
Plasma and Urinary Metabolomic Analysis of Gout and Asymptomatic Hyperuricemia and Profiling of Potential Biomarkers: A Pilot Study

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Participants
2.2. Data Collection
2.3. Sample Preparation and Metabolomic Measurements Using Gas Chromatography–Tandem Mass Spectrometry (GC-MS/MS)
2.4. Genetic Analysis
2.5. Statistical Analysis
3. Results
3.1. Demographic, Clinical, and Genetic Characteristics of the Enrolled Participants
3.2. Multivariate PLS Discriminant Analysis (PLS-DA) to Select the Candidate Metabolites That Distinguish Gout from Asymptomatic Hyperuricemia
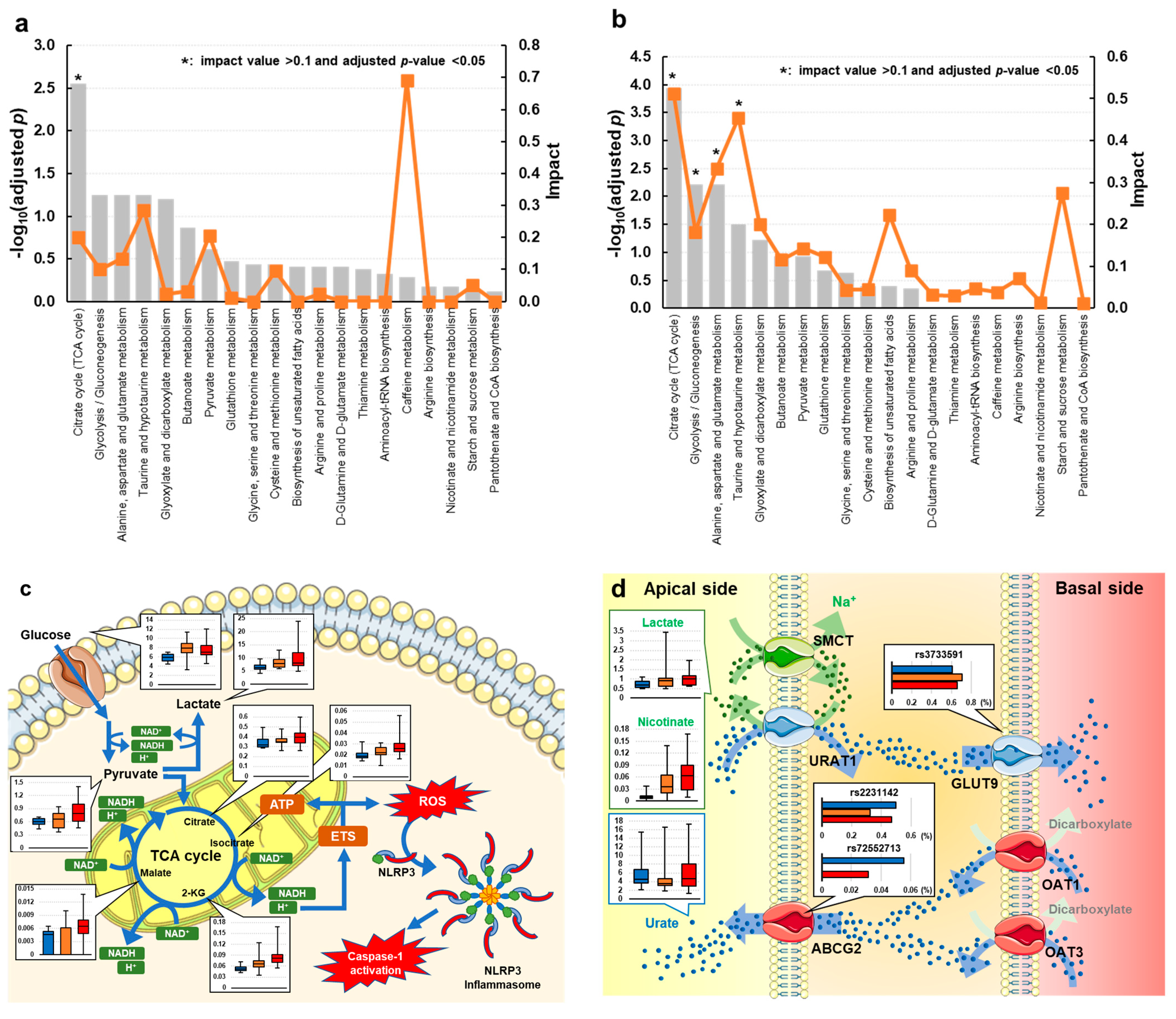
3.3. Univariate and Metabolic Pathway Analyses of Gout and Asymptomatic Hyperuricemia
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dehlin, M.; Jacobsson, L.; Roddy, E. Global epidemiology of gout: Prevalence, incidence, treatment patterns and risk factors. Nat. Rev. Rheumatol. 2020, 16, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.J.; Fransen, J.; Jansen, T.L.; Dalbeth, N.; Schumacher, H.R.; Brown, M.; Louthrenoo, W.; Vazquez-Mellado, J.; Eliseev, M.; McCarthy, G.; et al. Study for Updated Gout Classification Criteria: Identification of Features to Classify Gout. Arthritis Care Res. 2015, 67, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Campion, E.W.; Glynn, R.J.; DeLabry, L.O. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am. J. Med. 1987, 82, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Phipps-Green, A.; Frampton, C.; Neogi, T.; Taylor, W.J.; Merriman, T.R. Relationship between serum urate concentration and clinically evident incident gout: An individual participant data analysis. Ann. Rheum. Dis. 2018, 77, 1048–1052. [Google Scholar] [CrossRef]
- Dalbeth, N.; House, M.E.; Aati, O.; Tan, P.; Franklin, C.; Horne, A.; Gamble, G.D.; Stamp, L.K.; Doyle, A.J.; McQueen, F.M. Urate crystal deposition in asymptomatic hyperuricaemia and symptomatic gout: A dual energy CT study. Ann. Rheum. Dis. 2015, 74, 908–911. [Google Scholar] [CrossRef]
- Wang, P.; Smith, S.E.; Garg, R.; Lu, F.; Wohlfahrt, A.; Campos, A.; Vanni, K.; Yu, Z.; Solomon, D.H.; Kim, S.C. Identification of monosodium urate crystal deposits in patients with asymptomatic hyperuricemia using dual-energy CT. RMD Open 2018, 4, e000593. [Google Scholar] [CrossRef]
- Li, Y.; Ju, L.; Hou, Z.; Deng, H.; Zhang, Z.; Wang, L.; Yang, Z.; Yin, J.; Zhang, Y. Screening, verification, and optimization of biomarkers for early prediction of cardiotoxicity based on metabolomics. J. Proteome Res. 2015, 14, 2437–2445. [Google Scholar] [CrossRef]
- Albrecht, E.; Waldenberger, M.; Krumsiek, J.; Evans, A.M.; Jeratsch, U.; Breier, M.; Adamski, J.; Koenig, W.; Zeilinger, S.; Fuchs, C.; et al. Metabolite profiling reveals new insights into the regulation of serum urate in humans. Metabolomics 2014, 10, 141–151. [Google Scholar] [CrossRef]
- Li, Q.; Wei, S.; Wu, D.; Wen, C.; Zhou, J. Urinary Metabolomics Study of Patients with Gout Using Gas Chromatography-Mass Spectrometry. BioMed Res. Int. 2018, 2018, 3461572. [Google Scholar] [CrossRef]
- Huang, Y.; Xiao, M.; Ou, J.; Lv, Q.; Wei, Q.; Chen, Z.; Wu, J.; Tu, L.; Jiang, Y.; Zhang, X.; et al. Identification of the urine and serum metabolomics signature of gout. Rheumatology 2020, 59, 2960–2969. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Chang, D.; Guo, F.; Pan, H.; Yang, Y. Metabolomics approach by 1H NMR spectroscopy of serum reveals progression axes for asymptomatic hyperuricemia and gout. Arthritis Res. Ther. 2018, 20, 111. