
Therapeutic Opportunities of Disrupting Genome Integrity in Adult Diffuse Glioma

 , , , , and
, , , , and
Abstract
1. Introduction
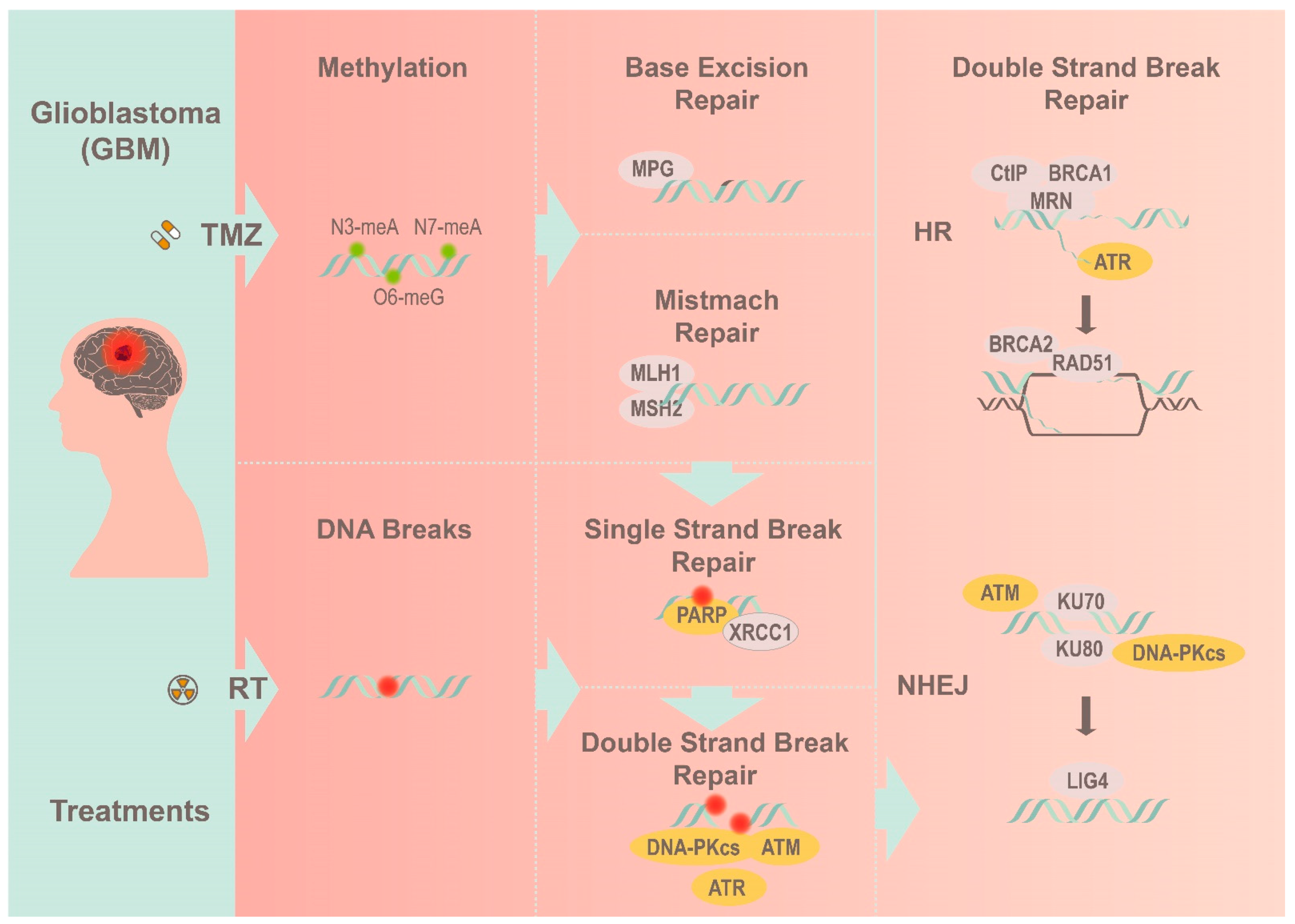
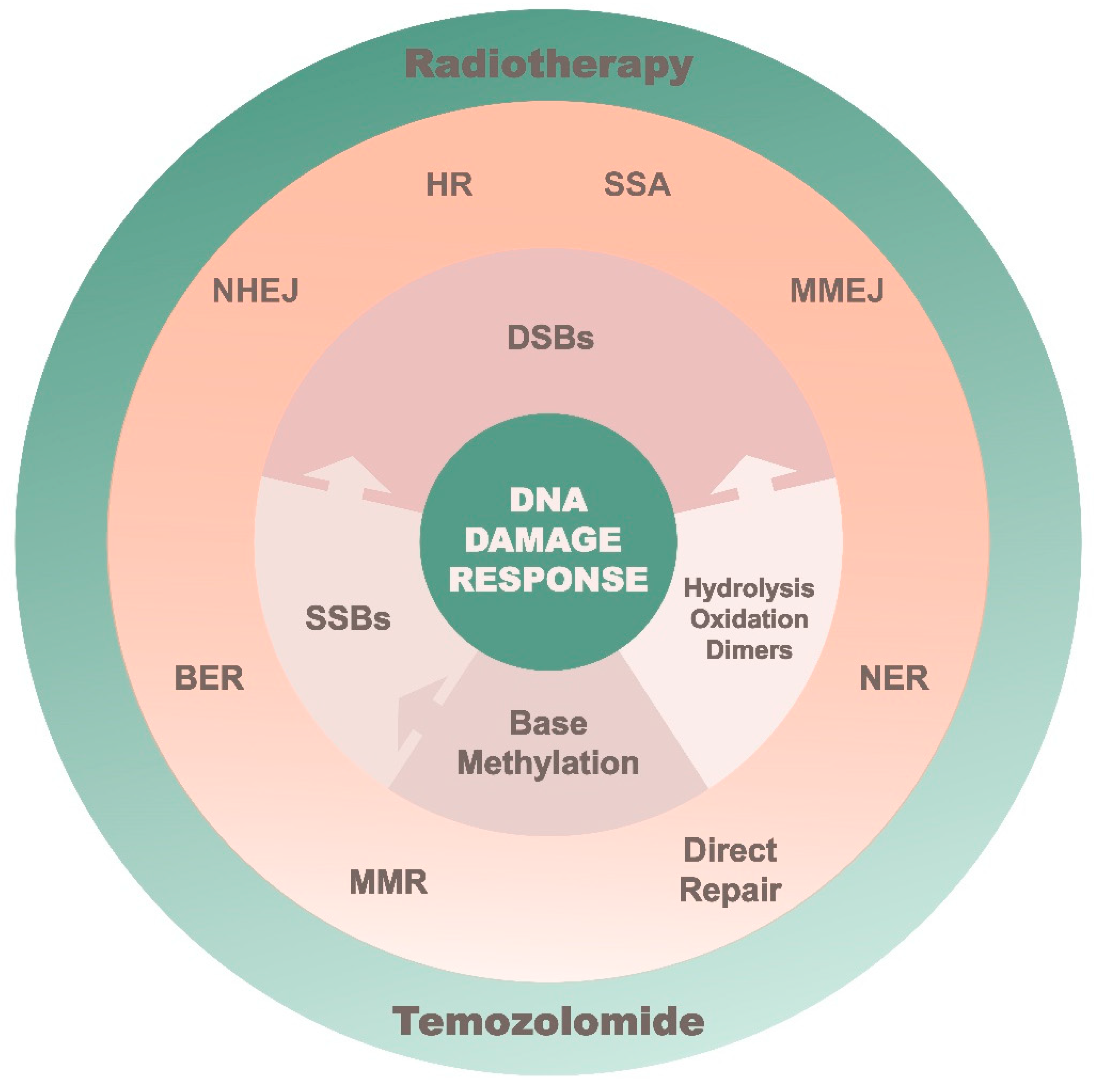
2. Implications of the DNA Damage Response Cascade
3. Therapeutics against the DNA Damage Response
4. Alternate Strategy in Achieving Therapeutic Susceptibility
5. Mutational Signature in Predicting DNA Damage Response
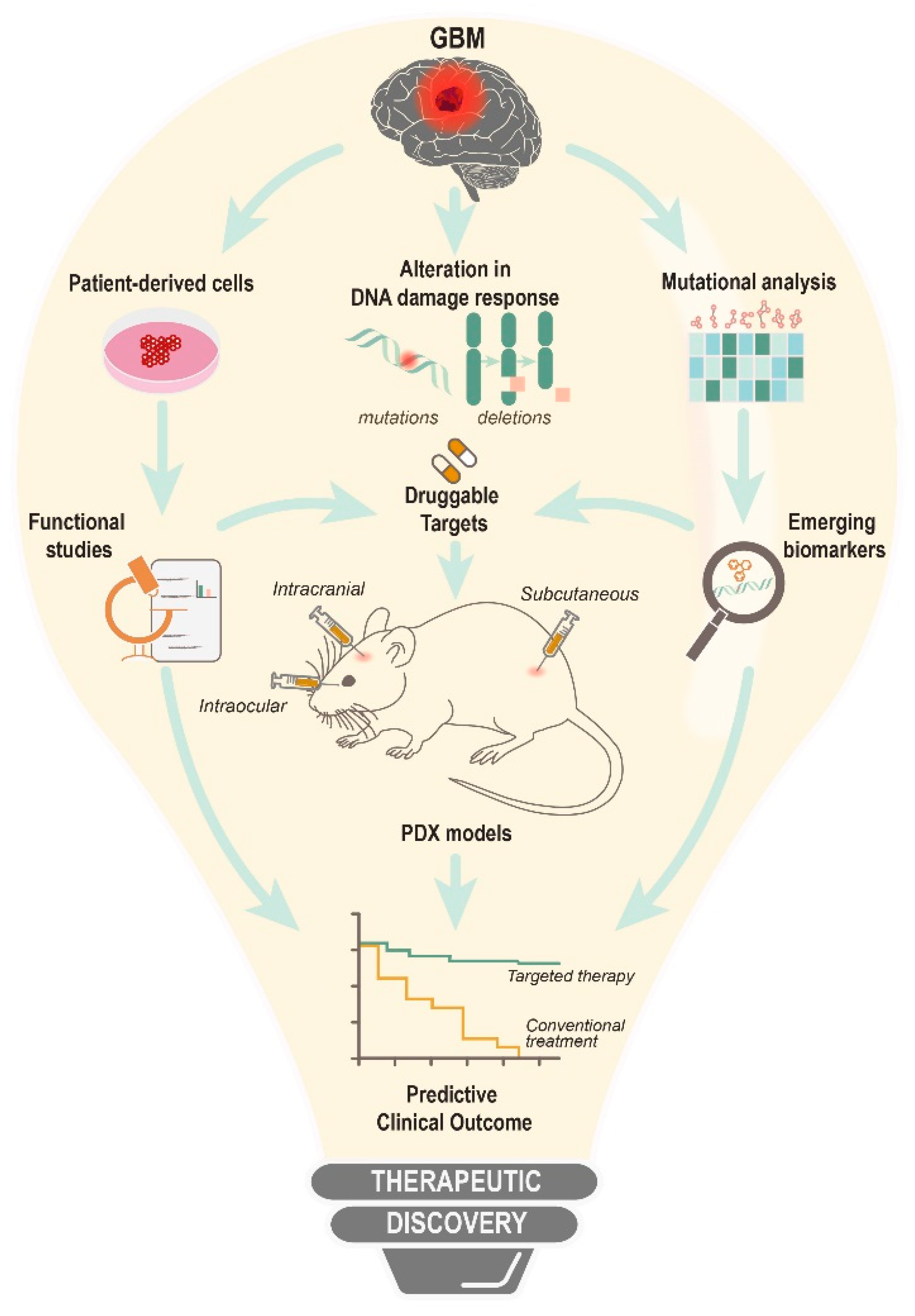
6. Targeting DNA Damage Response from Preclinical Models to Clinical Trials
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 53BP1 | p53-Binding Protein 1 |
| AMBRA1 | Autophagy and Beclin 1 Regulator 1 |
| ATM | Ataxia-Telangiectasia Mutated |
| ATR | Ataxia-Telangiectasia and Rad3-related |
| BBB | Blood-Brain Barrier |
| BCL-2 | B-cell Lymphoma 2 |
| BCR-ABL | Breakpoint cluster region—Abelson murine leukemia viral oncogene homolog 1 |
| BER | Base Excision Repair |
| BRAF | B-Raf proto-oncogene |
| BRCA1 | Breast Cancer 1 |
| BRCA2 | Breast Cancer 2 |
| BRCT | BRCA1 C-terminal domain |
| C–Abl | Abelson murine leukemia viral oncogene homolog 1, variant c |
| CDC25 | Cell Division Cycle 25 |
| CDC25A | Cell Division Cycle 25A |
| CDK2 | Cyclin Dependent Kinase 2 |
| CDK18 | Cyclin Dependent Kinase 18 |
| CDKN2A | Cyclin Dependent Kinase Inhibitor 2A |
| CHK1 | Checkpoint Kinase 1 |
| CHK2 | Checkpoint Kinase 2 |
| DDR | DNA Damage Response |
| DNA-PKcs | DNA-dependent Protein Kinase catalytic subunit |
| DSB | Double Strand Break |
| EGFR | Epidermal Growth Factor Receptor |
| EGFRvIII | Epidermal Growth Factor Receptor variant III |
| FANCD2 | Fanconi Anemia Complementation Group D2 |
| GNS | Glioma-derived neural stem-like |
| H2AX | H2A Histone family member X |
| HR | Homologous Recombination |
| H3k9me3 | Histone 3 lysine 9 trimethylation |
| IDH | Isocitrate Dehydrogenase |
| KDM4B | Histone Lysine Demethylase subfamily 4B |
| NER | Nucleotide Excision Repair |
| NBS1 | Nijmegen Breakage Syndrome 1 |
| NF1 | Neurofibromin 1 |
| NHEJ | Non-Homologous End Joining |
| MDC1 | Mediator of DNA Damage Checkpoint |
| MMEJ | Microhomology-mediated End Joining |
| MMR | Mismatch Repair |
| MRN | MRE11-RAD50-NBS1 protein complex |
| MYC | Myelocytomatosis viral related oncogene |
| MYCN | Myelocytomatosis viral related oncogene member N of the MYC family |
| P53 | Tumour Suppressor Protein 53-kDa |
| PARP | Poly (ADP-ribose) Polymerase |
| PDGFRA | Platelet Derived Growth Factor Receptor Alpha |
| PDX | Patient-Derived Xenografts |
| POLD1 | DNA Polymerase Delta 1 |
| Pol θ | DNA Polymerase θ |
| PTEN | Phosphatase and Tensin Homolog |
| RAC1 | RAS related C3 botulinum Toxin Substrate 1 |
| RAD51 | DNA Repair Protein RAD51 homolog |
| RAD52 | DNA Repair Protein RAD52 homolog |
| RAP80 | Receptor-Associated Protein 80-kDa |
| RAS | Rat Sarcoma Virus, small GTPase family |
| RNF8 | Ring Finger Protein 8 |
| SIRT6 | Sirtuin 6 |
| SMC1 | Structural Maintenance of Chromosomes 1 |
| SSA | Single Strand Annealing |
