 0.15 | 0.03 | −0.03 ± 0.13 | 0.56 ± 0.26 | 0.03 |
| ∆ BW-BMI SDS | 0.67 ± 0.25 | −0.13 ± 0.19 | 0.01 | −0.13 ± 0.16 | 0.51 ± 0.29 | 0.04 |
| Waist | 58.8 ± 1.4 | 56.1 ± 1.1 | Ns | 55.0 ±0.8 | 58.6 ± 1.6 | 0.04 |
| SBP | 101.0 ± 2.1 | 92.3 ± 1.6 | 0.002 | 94.3 ± 1.9 | 98.8 ± 2.1 | Ns |
| DBP | 56.9 ± 1.2 | 55.4 ± 1.4 | Ns | 55.6 ± 1.3 | 56.3 ± 1.4 | Ns |
| HDL-cholesterol | 56.4 ± 1.8 | 52.9 ± 1.7 | Ns | 54.5 ± 1.9 | 54.5 ± 1.5 | Ns |
| Triglycerides | 53.0 ± 3.4 | 48.8 ± 2.6 | Ns | 51.0 ± 3.0 | 50.4 ± 2.9 | Ns |
| Glucose | 83.7 ± 1.3 | 81.2 ± 1.4 | Ns | 82.0 ± 1.3 | 82.9 ± 1.5 | Ns |
| Insulin | 6.1 ± 0.3 | 5.0 ± 0.4 | 0.04 | 4.87 ± 0.32 | 6.10 ± 0.48 | 0.03 |
| HOMA-IR | 1.28 ± 0.10 | 1.03 ± 0.09 | 0.04 | 0.95 ± 0.07 | 1.21 ± 0.11 | 0.04 |
| Perirenal fat | 0.10 ±0.01 | 0.11 ± 0.01 | Ns | 0.11 ± 0.01 | 0.10 ± 0.01 | Ns |
| FM SDS | 1.11 ± 0.39 | 0.01 ± 0.23 | 0.01 | 0.02 ± 0.26 | 1.12 ± 0.39 | 0.02 |
| (B) Umbilical Cord | ||||||
| HADHA Methylation | HADHA Expression | |||||
| Low Levels (<p50) | High Levels (>p50) | p Value | Low Levels (<p50) | High Levels (>p50) | p Value | |
| Weight SDS | 0.16 ± 0.28 | 0.03 ± 0.17 | Ns | 0.49 ± 0.19 | −0.09 ± 0.21 | 0.04 |
| Height SDS | −0.26 ± 0.26 | 0.04 ± 0.23 | Ns | 0.27 ± 0.19 | −0.41 ± 0.20 | 0.01 |
| BMI SDS | 0.36 ± 0.25 | 0.03 ± 0.18 | Ns | 0.29 ± 0.19 | 0.15 ± 0.19 | Ns |
| ∆ BW-BMI SDS | 0.34 ± 0.26 | −0.07 ± 0.24 | Ns | 0.15 ± 0.23 | 0.27 ± 0.21 | Ns |
| Waist | 55.6 ± 1.2 | 56.1 ± 1.2 | Ns | 57.3 ± 1.1 | 56.8 ± 1.4 | Ns |
| SBP | 97.4 ± 2.4 | 92.8 ± 2.1 | Ns | 97.0 ± 1.6 | 96.0 ± 2.2 | Ns |
| DBP | 54.9 ± 1.3 | 54.2 ± 1.5 | Ns | 57.8 ± 1.3 | 54.7 ± 1.3 | Ns |
| HDL-cholesterol | 55.85 ± 1.92 | 53.56 ± 1.97 | Ns | 53.6 ± 1.6 | 54.9 ± 1.8 | Ns |
| Triglycerides | 46.41 ± 2.43 | 54.56 ± 3.96 | Ns | 52.1 ± 3.2 | 49.3 ± 2.5 | Ns |
| Glucose | 82.07 ± 1.53 | 80.68 ± 1.57 | Ns | 82.67 ± 1.47 | 82.36 ± 1.28 | Ns |
| Insulin | 5.13 ± 0.39 | 5.12 ± 0.43 | Ns | 5.71 ± 0.41 | 5.36 ± 0.38 | Ns |
| HOMA-IR | 1.07 ± 0.09 | 1.01 ± 0.08 | Ns | 1.18 ± 0.10 | 1.11 ± 0.08 | Ns |
| Perirenal fat | 0.11 ± 0.01 | 0.10 ± 0.01 | Ns | 0.10 ± 0.01 | 0.11 ± 0.01 | Ns |
| FM SDS | 0.51 ± 0.39 | 0.28 ± 0.33 | Ns | 0.61 ± 0.34 | 0.48 ± 0.32 | Ns |