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, L.; Liu, X.Y.; Chen, X.; Song, Y.X.; Li, X.H.; Jiang, C.; Peng, A.; Liu, J.Y. Plasma profiling of amino acids distinguishes acute gout from asymptomatic hyperuricemia. Amino Acids 2018, 50, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wang, C.; Liang, N.; Liu, Z.; Li, X.; Zhu, Z.J.; Merriman, T.R.; Dalbeth, N.; Terkeltaub, R.; Li, C.; et al. Serum Metabolomics Identifies Dysregulated Pathways and Potential Metabolic Biomarkers for Hyperuricemia and Gout. Arthritis Rheumatol. 2021, 73, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Kottgen, A.; Albrecht, E.; Teumer, A.; Vitart, V.; Krumsiek, J.; Hundertmark, C.; Pistis, G.; Ruggiero, D.; O’Seaghdha, C.M.; Haller, T.; et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 2013, 45, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, H.; Yamamoto, K.; Nakaoka, H.; Nakayama, A.; Sakiyama, M.; Chiba, T.; Takahashi, A.; Nakamura, T.; Nakashima, H.; Takada, Y.; et al. Genome-wide association study of clinically defined gout identifies multiple risk loci and its association with clinical subtypes. Ann. Rheum. Dis. 2016, 75, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Hisatome, I.; Ichida, K.; Mineo, I.; Ohtahara, A.; Ogino, K.; Kuwabara, M.; Ishizaka, N.; Uchida, S.; Kurajoh, M.; Kohagura, K.; et al. Japanese Society of Gout and Uric & Nucleic Acids 2019 Guidelines for Management of Hyperuricemia and Gout 3rd edition. Gout Uric Nucleic Acids 2020, 44, sp-1–sp-40. [Google Scholar] [CrossRef]
- Worley, B.; Powers, R. Multivariate Analysis in Metabolomics. Curr. Metabolomics 2013, 1, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef]
- Dalbeth, N.; Stamp, L. Hyperuricaemia and gout: Time for a new staging system? Ann. Rheum. Dis. 2014, 73, 1598–1600. [Google Scholar] [CrossRef]
- Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Nakayama, A.; Ikebuchi, Y.; Ito, K.; Kusanagi, Y.; Chiba, T.; Tadokoro, S.; et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: A function-based genetic analysis in a Japanese population. Sci. Transl. Med. 2009, 1, 5ra11. [Google Scholar] [CrossRef]
- Choi, H.K.; Atkinson, K.; Karlson, E.W.; Curhan, G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: The health professionals follow-up study. Arch. Intern. Med. 2005, 165, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Atkinson, K.; Karlson, E.W.; Willett, W.; Curhan, G. Alcohol intake and risk of incident gout in men: A prospective study. Lancet 2004, 363, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Liberopoulos, E.N.; Miltiadous, G.A.; Elisaf, M.S. Alcohol intake, serum uric acid concentrations, and risk of gout. Lancet 2004, 364, 246–247. [Google Scholar] [CrossRef]
- Williams, P.T. Effects of diet, physical activity and performance, and body weight on incident gout in ostensibly healthy, vigorously active men. Am. J. Clin. Nutr. 2008, 87, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, Y.; Nakaoka, H.; Nakayama, A.; Okada, Y.; Yamamoto, K.; Higashino, T.; Sakiyama, M.; Shimizu, T.; Ooyama, H.; Ooyama, K.; et al. Genome-wide association study revealed novel loci which aggravate asymptomatic hyperuricaemia into gout. Ann. Rheum. Dis. 2019, 78, 1430–1437. [Google Scholar] [CrossRef]
- Park, J.W.; Noh, J.H.; Kim, J.M.; Lee, H.Y.; Kim, K.A.; Park, J.Y. Gene Dose-Dependent and Additive Effects of ABCG2 rs2231142 and SLC2A9 rs3733591 Genetic Polymorphisms on Serum Uric Acid Levels. Metabolites 2022, 12, 1192. [Google Scholar] [CrossRef]
- Wan Rohani, W.T.; Mahfudzah, A.; Nazihah, M.Y.; Tan, H.L.; Wan Syamimee, W.G.; Amanda Jane, P.G.; Tony Richard, M. Association of solute carrier family 2, member 9 (SLC2A9) genetic variant rs3733591 with gout in a Malay sample set. Med. J. Malaysia 2018, 73, 307–310. [Google Scholar]