| SSB | Single Strand Break |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| Tip60 | Tat-Interactive Protein 60-kDa |
| TMZ | Temozolomide |
| WEE1 | Wee1-like protein kinase |
References
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.C.; Roberts, T.L.; Day, B.W.; Stringer, B.W.; Kozlov, S.; Fazry, S.; Bruce, Z.C.; Ensbey, K.S.; Walker, D.G.; Boyd, A.W.; et al. Increased sensitivity to ionizing radiation by targeting the homologous recombination pathway in glioma initiating cells. Mol. Oncol. 2014, 8, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, B.; McEllin, B.; Camacho, C.; Tomimatsu, N.; Sirasanagandala, S.; Nannepaga, S.; Hatanpaa, K.J.; Mickey, B.; Madden, C.; Maher, E.; et al. EGFRvIII and DNA Double-Strand Break Repair: A Molecular Mechanism for Radioresistance in Glioblastoma. Cancer Res. 2009, 69, 4252–4259. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef]
- Barthel, F.P.; Johnson, K.C.; Varn, F.S.; Moskalik, A.D.; Tanner, G.; Kocakavuk, E.; Anderson, K.J.; Abiola, O.; Aldape, K.; Alfaro, K.D.; et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019, 576, 112–120. [Google Scholar] [CrossRef]
- Kocakavuk, E.; Anderson, K.J.; Varn, F.S.; Johnson, K.C.; Amin, S.B.; Sulman, E.P.; Lolkema, M.P.; Barthel, F.P.; Verhaak, R.G.W. Radiotherapy is associated with a deletion signature that contributes to poor outcomes in patients with cancer. Nat. Genet. 2021, 53, 1088–1096. [Google Scholar] [CrossRef]
- Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246. [Google Scholar] [CrossRef]
- Sedletska, Y.; Radicella, J.P.; Sage, E. Replication fork collapse is a major cause of the high mutation frequency at three-base lesion clusters. Nucleic Acids Res. 2013, 41, 9339–9348. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Dahan, P.; Gala, J.M.; Delmas, C.; Monferran, S.; Malric, L.; Zentkowski, D.; Lubrano, V.; Toulas, C.; Moyal, E.C.-J.; Lemarie, A. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: Possible involvement in radioresistance. Cell Death Dis. 2014, 5, e1543. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Xu, Y.; Ayrapetov, M.K.; Moreau, L.A.; Whetstine, J.R.; Price, B.D. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 2009, 11, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 Directly Binds Phosphorylated Histone H2AX to Regulate Cellular Responses to DNA Double-Strand Breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef]
- Lou, Z.; Minter-Dykhouse, K.; Franco, S.; Gostissa, M.; Rivera, M.A.; Celeste, A.; Manis, J.P.; van Deursen, J.; Nussenzweig, A.; Paull, T.T.; et al. MDC1 Maintains Genomic Stability by Participating in the Amplification of ATM-Dependent DNA Damage Signals. Mol. Cell 2006, 21, 187–200. [Google Scholar] [CrossRef]
- Huen, M.S.-Y.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 Transduces the DNA-Damage Signal via Histone Ubiquitylation and Checkpoint Protein Assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 Ubiquitylates Histones at DNA Double-Strand Breaks and Promotes Assembly of Repair Proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef]