| (A) Placenta | ||||||
| SLC2A8 Methylation | SLC2A8 Expression | |||||
| Low Levels (<p50) | High Levels (>p50) | p Value | Low Levels (<p50) | High Levels (>p50) | p Value | |
| Weight SDS | 0.80 ± 0.31 | 0.01 ± 0.18 | 0.02 | 0.19 ± 0.21 | −0.01 ± 0.19 | Ns |
| Height SDS | 0.75 ± 0.19 | −0.13 ± 0.20 | 0.007 | −0.19 ± 0.18 | 0.01 ± 0.24 | Ns |
| BMI SDS | 0.50 ± 0.31 | 0.23 ± 0.20 | Ns | 0.37 ± 0.20 | 0.01 ± 0.18 | Ns |
| ∆ BW-BMI SDS | 0.51 ± 0.35 | 0.18 ± 0.22 | Ns | 0.47 ± 0.22 | −0.13 ± 0.21 | Ns |
| Waist | 58.4 ± 2.0 | 57.6 ± 1.3 | Ns | 56.0 ± 1.2 | 57.4 ± 1.3 | Ns |
| SBP | 96.0 ± 2.1 | 95.9 ± 1.8 | Ns | 96.3 ± 2.3 | 96.4 ± 1.6 | Ns |
| DBP | 56.5 ± 1.7 | 55.4 ± 1.5 | Ns | 55.8 ± 1.2 | 56.1 ± 1.5 | Ns |
| HDL-cholesterol | 55.1 ± 3.0 | 55.9 ± 1.4 | Ns | 52.9 ± 1.7 | 56.2 ± 1.8 | Ns |
| Triglycerides | 47.8 ± 3.5 | 50.7 ± 2.7 | Ns | 50.8 ± 3.1 | 50.7 ± 2.8 | Ns |
| Glucose | 80.9 ± 1.5 | 82.8 ± 1.6 | Ns | 81.8 ± 1.2 | 83.1 ± 1.6 | Ns |
| Insulin | 5.6 ± 0.4 | 5.5 ± 0.4 | Ns | 5.3 ± 0.4 | 5.6 ± 0.3 | Ns |
| HOMA-IR | 1.14 ± 0.1 | 1.17 ± 0.1 | Ns | 1.10 ± 0.09 | 1.17 ± 0.10 | Ns |
| Perirenal fat | 0.13 ± 0.01 | 0.10 ± 0.01 | 0.02 | 0.12 ± 0.01 | 0.10 ± 0.01 | Ns |
| FM SDS | 1.02 ± 0.47 | 0.74 ± 0.36 | Ns | −0.15 ± 0.17 | 0.78 ± 0.36 | 0.02 |
| (B) Umbilical Cord | ||||||
| SLC2A8 Methylation | SLC2A8 Expression | |||||
| Low Levels (<p50) | High Levels (>p50) | p Value | Low Levels (<p50) | High Levels (>p50) | p Value | |
| Weight SDS | 0.75 ± 0.28 | 0.01 ± 0.18 | 0.03 | 0.05 ± 0.19 | 0.20 ± 0.21 | Ns |
| Height SDS | 0.50 ± 0.23 | −0.16 ± 0.18 | 0.02 | 0.04 ± 0.21 | −0.17 ± 0.20 | Ns |
| BMI SDS | 0.67 ± 0.27 | 0.10 ± 0.18 | 0.04 | 0.07 ± 0.16 | 0.37 ± 0.22 | Ns |
| ∆ BW-BMI SDS | 0.59 ± 0.32 | 0.14 ± 0.23 | Ns | 0.04 ± 0.20 | 0.38 ± 0.23 | Ns |
| Waist | 58.3 ± 1.9 | 57.7 ± 1.4 | Ns | 56.0 ± 1.2 | 58.0 ± 1.2 | Ns |
| SBP | 95.0 ± 2.0 | 96.3 ± 2.0 | Ns | 94.4 ± 1.8 | 98.6 ± 2.12 | Ns |
| DBP | 55.6 ± 1.9 | 56.1 ± 1.4 | Ns | 57.0 ± 1.4 | 55.5 ± 1.2 | Ns |
| HDL-cholesterol | 56.9 ± 2.7 | 54.7 ± 1.5 | Ns | 53.9 ± 1.8 | 54.7 ± 1.7 | Ns |
| Triglycerides | 45.6 ± 3.4 | 51.6 ± 2.8 | Ns | 45.7 ± 2.3 | 55.3 ± 3.1 | 0.02 |