- Yang, H.J.; Liu, M.; Kim, M.J.; Park, S. The haplotype of SLC2A9_rs3733591, PKD2_rs2725220 and ABCG2_rs2231142 increases the hyperuricaemia risk and alcohol, chicken and processed meat intakes and smoking interact with its risk. Int. J. Food Sci. Nutr. 2021, 72, 391–401. [Google Scholar] [CrossRef]
- Ichida, K.; Matsuo, H.; Takada, T.; Nakayama, A.; Murakami, K.; Shimizu, T.; Yamanashi, Y.; Kasuga, H.; Nakashima, H.; Nakamura, T.; et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 2012, 3, 764. [Google Scholar] [CrossRef]
- Ohashi, Y.; Toyoda, M.; Saito, N.; Koizumi, M.; Kanai, G.; Komaba, H.; Kimura, M.; Wada, T.; Takahashi, H.; Takahashi, Y.; et al. Evaluation of ABCG2-mediated extra-renal urate excretion in hemodialysis patients. Sci. Rep. 2023, 13, 93. [Google Scholar] [CrossRef]
- Eckenstaler, R.; Benndorf, R.A. The Role of ABCG2 in the Pathogenesis of Primary Hyperuricemia and Gout-An Update. Int. J. Mol. Sci. 2021, 22, 6678. [Google Scholar] [CrossRef]
- Matsuo, H.; Chiba, T.; Nagamori, S.; Nakayama, A.; Domoto, H.; Phetdee, K.; Wiriyasermkul, P.; Kikuchi, Y.; Oda, T.; Nishiyama, J.; et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am. J. Hum. Genet. 2008, 83, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.G.; Shaper, A.G.; Thelle, D.S.; Whitehead, T.P. Serum uric acid, serum glucose and diabetes: Relationships in a population study. Postgrad Med. J. 1986, 62, 1001–1006. [Google Scholar] [CrossRef]
- Han, T.; Lan, L.; Qu, R.; Xu, Q.; Jiang, R.; Na, L.; Sun, C. Temporal relationship between hyperuricemia and insulin resistance and its impact on future risk of hypertension. Hypertension 2017, 70, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Bombelli, M.; Quarti-Trevano, F.; Tadic, M.; Facchetti, R.; Cuspidi, C.; Mancia, G.; Grassi, G. Uric acid and risk of new-onset metabolic syndrome, impaired fasting glucose and diabetes mellitus in a general Italian population: Data from the Pressioni Arteriose Monitorate E Loro Associazioni study. J. Hypertens. 2018, 36, 1492–1498. [Google Scholar] [CrossRef]
- Facchini, F.; Chen, Y.D.; Hollenbeck, C.B.; Reaven, G.M. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA 1991, 266, 3008–3011. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; Enerback, S. Lactate: The ugly duckling of energy metabolism. Nat. Metab. 2020, 2, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Dalbeth, N.; Gosling, A.L.; Gaffo, A.; Abhishek, A. Gout. Lancet 2021, 397, 1843–1855. [Google Scholar] [CrossRef]
- Masulli, M.; D’Elia, L.; Angeli, F.; Barbagallo, C.M.; Bilancio, G.; Bombelli, M.; Bruno, B.; Casiglia, E.; Cianci, R.; Cicero, A.F.G.; et al. Serum uric acid levels threshold for mortality in diabetic individuals: The URic acid Right for heArt Health (URRAH) project. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 1245–1252. [Google Scholar] [CrossRef]
- Maloberti, A.; Mengozzi, A.; Russo, E.; Cicero, A.F.G.; Angeli, F.; Agabiti Rosei, E.; Barbagallo, C.M.; Bernardino, B.; Bombelli, M.; Cappelli, F.; et al. The Results of the URRAH (Uric Acid Right for Heart Health) Project: A Focus on Hyperuricemia in Relation to Cardiovascular and Kidney Disease and its Role in Metabolic Dysregulation. High Blood Press Cardiovasc. Prev. 2023, 30, 411–425. [Google Scholar] [CrossRef]
- Palatini, P.; Virdis, A.; Masi, S.; Mengozzi, A.; Casiglia, E.; Tikhonoff, V.; Cicero, A.F.G.; Ungar, A.; Parati, G.; Rivasi, G.; et al. Hyperuricemia increases the risk of cardiovascular mortality associated with very high HdL-cholesterol level. Nutr. Metab. Cardiovasc. Dis. 2023, 33, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Cipolletta, E.; Tata, L.J.; Nakafero, G.; Avery, A.J.; Mamas, M.A.; Abhishek, A. Association between Gout Flare and Subsequent Cardiovascular Events among Patients with Gout. JAMA 2022, 328, 440–450. [Google Scholar] [CrossRef]