- Botuyan, M.V.; Lee, J.; Ward, I.M.; Kim, J.-E.; Thompson, J.R.; Chen, J.; Mer, G. Structural Basis for the Methylation State-Specific Recognition of Histone H4-K20 by 53BP1 and Crb2 in DNA Repair. Cell 2006, 127, 1361–1373. [Google Scholar] [CrossRef]
- Sims, J.J.; Cohen, R.E. Linkage-Specific Avidity Defines the Lysine 63-Linked Polyubiquitin-Binding Preference of Rap80. Mol. Cell 2009, 33, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Mailand, N.; Syljuåsen, R.G.; Bartek, J.; Lukas, J. The ATM–Chk2–Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.-S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef]
- Yazdi, P.T.; Wang, Y.; Zhao, S.; Patel, N.; Lee, E.Y.-H.; Qin, J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002, 16, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Fong, C.S.; Ohtsuki, C.; Haraguchi, T.; Hiraoka, Y. Spatiotemporal regulations of Wee1 at the G2/M transition. Mol. Biol. Cell 2011, 22, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Khoronenkova, S.V.; Dianov, G.L. ATM prevents DSB formation by coordinating SSB repair and cell cycle progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3997–4002. [Google Scholar] [CrossRef]
- Chen, L.; Nievera, C.J.; Lee, A.Y.-L.; Wu, X. Cell Cycle-dependent Complex Formation of BRCA1·CtIP·MRN Is Important for DNA Double-strand Break Repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Agarwal, H.; Priya, S.; Batra, H.; Modi, P.; Pandey, M.; Saha, D.; Raghavan, S.C.; Sengupta, S. MRN complex-dependent recruitment of ubiquitylated BLM helicase to DSBs negatively regulates DNA repair pathways. Nat. Commun. 2018, 9, 1016. [Google Scholar] [CrossRef]
- Onaka, A.T.; Su, J.; Katahira, Y.; Tang, C.; Zafar, F.; Aoki, K.; Kagawa, W.; Niki, H.; Iwasaki, H.; Nakagawa, T. DNA replication machinery prevents Rad52-dependent single-strand annealing that leads to gross chromosomal rearrangements at centromeres. Commun. Biol. 2020, 3, 202. [Google Scholar] [CrossRef]
- Black, S.J.; Ozdemir, A.Y.; Kashkina, E.; Kent, T.; Rusanov, T.; Ristic, D.; Shin, Y.; Suma, A.; Hoang, T.; Chandramouly, G.; et al. Molecular basis of microhomology-mediated end-joining by purified full-length Polθ. Nat. Commun. 2019, 10, 4423. [Google Scholar] [CrossRef]
- Ning, J.-F.; Stanciu, M.; Humphrey, M.R.; Gorham, J.; Wakimoto, H.; Nishihara, R.; Lees, J.; Zou, L.; Martuza, R.L.; Rabkin, S.D. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat. Commun. 2019, 10, 2910. [Google Scholar] [CrossRef] [PubMed]
- Salles, D.; Mencalha, A.L.; Ireno, I.C.; Wiesmüller, L.; Abdelhay, E. BCR-ABL stimulates mutagenic homologous DNA double-strand break repair via the DNA-end-processing factor CtIP. Carcinogenesis 2010, 32, 27–34. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, L.; Ge, J.; Lan, Y.; Shi, Y.; Luo, Y.; Tan, Y.; Liang, M.; Deng, S.; Zhang, X.; Wang, W.; et al. Tumor mutational burden is associated with poor outcomes in diffuse glioma. BMC Cancer 2020, 20, 213. [Google Scholar] [CrossRef]