| Glucose | 80.0 ± 1.4 | 83.6 ± 1.6 | Ns | 80.72 ± 1.30 | 84.21 ± 1.38 | 0.05 |
| Insulin | 5.2 ± 0.4 | 5.7 ± 0.4 | Ns | 5.32 ± 0.41 | 5.73 ± 0.38 | Ns |
| HOMA-IR | 1.0 ± 0.1 | 1.2 ± 0.1 | Ns | 1.08 ± 0.09 | 1.21 ± 0.09 | Ns |
| Perirenal fat | 0.13 ± 0.01 | 0.10 ± 0.01 | 0.006 | 0.10 ± 0.01 | 0.11 ± 0.01 | Ns |
| FM SDS | 0.75 ± 0.42 | 0.84 ± 0.38 | Ns | 0.10 ± 0.26 | 1.02 ± 0.37 | 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Vilarrubla, A.; Mas-Parés, B.; Carreras-Badosa, G.; Bonmatí-Santané, A.; Martínez-Calcerrada, J.-M.; Niubó-Pallàs, M.; de Zegher, F.; Ibáñez, L.; López-Bermejo, A.; Bassols, J. DNA Methylation Signatures in Paired Placenta and Umbilical Cord Samples: Relationship with Maternal Pregestational Body Mass Index and Offspring Metabolic Outcomes. Biomedicines 2024, 12, 301. https://doi.org/10.3390/biomedicines12020301
Gómez-Vilarrubla A, Mas-Parés B, Carreras-Badosa G, Bonmatí-Santané A, Martínez-Calcerrada J-M, Niubó-Pallàs M, de Zegher F, Ibáñez L, López-Bermejo A, Bassols J. DNA Methylation Signatures in Paired Placenta and Umbilical Cord Samples: Relationship with Maternal Pregestational Body Mass Index and Offspring Metabolic Outcomes. Biomedicines. 2024; 12(2):301. https://doi.org/10.3390/biomedicines12020301
Chicago/Turabian StyleGómez-Vilarrubla, Ariadna, Berta Mas-Parés, Gemma Carreras-Badosa, Alexandra Bonmatí-Santané, Jose-Maria Martínez-Calcerrada, Maria Niubó-Pallàs, Francis de Zegher, Lourdes Ibáñez, Abel López-Bermejo, and Judit Bassols. 2024. "DNA Methylation Signatures in Paired Placenta and Umbilical Cord Samples: Relationship with Maternal Pregestational Body Mass Index and Offspring Metabolic Outcomes" Biomedicines 12, no. 2: 301. https://doi.org/10.3390/biomedicines12020301
APA StyleGómez-Vilarrubla, A., Mas-Parés, B., Carreras-Badosa, G., Bonmatí-Santané, A., Martínez-Calcerrada, J.-M., Niubó-Pallàs, M., de Zegher, F., Ibáñez, L., López-Bermejo, A., & Bassols, J. (2024). DNA Methylation Signatures in Paired Placenta and Umbilical Cord Samples: Relationship with Maternal Pregestational Body Mass Index and Offspring Metabolic Outcomes. Biomedicines, 12(2), 301. https://doi.org/10.3390/biomedicines12020301