- Kumar, A.A.; Kelly, D.P.; Chirinos, J.A. Mitochondrial Dysfunction in Heart Failure with Preserved Ejection Fraction. Circulation 2019, 139, 1435–1450. [Google Scholar] [CrossRef] [PubMed]
- Ranjbarvaziri, S.; Kooiker, K.B.; Ellenberger, M.; Fajardo, G.; Zhao, M.; Vander Roest, A.S.; Woldeyes, R.A.; Koyano, T.T.; Fong, R.; Ma, N.; et al. Altered Cardiac Energetics and Mitochondrial Dysfunction in Hypertrophic Cardiomyopathy. Circulation 2021, 144, 1714–1731. [Google Scholar] [CrossRef]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.D. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol. Med. 2015, 21, 193–201. [Google Scholar] [CrossRef]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Perez-Ruiz, F.; Calabozo, M.; Erauskin, G.G.; Ruibal, A.; Herrero-Beites, A.M. Renal underexcretion of uric acid is present in patients with apparent high urinary uric acid output. Arthritis Rheum. 2002, 47, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.K.; Mount, D.B. The molecular physiology of uric acid homeostasis. Annu. Rev. Physiol. 2015, 77, 323–345. [Google Scholar] [CrossRef]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef]
- Komoda, F.; Sekine, T.; Inatomi, J.; Enomoto, A.; Endou, H.; Ota, T.; Matsuyama, T.; Ogata, T.; Ikeda, M.; Awazu, M.; et al. The W258X mutation in SLC22A12 is the predominant cause of Japanese renal hypouricemia. Pediatr. Nephrol. 2004, 19, 728–733. [Google Scholar] [CrossRef]
- Iwai, N.; Mino, Y.; Hosoyamada, M.; Tago, N.; Kokubo, Y.; Endou, H. A high prevalence of renal hypouricemia caused by inactive SLC22A12 in Japanese. Kidney Int. 2004, 66, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Cachau, R.; David, V.A.; Thompson, M.; Jung, W.; Jee, S.H.; Daar, I.O.; Winkler, C.A.; Cho, S.K. Characterization of a Compound Heterozygous SLC2A9 Mutation That Causes Hypouricemia. Biomedicines 2021, 9, 1172. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, S.; Gopal, E.; Fei, Y.J.; Ganapathy, V. Functional identification of SLC5A8, a tumor suppressor down-regulated in colon cancer, as a Na+-coupled transporter for short-chain fatty acids. J. Biol. Chem. 2004, 279, 13293–13296. [Google Scholar] [CrossRef]
- Drabkin, M.; Yogev, Y.; Zeller, L.; Zarivach, R.; Zalk, R.; Halperin, D.; Wormser, O.; Gurevich, E.; Landau, D.; Kadir, R.; et al. Hyperuricemia and gout caused by missense mutation in d-lactate dehydrogenase. J. Clin. Investig. 2019, 129, 5163–5168. [Google Scholar] [CrossRef] [PubMed]
- White, W.B.; Saag, K.G.; Becker, M.A.; Borer, J.S.; Gorelick, P.B.; Whelton, A.; Hunt, B.; Castillo, M.; Gunawardhana, L.; Investigators, C. Cardiovascular Safety of Febuxostat or Allopurinol in Patients with Gout. N. Engl. J. Med. 2018, 378, 1200–1210. [Google Scholar] [CrossRef]
- Kojima, S.; Matsui, K.; Hiramitsu, S.; Hisatome, I.; Waki, M.; Uchiyama, K.; Yokota, N.; Tokutake, E.; Wakasa, Y.; Jinnouchi, H.; et al. Febuxostat for Cerebral and CaRdiorenovascular Events PrEvEntion StuDy. Eur. Heart J. 2019, 40, 1778–1786. [Google Scholar] [CrossRef]
- Cicero, A.F.G.; Fogacci, F.; Kuwabara, M.; Borghi, C. Therapeutic Strategies for the Treatment of Chronic Hyperuricemia: An Evidence-Based Update. Medicina 2021, 57, 58. [Google Scholar] [CrossRef]
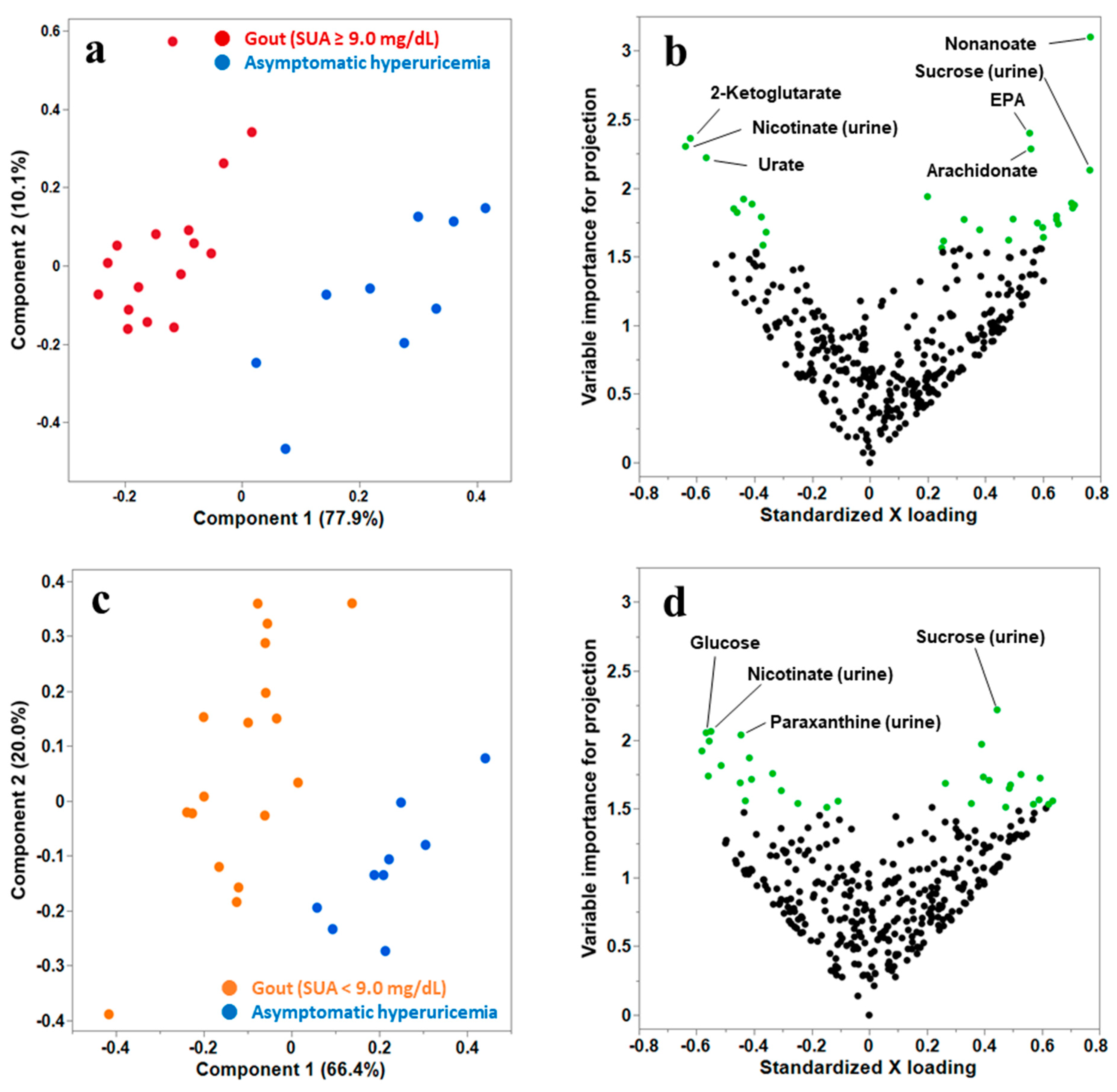

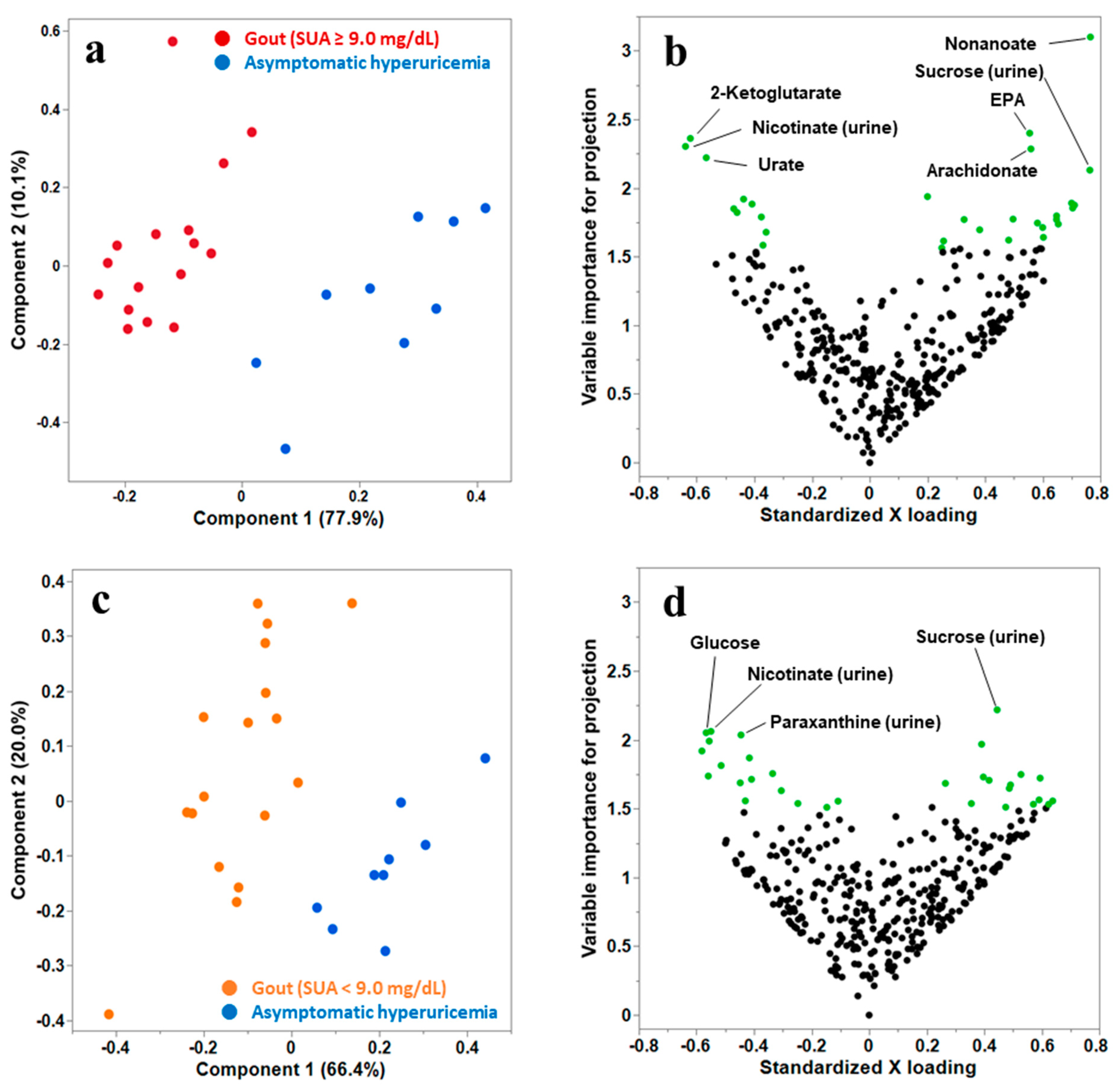{kind=link}
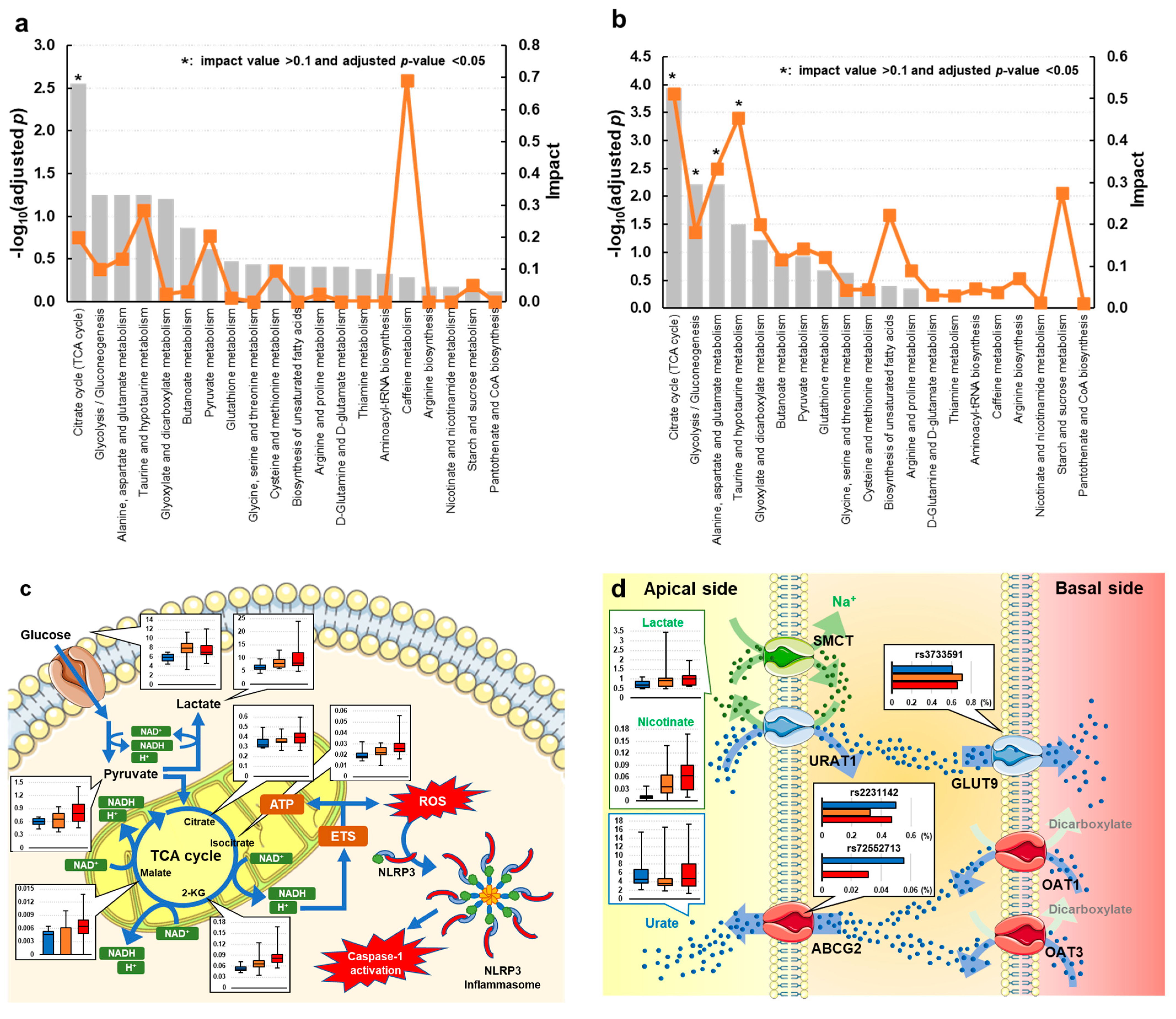{kind=link}
| Gout (n = 33) | p | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| All (n = 42) | AHU (n = 9) | All Gout | Low-Risk Gout (SUA < 9.0 mg/dL) | High-Risk Gout (SUA ≥ 9.0 mg/dL) | |||||||
| n | % | n | % | n | % | n | % | n | % | ||
| Demographic data | |||||||||||
| Family history of gout 1 | 13 | 31.0 | 5 | 55.6 | 8 | 24.2 | 2 | 11.8 | 6 | 37.5 | 0.056 |
| Uric acid-lowering treatment 2 | 6 | 14.3 | 0 | 0 | 6 | 18.2 | 2 | 11.8 | 4 | 25 | 0.232 |