- Yang, L.; Shen, C.; Estrada-Bernal, A.; Robb, R.; Chatterjee, M.; Sebastian, N.; Webb, A.; Mo, X.; Chen, W.; Krishnan, S.; et al. Oncogenic KRAS drives radioresistance through upregulation of NRF2-53BP1-mediated non-homologous end-joining repair. Nucleic Acids Res. 2021, 49, 11067–11082. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.G.; Yeh, R.-F.; Ray, A.; Wang, N.J.; Smirnov, I.; Yu, M.; Hariono, S.; Silber, J.; Feiler, H.S.; Gray, J.W.; et al. Comparative analyses of gene copy number and mRNA expression in glioblastoma multiforme tumors and xenografts. Neuro-Oncology 2009, 11, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Petroni, M.; Sardina, F.; Infante, P.; Bartolazzi, A.; Locatelli, E.; Fabretti, F.; Di Giulio, S.; Capalbo, C.; Cardinali, B.; Coppa, A.; et al. MRE11 inhibition highlights a replication stress-dependent vulnerability of MYCN-driven tumors. Cell Death Dis. 2018, 9, 895. [Google Scholar] [CrossRef] [PubMed]
- Bajrami, I.; Walker, C.; Krastev, D.B.; Weekes, D.; Song, F.; Wicks, A.J.; Alexander, J.; Haider, S.; Brough, R.; Pettitt, S.J.; et al. Sirtuin inhibition is synthetic lethal with BRCA1 or BRCA2 deficiency. Commun. Biol. 2021, 4, 1270. [Google Scholar] [CrossRef]
- Weterings, E.; Gallegos, A.C.; Dominick, L.N.; Cooke, L.S.; Bartels, T.N.; Vagner, J.; Matsunaga, T.O.; Mahadevan, D. A novel small molecule inhibitor of the DNA repair protein Ku70/80. DNA Repair 2016, 43, 98–106. [Google Scholar] [CrossRef]
- Jones, P.; Altamura, S.; Boueres, J.; Ferrigno, F.; Fonsi, M.; Giomini, C.; Lamartina, S.; Monteagudo, E.; Ontoria, J.M.; Orsale, M.V.; et al. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): A novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J. Med. Chem. 2009, 52, 7170–7185. [Google Scholar] [CrossRef]
- Cea, M.; Cagnetta, A.; Adamia, S.; Acharya, C.; Tai, Y.-T.; Fulciniti, M.; Ohguchi, H.; Munshi, A.; Acharya, P.; Bhasin, M.K.; et al. Evidence for a role of the histone deacetylase SIRT6 in DNA damage response of multiple myeloma cells. Blood 2016, 127, 1138–1150. [Google Scholar] [CrossRef]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.B.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.; Smith, G.C.M. Identification and Characterization of a Novel and Specific Inhibitor of the Ataxia-Telangiectasia Mutated Kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; Luken, M.J.D.M.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef] [PubMed]
- Dietlein, F.; Thelen, L.; Jokic, M.; Jachimowicz, R.D.; Ivan, L.; Knittel, G.; Leeser, U.; Van Oers, J.; Edelmann, W.; Heukamp, L.; et al. A Functional Cancer Genomics Screen Identifies a Druggable Synthetic Lethal Interaction between MSH3 and PRKDC. Cancer Discov. 2014, 4, 592–605. [Google Scholar] [CrossRef]
- Signore, M.; Pelacchi, F.; di Martino, S.; Runci, D.; Biffoni, M.; Giannetti, S.; Morgante, L.; De Majo, M.; Petricoin, E.F.; Stancato, L.; et al. Combined PDK1 and CHK1 inhibition is required to kill glioblastoma stem-like cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1223. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.E.; Walton, M.I.; Eve, P.D.; Boxall, K.J.; Antoni, L.; Caldwell, J.J.; Aherne, W.; Pearl, L.H.; Oliver, A.W.; Collins, I.; et al. CCT241533 is a potent and selective inhibitor of CHK2 that potentiates the cytotoxicity of PARP inhibitors. Cancer Res. 2011, 71, 463–472. [Google Scholar] [CrossRef]