| Urolithiasis | 6 | 14.3 | 0 | 0 | 6 | 18.2 | 4 | 23.5 | 2 | 12.5 | 0.339 |
| Hypertension | 6 | 14.3 | 0 | 0 | 6 | 18.2 | 4 | 23.5 | 2 | 12.5 | 0.339 |
| Hyperlipidemia | 4 | 38 | 1 | 11.1 | 3 | 9.1 | 2 | 11.8 | 1 | 6.3 | 1 |
| Gout | Kruskal–Wallis | |||||
|---|---|---|---|---|---|---|
| Median (IQR: Q1–Q3) | All | AHU | All Gout | Low-Risk Gout (SUA < 9.0 mg/dL) | High-Risk Gout (SUA ≥ 9.0 mg/dL) | p |
| Basic characteristics | ||||||
| Age (years) | 45 (39–55) | 34 (28–44) | 50 (40–57) | 50 (43–58) a | 47 (39–57) a | 0.005 |
| Duration of hyperuricemia (years) | 7.0 (4.0–10.0) | 4.0 (3.5–9.0) | 8.0 (4.7–10.0) | 7.0 (5.0–10) | 9.0 (4.1–13.8) | 0.357 |
| Body mass index (kg/m2) | 25.6 (23.6–28.3) | 26.2 (23.6–29.6) | 25.6 (23.6–27.6) | 24.6 (23.2–26.1) | 26.9 (24.0–30.1) | 0.117 |
| Clinical characteristics | ||||||
| SBP (mmHg) | 124 (120–134) | 122 (119–134) | 124 (120–130) | 122 (120–129) | 127 (119–140) | 0.487 |
| DBP (mmHg) | 82 (76–87) | 78 (71–85) | 82 (77–88) | 82 (77–86) | 82 (77–94) | 0.697 |
| WBCs (103/μL) | 66 (57–79) | 61 (51–69) | 68 (57–81) | 63 (55–77) | 71 (58–88) | 0.229 |
| RBCs (10⁶/μL) | 483 (463–520) | 522 (517–533) | 477 (449–490) | 479 (454–496) a | 475 (444–523) a | <0.001 |
| Hematocrit (%) | 45.2 (42.8–47.0) | 46.1 (45.7–48.4) | 45 (42.0–46.5) | 45.0 (42.6–46.7) | 43.1 (41.5–46.5) a | 0.034 |
| PLTs (103/μL) | 24.7 (22.5–29.8) | 23.7 (20.8–25.0) | 27.6 (23.2–31.1) | 23.8 (22.2–30.1) | 28.8 (23.3–32.7) | 0.107 |
| AST (IU/L) | 25 (20–32) | 30 (20–37) | 25 (20–32) | 27 (24–25) | 21 (19–28) | 0.115 |
| ALT (IU/L) | 29 (21–42) | 40 (21–68) | 28 (21–39) | 25 (22–38) | 29 (21–44) | 0.371 |
| γ-GTP (IU/L) | 53 (29–81) | 77 (35–186) | 49 (28–71) | 57 (27–68) | 45 (37–76) | 0.334 |
| LDL-C (mg/dL) | 130 (104–159) | 122 (95–190) | 130 (108–152) | 132 (106–145) | 128 (108–167) | 0.892 |
| HDL-C (mg/dL) | 53 (46–65) | 49 (42–55) | 53 (46–67) | 57 (46–72) | 53 (46–65) | 0.184 |
| TG (mg/dL) | 147 (88–201) | 178 (131–257) | 138 (84–183) | 102 (81–181) | 151 (122–200) | 0.149 |
| BUN (mg/dL) | 12.5 (10.1–14.9) | 13.1 (12.4–14.8) | 12.3 (9.8–15.1) | 12.1 (9.6–14.3) | 12.9 (11.3–15.8) | 0.271 |
| HbA1c | 5.4 (5.2–5.6) | 5.3 (4.9–5.6) | 5.4 (5.2–5.7) | 5.4 (5.2–5.6) | 5.5 (5.2–5.8) | 0.207 |
| Sodium (mEq/L) | 140 (138–141) | 140 (138–142) | 140 (138–141) | 139 (138–140) | 141 (138–142) | 0.239 |
| Potassium (mEq/L) | 4.2 (4.0–4.4) | 4.3 (4.1–4.4) | 4.2 (4.0–4.5) | 4.2 (4.0–4.4) | 4.4 (4.0–4.6) | 0.495 |
| Chloride (mEq/L) | 102 (101–104) | 101 (100–103) | 102 (102–105) | 102 (100–105) | 103 (102–104) | 0.144 |
| eGFR (mL/min/1.73 m2) | 76.5 (63.8–83.7) | 78.9 (70.8–97.3) | 72.8 (63.0–81.0) | 76.4 (64.7–84.1) | 71.6 (60.2–78.7) | 0.143 |
| SUA (mg/dL) | 8.8 (8.5–9.5) | 8.8 (8.6–9.4) | 8.8 (8.4–9.7) | 8.4 (8.2–8.6) a | 9.7 (9.4–10.5) a,b | <0.001 |
| UUA (mg/dL) | 9 (7–13) | 9 (8–31) | 9 (7–13) | 8 (6–12) | 10 (8–14) | 0.367 |
| UUA/UCr ratio | 0.42 (0.33–0.50) | 0.46 (0.39–0.50) | 0.39 (0.31–0.50) | 0.41 (0.32–0.50) | 0.38 (0.31–0.50) | 0.51 |
| FEUA (%) | 4.1 (3.3–4.8) | 4.4 (3.9–4.8) | 4.0 (3.2–5.1) | 4.2 (3.3–5.4) | 3.6 (2.9–4.8) | 0.33 |
| Gout | ||||||
|---|---|---|---|---|---|---|
| AHU | Overall Gout | Low-Risk Gout (SUA < 9.0 mg/dL) | High-Risk Gout (SUA ≥ 9.0 mg/dL) | |||
| Variant | rsID | VAF | VAF | VAF | VAF | Ref. VAF |
| ABCG2 SNPs | ||||||
| Non-synonymous variants | ||||||
| p.Q126X, c.376C>T | rs72552713 | 0.056 | 0.015 | 0 | 0.031 | 0.009 |
| p.Q141K, c.421C>A | rs2231142 | 0.500 | 0.394 | 0.324 | 0.469 | 0.291 |
| SLC2A9 SNPs | ||||||
| Non-synonymous variants | ||||||
| p.A17T, c.49G>A | rs6820230 | 0.222 | 0.091 | 0.118 | 0.063 | 0.074 |
| p.G25R, c.73G>A | rs2276961 | 0.333 | 0.424 | 0.412 | 0.438 | 0.451 |