- Willmore, E.; De Caux, S.; Sunter, N.J.; Tilby, M.J.; Jackson, G.H.; Austin, C.; Durkacz, B.W. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 2004, 103, 4659–4665. [Google Scholar] [CrossRef]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Pol theta inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Motlekar, N.A.; Burgwin, C.M.; Napper, A.D.; Diamond, S.L.; Mazin, A.V. Identification of Specific Inhibitors of Human RAD51 Recombinase Using High-Throughput Screening. ACS Chem. Biol. 2011, 6, 628–635. [Google Scholar] [CrossRef]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef]
- Cornwell, M.J.; Thomson, G.J.; Coates, J.; Belotserkovskaya, R.; Waddell, I.D.; Jackson, S.P.; Galanty, Y. Small-Molecule Inhibition of UBE2T/FANCL-Mediated Ubiquitylation in the Fanconi Anemia Pathway. ACS Chem. Biol. 2019, 14, 2148–2154. [Google Scholar] [CrossRef]
- McCord, M.; Steffens, A.; Javier, R.; Kam, K.-L.; McCortney, K.; Horbinski, C. The efficacy of DNA mismatch repair enzyme immunohistochemistry as a screening test for hypermutated gliomas. Acta Neuropathol. Commun. 2020, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Jucaite, A.; Stenkrona, P.; Cselényi, Z.; De Vita, S.; Buil-Bruna, N.; Varnäs, K.; Savage, A.; Varrone, A.; Johnström, P.; Schou, M.; et al. Brain exposure of the ATM inhibitor AZD1390 in humans—A positron emission tomography study. Neuro-Oncology 2021, 23, 687–696. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Hanna, C.; Walker, S.M.; Gonda, D.; Li, J.; Wikstrom, K.; Savage, K.; Butterworth, K.; Chen, C.; Harkin, D.P.; et al. Mechanistic Rationale to Target PTEN-Deficient Tumor Cells with Inhibitors of the DNA Damage Response Kinase ATM. Cancer Res. 2015, 75, 2159–2165. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Yuan, G.; Liu, X.; Ren, R.; Li, J.; Zhang, W.; Wu, J.; Xu, X.; Fuchou, T.; Li, Y.; et al. PTEN deficiency reprogrammes human neural stem cells towards a glioblastoma stem cell-like phenotype. Nat. Commun. 2015, 6, 10068. [Google Scholar] [CrossRef] [PubMed]
- Charrier, J.-D.; Durrant, S.J.; Golec, J.M.C.; Kay, D.P.; Knegtel, R.M.A.; MacCormick, S.; Mortimore, M.; O’Donnell, M.E.; Pinder, J.L.; Reaper, P.M.; et al. Discovery of Potent and Selective Inhibitors of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Protein Kinase as Potential Anticancer Agents. J. Med. Chem. 2011, 54, 2320–2330. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; MacCormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Galban, S.; Li, F.; Heist, K.A.; Galbán, C.J.; Lawrence, T.S.; Holland, E.C.; Thomae, T.L.; Chenevert, T.L.; Rehemtulla, A.; et al. DW-MRI as a Predictive Biomarker of Radiosensitization of GBM through Targeted Inhibition of Checkpoint Kinases. Transl. Oncol. 2013, 6, 133–142. [Google Scholar] [CrossRef]
- Willis, S.G.; Lange, T.; Demehri, S.; Otto, S.; Crossman, L.; Niederwieser, D.; Stoffregen, E.P.; McWeeney, S.; Kovacs, I.; Park, B.; et al. High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: Correlation with clonal cytogenetic evolution but not response to therapy. Blood 2005, 106, 2128–2137. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Li, S.; Topatana, W.; Juengpanich, S.; Cao, J.; Hu, J.; Zhang, B.; Ma, D.; Cai, X.; Chen, M. Development of synthetic lethality in cancer: Molecular and cellular classification. Signal Transduct. Target. Ther. 2020, 5, 241. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; Van Der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Frankum, J.R.; Punta, M.; Lise, S.; Alexander, J.; Chen, Y.; Yap, T.A.; Haider, S.; Tutt, A.N.J.; Lord, C.J. Clinical BRCA1/2 reversion analysis identifies hotspot mutations and predicted neoantigens associated with therapy resistance. Cancer Discov. 2020, 10, 1475–1488. [Google Scholar] [CrossRef]
- Maiani, E.; Milletti, G.; Nazio, F.; Holdgaard, S.G.; Bartkova, J.; Rizza, S.; Cianfanelli, V.; Lorente, M.; Simoneschi, D.; Di Marco, M.; et al. AMBRA1 regulates cyclin D to guard S-phase entry and genomic integrity. Nature 2021, 592, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Hocke, S.; Guo, Y.; Job, A.; Orth, M.; Ziesch, A.; Lauber, K.; De Toni, E.N.; Gress, T.; Herbst, A.; Göke, B.; et al. A synthetic lethal screen identifies ATR-inhibition as a novel therapeutic approach for POLD1-deficient cancers. Oncotarget 2016, 7, 7080–7095. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Kass, E.M.; Yen, W.-F.; Ludwig, T.; Moynahan, M.E.; Chaudhuri, J.; Jasin, M. ATM loss leads to synthetic lethality in BRCA1 BRCT mutant mice associated with exacerbated defects in homology-directed repair. Proc. Natl. Acad. Sci. USA 2017, 114, 7665–7670. [Google Scholar] [CrossRef] [PubMed]
- Albarakati, N.; Abdel-Fatah, T.M.; Doherty, R.; Russell, R.; Agarwal, D.; Moseley, P.; Perry, C.; Arora, A.; Alsubhi, N.; Seedhouse, C.; et al. Targeting BRCA1-BER deficient breast cancer by ATM or DNA-PKcs blockade either alone or in combination with cisplatin for personalized therapy. Mol. Oncol. 2015, 9, 204–217. [Google Scholar] [CrossRef]
- Mengwasser, K.E.; Adeyemi, R.O.; Leng, Y.; Choi, M.Y.; Clairmont, C.; D’Andrea, A.D.; Elledge, S.J. Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol. Cell 2019, 73, 885–899.e6. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Zimmermann, M.; Murina, O.; Reijns, M.; Agathanggelou, A.; Challis, R.; Žygimantė, T.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018, 559, 285–289. [Google Scholar] [CrossRef]
- Brough, R.; Gulati, A.; Haider, S.; Kumar, R.; Campbell, J.; Knudsen, E.; Pettitt, S.J.; Ryan, C.J.; Lord, C.J. Identification of highly penetrant Rb-related synthetic lethal interactions in triple negative breast cancer. Oncogene 2018, 37, 5701–5718. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhao, H.; Diplas, B.H.; Liu, S.; Liu, J.; Wang, D.; Lu, Y.; Zhu, Q.; Wu, J.; Wang, W.; et al. Genome-Wide CRISPR-Cas9 Screen Reveals Selective Vulnerability of ATRX-Mutant Cancers to WEE1 Inhibition. Cancer Res. 2019, 80, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Oeck, S.; Dow, J.; Economos, N.G.; Mirfakhraie, L.; Liu, Y.; Noronha, K.; Bao, X.; Li, J.; Shuch, B.M.; et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 2020, 582, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Garama, D.J.; Harris, T.J.; White, C.L.; Rossello, F.; Abdul-Hay, M.; Gough, D.; Levy, D.E. A Synthetic Lethal Interaction between Glutathione Synthesis and Mitochondrial Reactive Oxygen Species Provides a Tumor-Specific Vulnerability Dependent on STAT3. Mol. Cell. Biol. 2015, 35, 3646–3656. [Google Scholar] [CrossRef] [PubMed]
- Ogunrinu, T.A.; Sontheimer, H. Hypoxia Increases the Dependence of Glioma Cells on Glutathione. J. Biol. Chem. 2010, 285, 37716–37724. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, S.; Li, Y.; Liu, Z.; Mi, L.; Cai, Y.; Wang, X.; Chen, L.; Ran, H.; Xiao, D.; et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat. Commun. 2021, 12, 3720. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lim, K.Y.; Park, J.W.; Kang, J.; Won, J.K.; Lee, K.; Shim, Y.; Park, C.-K.; Kim, S.-K.; Choi, S.-H.; et al. Sporadic and Lynch syndrome-associated mismatch repair-deficient brain tumors. Lab. Investig. 2021, 102, 160–171. [Google Scholar] [CrossRef] [PubMed]
- McFaline-Figueroa, J.L.; Braun, C.J.; Stanciu, M.; Nagel, Z.D.; Mazzucato, P.; Sangaraju, D.; Cerniauskas, E.; Barford, K.; Vargas, A.; Chen, Y.; et al. Minor Changes in Expression of the Mismatch Repair Protein MSH2 Exert a Major Impact on Glioblastoma Response to Temozolomide. Cancer Res. 2015, 75, 3127–3138. [Google Scholar] [CrossRef]
- Stritzelberger, J.; Distel, L.; Buslei, R.; Fietkau, R.; Putz, F. Acquired temozolomide resistance in human glioblastoma cell line U251 is caused by mismatch repair deficiency and can be overcome by lomustine. Clin. Transl. Oncol. 2017, 20, 508–516. [Google Scholar] [CrossRef]
- Martin, S.A.; McCabe, N.; Mullarkey, M.; Cummins, R.; Burgess, D.J.; Nakabeppu, Y.; Oka, S.; Kay, E.; Lord, C.J.; Ashworth, A. DNA Polymerases as Potential Therapeutic Targets for Cancers Deficient in the DNA Mismatch Repair Proteins MSH2 or MLH1. Cancer Cell 2010, 17, 235–248. [Google Scholar] [CrossRef]
- Hu, K.; Li, K.; Lv, J.; Feng, J.; Chen, J.; Wu, H.; Cheng, F.; Jiang, W.; Wang, J.; Pei, H.; et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Investig. 2020, 130, 1752–1766. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jo, D.H.; Kim, J.H.; Cho, C.S.; Han, J.E.; Kim, Y.; Park, H.; Yoo, S.H.; Yu, Y.S.; Moon, H.E.; et al. Development of a patient-derived xenograft model of glioblastoma via intravitreal injection in mice. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Burt, T.; Young, G.; Lee, W.; Kusuhara, H.; Langer, O.; Rowland, M.; Sugiyama, Y. Phase 0/microdosing approaches: Time for mainstream application in drug development? Nat. Rev. Drug Discov. 2020, 19, 801–818. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| DNA Damage Response Inhibitors | ||||
| Signaling Pathway | Therapeutic Agent | Molecular Target | Ref | |