| p.R294H, c.881G>A | rs3733591 | 0.611 | 0.682 | 0.706 | 0.656 | 0.686 |
| p.P350L, c.1049C>T | rs2280205 | 0.111 | 0.288 | 0.176 | 0.406 | 0.286 |
| Synonymous variants | ||||||
| p.L108L, c.322T>C | rs13113918 | 1 | 0.970 | 0.941 | 1 | 0.975 |
| p.T125T, c375G>A | rs10939650 | 0.611 | 0.515 | 0.559 | 0.469 | 0.488 |
| p.I168I, c.504C>T | rs3733589 | 0.333 | 0.470 | 0.471 | 0.469 | 0.401 |
| p.L189L, c.567T>C | rs13125646 | 1 | 1 | 1 | 1 | 0.977 |
| Median (IQR: Q1–Q3) | |||||||
|---|---|---|---|---|---|---|---|
| Metabolites (Arbitrary Units) | Sample | All (n = 42) | AHU (n = 9) | Gout (n = 33) | Trend | Raw p | Adjusted p |
| Glucose metabolites | |||||||
| Glucose | Plasma | 7.02 (5.98–8.67) | 5.88 (4.93–6.75) | 7.43 (6.35–9.07) | ↑ | 0.006 | 0.074 |
| Pyruvate | Plasma | 0.67 (0.57–0.81) | 0.60 (0.54–0.67) | 0.70 (0.60–0.91) | ↑ | 0.048 | 0.124 |
| Lactate | Plasma | 7.42 (6.38–9.41) | 6.28 (5.63–7.71) | 7.86 (6.77–10.00) | ↑ | 0.018 | 0.080 |
| 1,5-Anhydroglucitol (×10−2) | Plasma | 0.63 (0–1.00) | 0.86 (0.67–1.48) | 0.54 (0–0.95) | ↓ | 0.037 | 0.121 |
| 2-Phosphoglycerate (×10−3) | Urinary | 0 (0–0) | 0 (0–6.60) | 0 (0–0) | - | 0.125 | 0.180 |
| TCA cycle intermediates | |||||||
| Aconitate (×10−3) | Plasma | 0 (0–1.70) | 0 (0–0) | 0 (0–7.46) | - | 0.065 | 0.127 |
| Isocitrate (×10−2) | Plasma | 2.18 (1.96–2.73) | 1.89 (1.62–2.13) | 2.34 (2.07–2.86) | ↑ | 0.015 | 0.092 |
| 2-Ketoglutarate (×10−2) | Plasma | 7.04 (5.53–8.62) | 5.18 (4.61–5.85) | 7.33 (6.45–9.01) | ↑ | <0.001 | 0.039 |
| Malate (×10−3) | Plasma | 5.02 (0–6.70) | 4.62 (0–5.33) | 5.51 (0–7.05) | - | 0.349 | 0.372 |
| Lipid metabolites | |||||||
| Arachidonate (×10−1) | Plasma | 0.72 (0.52–0.86) | 0.85 (0.73–1.02) | 0.67 (0.43–0.83) | ↓ | 0.015 | 0.082 |
| Eicosapentaenoate (×10−2) | Plasma | 7.33 (5.33–8.83) | 9.31 (7.38–9.53) | 7.18 (4.92–8.55) | ↓ | 0.013 | 0.091 |
| Docosahexaenoate (×10−2) | Plasma | 1.20 (0–1.78) | 0.55 (0–1.45) | 1.25 (0.38–1.93) | - | 0.117 | 0.179 |
| Caproate (×10−1) | Plasma | 1.18 (1.03–1.39) | 1.30 (1.07–1.41) | 1.17 (0.95–1.41) | - | 0.350 | 0.365 |
| Nonanoate (×10−2) | Plasma | 3.63 (2.98–6.88) | 6.84 (6.27–7.29) | 3.31 (2.67–5.93) | ↓ | 0.004 | 0.065 |
| Amino acid metabolites | |||||||
| Tryptophan | Plasma | 1.22 (0.93–1.74) | 1.58 (1.19–2.20) | 1.11 (0.85–1.70) | - | 0.055 | 0.128 |
| 5-Oxoproline | Plasma | 7.17 (5.85–7.84) | 7.77 (6.71–8.75) | 6.86 (5.72–7.71) | - | 0.064 | 0.131 |
| Serine | Plasma | 0.48 (0.19–1.02) | 0.98 (0.46–1.12) | 0.42 (0.18–0.87) | - | 0.083 | 0.151 |
| Cysteine (×10−2) | Plasma | 3.12 (2.46–4.51) | 2.58 (1.62–3.01) | 3.22 (2.59–5.06) | - | 0.055 | 0.123 |
| Lysine | Plasma | 0.57 (0.36–0.86) | 0.59 (0.51–0.97) | 0.55 (0.35–0.81) | - | 0.319 | 0.364 |
| Tyramine | Plasma | 1.20 (0.71–1.67) | 1.27 (0.97–1.91) | 1.15 (0.68–1.63) | - | 0.366 | 0.374 |
| Aspartate (×10−2) | Urinary | 4.35 (2.59–7.91) | 6.32 (4.82–9.39) | 3.85 (2.47–7.50) | - | 0.122 | 0.181 |
| Tyramine (×10−1) | Urinary | 2.36 (1.69–3.80) | 3.74 (1.94–7.25) | 2.34 (1.63–3.61) | - | 0.145 | 0.197 |
| 4-Aminobutyrate (×10−2) | Urinary | 4.24 (0–7.43) | 7.35 (3.82–9.97) | 3.63 (0–6.28) | - | 0.051 | 0.125 |
| 5-Aminolevulinate (×10−2) | Urinary | 1.33 (0.87–1.82) | 1.72 (1.14–2.39) | 1.10 (0.42–1.62) | - | 0.106 | 0.173 |
| 3-Aminoglutarate (×10−2) | Urinary | 4.97 (3.03–8.78) | 7.64 (5.35–10.38) | 4.31 (2.84–8.18) | - | 0.095 | 0.161 |
| Hypotaurine (×10−2) | Urinary | 0 (0–5.66) | 0 (0–0.68) | 1.94 (0–6.37) | ↑ | 0.043 | 0.132 |
| Purine and pyrimidine metabolites | |||||||
| Adenosine (×10−1) | Urinary | 0.91 (0.67–1.51) | 1.31 (0.78–2.39) | 0.90 (0.61–1.28) | - | 0.108 | 0.171 |
| Guanine (×10−2) | Urinary | 6.55 (3.14–9.89) | 7.87 (4.98–11.56) | 6.05 (2.38–7.93) | - | 0.129 | 0.181 |