| Sensor | MRN | Mirin | MRE11 | [36] |
| KU70/80 | STL127705 | KU70/80 and DNA-PK | [38] | |
| PARP1 | Niraparib | PARP1 and 2 | [39] | |
| SIRT6 | SIRT6-IN-1 | SIRT6,1 and 2 | [40] | |
| Transducers | ATM | KU-55933 | ATM | [41] |
| ATR | VX-970 | ATR | [42] | |
| DNA-PK | KU-0060648 | DNA-PK and PI3K | [43] | |
| Effectors | CHK1 | UCN-01 | CHK1 | [44] |
| CHK2 | CCT241533 | CHK2 | [45] | |
| DNA Repair Inhibitors | ||||
| Signaling Pathway | Therapeutic Agent | Molecular Target | Ref | |
| Double strand break repair | NHEJ | NU7026 | DNA-PK | [46] |
| MMEJ | ART558 | Pol θ | [47] | |
| HR | B02 | RAD51 | [48] | |
| SSA | D-I03 | RAD52 | [49] | |
| FA | CU2 | FANCL | [50] | |
| Biological Process | Molecular Target | Therapeutic Inhibitor | Deficient Gene | Ref |
|---|---|---|---|---|
| DDR activation | ATR | VE-821 | POLD1 | [65] |
| DNA repair | PARP1 | Niraparib | BRCA1, BRCA2 | [61] |
| DNA repair | Pol θ | ART558 | BRCA1, BRCA2, FANCD2 | [47] |
| DNA repair | ATM | KU55933 | BRCA1 | [66] |
| DNA repair | DNA-PK | NU7441 | BRCA1 | [67] |
| DNA repair | FEN1 | FEN1-IN-3 | BRCA2 | [68] |
| DNA sensor/repair | PARP1 | Niraparib | IDH1, IDH2 | [69] |
| DNA sensor/repair | PARP1 | Talazoparib | RNASEH2A, RNASEH2B | [70] |
| Checkpoint arrest | CHK1 | UCN-01 | AMBRA1 | [64] |
| Cell-cycle progression | SKP2 | SKPinC1 | RB1 | [71] |
| Cell-cycle progression | WEE1 | AZD1775 | ATRX | [72] |
| Molecular Target | Inhibitor | Combination Treatment | Disease Setting | Predictive Biomarker | Clinical Phase | Clinical Trial Number |
|---|---|---|---|---|---|---|
| PARP1/2 | Niraparib | Monotherapy | Primary GBM, recurrent GBM | IDH mut, ATRX loss | I | NCT05076513 |
| DNA-PK | Nedisertib | RT, TMZ | GBM, Gliosarcoma | Unmethylated MGMT | I | NCT04555577 |
| ATM | AZD1390 | RT | GBM | - | I | NCT03423628 |
| WEE1 | Adavosertib | RT, TMZ | Primary GBM, recurrent GBM | - | I | NCT01849146 |
| CDK4/6 | Abemaciclib | Monotherapy | Recurrent GBM, Gliosarcoma | RB | II | NCT01227434 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilar-Morante, D.; Gómez-Cabello, D.; Quek, H.; Liu, T.; Hamerlik, P.; Lim, Y.C. Therapeutic Opportunities of Disrupting Genome Integrity in Adult Diffuse Glioma. Biomedicines 2022, 10, 332. https://doi.org/10.3390/biomedicines10020332
Aguilar-Morante D, Gómez-Cabello D, Quek H, Liu T, Hamerlik P, Lim YC. Therapeutic Opportunities of Disrupting Genome Integrity in Adult Diffuse Glioma. Biomedicines. 2022; 10(2):332. https://doi.org/10.3390/biomedicines10020332
Chicago/Turabian StyleAguilar-Morante, Diana, Daniel Gómez-Cabello, Hazel Quek, Tianqing Liu, Petra Hamerlik, and Yi Chieh Lim. 2022. "Therapeutic Opportunities of Disrupting Genome Integrity in Adult Diffuse Glioma" Biomedicines 10, no. 2: 332. https://doi.org/10.3390/biomedicines10020332
APA StyleAguilar-Morante, D., Gómez-Cabello, D., Quek, H., Liu, T., Hamerlik, P., & Lim, Y. C. (2022). Therapeutic Opportunities of Disrupting Genome Integrity in Adult Diffuse Glioma. Biomedicines, 10(2), 332. https://doi.org/10.3390/biomedicines10020332