| Uracil (×10−2) | Urinary | 0.17 (0–0.85) | 0.59 (0–1.71) | 0 (0–0.78) | - | 0.232 | 0.291 |
| Saccharides | |||||||
| Mannose (×10−1) | Plasma | 0.59 (0.32–0.89) | 0.43 (0.20–0.69) | 0.64 (0.34–0.91) | - | 0.173 | 0.229 |
| Psicose (×10−2) | Plasma | 1.90 (1.27–4.20) | 3.16 (1.79–5.00) | 1.84 (0.93–3.37) | - | 0.078 | 0.147 |
| Sorbose (×10−2) | Plasma | 1.14 (0–2.18) | 2.14 (0.61–4.31) | 0.98 (0–1.89) | ↓ | 0.043 | 0.124 |
| Sucrose (×10−2) | Plasma | 0 (0–1.24) | 1.69 (0–6.92) | 0 (0–0.79) | ↓ | 0.029 | 0.109 |
| Sucrose | Urinary | 0.18 (0.10–0.38) | 1.08 (0.04–1.20) | 0.18 (0.11–0.33) | - | 0.250 | 0.306 |
| Tagatose (×10−1) | Urinary | 0.38 (0.18–1.19) | 0.93 (0.10–1.37) | 0.36 (0.18–0.89) | - | 0.334 | 0.372 |
| Xylose (×10−1) | Urinary | 1.17 (0.55–1.79) | 1.20 (0.36–1.65) | 1.16 (0.59–1.94) | - | 0.510 | 0.510 |
| Sugar alcohols | |||||||
| Mannitol | Plasma | 1.04 (0.87–1.26) | 0.94 (0.78–1.03) | 1.10 (0.91–1.35) | ↑ | 0.033 | 0.116 |
| Sorbitol (×10−1) | Plasma | 1.31 (0.94–1.97) | 1.95 (0.81–2.40) | 1.26 (0.95–1.90) | - | 0.305 | 0.356 |
| Galactitol (×10−1) | Urinary | 1.49 (0.79–2.44) | 1.64 (1.15–4.71) | 1.40 (0.78–2.37) | - | 0.214 | 0.276 |
| Urinary monocarboxylates | |||||||
| Lactate | Urinary | 0.86 (0.64–1.15) | 0.69 (0.55–0.91) | 0.97 (0.68–1.17) | ↑ | 0.045 | 0.123 |
| Nicotinate (×10−2) | Urinary | 3.42 (1.20–7.61) | 0.99 (0.35–1.37) | 5.42 (1.97–8.78) | ↑ | 0.001 | 0.025 |
| Others | |||||||
| 3-Hydroxypropionate (×10−2) | Plasma | 7.08 (6.32–8.13) | 6.45 (5.63–8.04) | 7.38 (6.42–8.13) | - | 0.334 | 0.364 |
| Phosphorate | Plasma | 20.8 (19.0–24.3) | 23.9 (20.6–27.6) | 20.2 (18.2–22.5) | ↓ | 0.024 | 0.098 |
| Pyrogallol (×10−3) | Plasma | 0 (0–0) | 0 (0–5.30) | 0 (0–0) | ↓ | 0.006 | 0.059 |
| 3-Methoxy-4-hydroxybenzoate (×10−2) | Urinary | 1.06 (0–1.80) | 0.77 (0–1.08) | 1.29 (0–1.95) | - | 0.094 | 0.165 |
| Creatinine | Urinary | 0.65 (0.41–1.03) | 1.02 (0.53–2.00) | 0.60 (0.40–0.91) | - | 0.055 | 0.117 |
| Hydroxylamine | Urinary | 0.84 (0.65–1.00) | 0.67 (0.50–0.79) | 0.90 (0.70–1.05) | ↑ | 0.007 | 0.057 |
| Malonate (×10−2) | Urinary | 1.63 (1.04–2.29) | 1.27 (1.01–2.03) | 1.68 (1.12–2.67) | - | 0.263 | 0.314 |
| Paraxanthine (×10−2) | Urinary | 3.78 (1.52–8.06) | 1.53 (0.33–3.38) | 4.67 (2.68–8.45) | ↑ | 0.016 | 0.078 |
| Overall Gout | Low-Risk Gout (SUA < 9.0 mg/dL) | High-Risk Gout (SUA ≥ 9.0 mg/dL) | |||||
|---|---|---|---|---|---|---|---|
| Sample | Fold Difference | Adjusted p | Fold Difference | Adjusted p | Fold Difference | Adjusted p | |
| Glucose metabolites | |||||||
| Glucose | Plasma | 1.264 | 0.074 | 1.346 | 0.387 | 1.199 | 0.129 |
| Pyruvate | Plasma | 1.167 | 0.124 | 1.113 | 0.596 | 1.327 | 0.116 |
| Lactate | Plasma | 1.252 | 0.080 | 1.250 | 0.389 | 1.291 | 0.137 |
| 1,5-Anhydroglucitol | Plasma | 0.628 | 0.121 | 0.698 | 0.470 | 0.616 | 0.170 |
| 2-Phosphoglycerate | Urinary | – | 0.180 | – | 0.445 | – | 0.526 |
| TCA cycle intermediates | |||||||
| Aconitate | Plasma | – | 0.127 | – | 0.497 | – | 0.128 |
| Isocitrate | Plasma | 1.238 | 0.092 | 1.153 | 0.477 | 1.369 | 0.111 |
| 2-Ketoglutarate | Plasma | 1.415 | 0.039 | 1.277 | 0.336 | 1.566 | 0.016 |
| Malate | Plasma | 1.193 | 0.372 | 1.198 | 0.910 | 1.398 | 0.149 |
| Lipid metabolites | |||||||
| Arachidonate | Plasma | 0.788 | 0.082 | 0.897 | 0.443 | 0.656 | 0.065 |
| Eicosapentaenoate | Plasma | 0.771 | 0.091 | 0.807 | 0.428 | 0.612 | 0.065 |
| Docosahexaenoate | Plasma | 2.273 | 0.179 | 2.818 | 0.469 | 1.927 | 0.552 |
| Caproate | Plasma | 0.900 | 0.365 | 0.985 | 0.962 | 0.838 | 0.164 |
| Nonanoate | Plasma | 0.484 | 0.065 | 0.675 | 0.394 | 0.432 | 0.031 |
| Amino acid metabolites | |||||||
| Tryptophan | Plasma | 0.703 | 0.128 | 0.797 | 0.443 | 0.696 | 0.185 |
| 5-Oxoproline | Plasma | 0.883 | 0.131 | 0.883 | 0.411 | 0.883 | 0.230 |
| Serine | Plasma | 0.429 | 0.151 | 0.434 | 0.408 | 0.421 | 0.327 |
| Cysteine | Plasma | 1.248 | 0.123 | 0.223 | 0.450 | 0.280 | 0.539 |
| Lysine | Plasma | 0.932 | 0.364 | 0.898 | 0.478 | 1.085 | 0.975 |
| Tyramine | Plasma | 0.906 | 0.374 | 0.866 | 0.500 | 1.016 | 0.983 |
| Aspartate | Urinary | 0.609 | 0.181 | 0.945 | 0.863 | 0.483 | 0.129 |
| Tyramine | Urinary | 0.626 | 0.197 | 0.791 | 0.796 | 0.495 | 0.180 |
| 4-Aminobutyrate | Urinary | 0.494 | 0.125 | 0.581 | 0.485 | 0.245 | 0.130 |
| 5-Aminolevulinate | Urinary | 0.640 | 0.173 | 0.804 | 0.798 | 0.5 | 0.143 |
| 3-Aminoglutarate | Urinary | 0.564 | 0.161 | 0.707 | 0.815 | 0.436 | 0.135 |
| Hypotaurine | Urinary | – | 0.132 | – | 0.463 | – | 0.169 |
| Purine and pyrimidine metabolites | |||||||
| Adenosine | Urinary | 0.687 | 0.171 | 0.763 | 0.715 | 0.542 | 0.172 |
| Guanine | Urinary | 0.769 | 0.181 | 0.848 | 0.789 | 0.555 | 0.171 |
| Uracil | Urinary | – | 0.291 | 0.983 | 0.891 | – | 0.243 |
| Saccharides | |||||||
| Mannose | Plasma | 1.488 | 0.229 | 1.430 | 0.479 | 1.280 | 0.490 |
| Psicose | Plasma | 0.582 | 0.147 | 0.661 | 0.526 | 0.544 | 0.178 |
| Sorbose | Plasma | 0.458 | 0.124 | 0.621 | 0.528 | 0.244 | 0.123 |
| Sucrose | Plasma | - | 0.109 | - | 0.359 | - | 0.288 |
| Sucrose | Urinary | 0.167 | 0.306 | 0.944 | 0.583 | 1.056 | 0.593 |
| Tagatose | Urinary | 0.387 | 0.372 | 0.398 | 0.801 | 0.290 | 0.534 |
| Xylose | Urinary | 0.967 | 0.510 | 1.208 | 0.462 | 0.667 | 0.988 |
| Sugar alcohols | |||||||
| Mannitol | Plasma | 1.170 | 0.116 | 1.213 | 0.353 | 1.074 | 0.336 |
| Sorbitol | Plasma | 0.646 | 0.356 | 0.913 | 0.962 | 0.579 | 0.281 |
| Galactitol | Urinary | 0.854 | 0.276 | 0.872 | 0.686 | 0.689 | 0.422 |
| Urinary monocarboxylates | |||||||
| Lactate | Urinary | 1.406 | 0.123 | 1.333 | 0.435 | 1.449 | 0.173 |
| Nicotinate | Urinary | 5.475 | 0.025 | 3.616 | 0.380 | 6.515 | 0.020 |
| Others | |||||||
| 3-Hydroxypropionate | Plasma | 1.144 | 0.364 | 1.220 | 0.426 | 1.051 | 0.999 |
| Phosphorate | Plasma | 0.845 | 0.098 | 0.851 | 0.432 | 0.831 | 0.146 |
| Pyrogallol | Plasma | – | 0.059 | – | 0.456 | – | 0.186 |
| 3-Methoxy-4-hydroxybenzoate | Urinary | 1.675 | 0.165 | 2.039 | 0.343 | 1.221 | 0.741 |
| Creatinine | Urinary | 0.588 | 0.117 | 0.627 | 0.429 | 0.559 | 0.211 |
| Hydroxylamine | Urinary | 1.343 | 0.057 | 1.322 | 0.412 | 1.371 | 0.067 |
| Malonate | Urinary | 1.323 | 0.314 | 1.724 | 0.380 | 1.024 | 0.984 |
| Paraxanthine | Urinary | 3.052 | 0.078 | 3.052 | 0.882 | 2.961 | 0.237 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohashi, Y.; Ooyama, H.; Makinoshima, H.; Takada, T.; Matsuo, H.; Ichida, K. Plasma and Urinary Metabolomic Analysis of Gout and Asymptomatic Hyperuricemia and Profiling of Potential Biomarkers: A Pilot Study. Biomedicines 2024, 12, 300. https://doi.org/10.3390/biomedicines12020300
Ohashi Y, Ooyama H, Makinoshima H, Takada T, Matsuo H, Ichida K. Plasma and Urinary Metabolomic Analysis of Gout and Asymptomatic Hyperuricemia and Profiling of Potential Biomarkers: A Pilot Study. Biomedicines. 2024; 12(2):300. https://doi.org/10.3390/biomedicines12020300
Chicago/Turabian StyleOhashi, Yuki, Hiroshi Ooyama, Hideki Makinoshima, Tappei Takada, Hirotaka Matsuo, and Kimiyoshi Ichida. 2024. "Plasma and Urinary Metabolomic Analysis of Gout and Asymptomatic Hyperuricemia and Profiling of Potential Biomarkers: A Pilot Study" Biomedicines 12, no. 2: 300. https://doi.org/10.3390/biomedicines12020300
APA StyleOhashi, Y., Ooyama, H., Makinoshima, H., Takada, T., Matsuo, H., & Ichida, K. (2024). Plasma and Urinary Metabolomic Analysis of Gout and Asymptomatic Hyperuricemia and Profiling of Potential Biomarkers: A Pilot Study. Biomedicines, 12(2), 300. https://doi.org/10.3390/biomedicines12020300