Therapeutic Strategies for Regulating Mitochondrial Oxidative Stress

 ,
,
Abstract
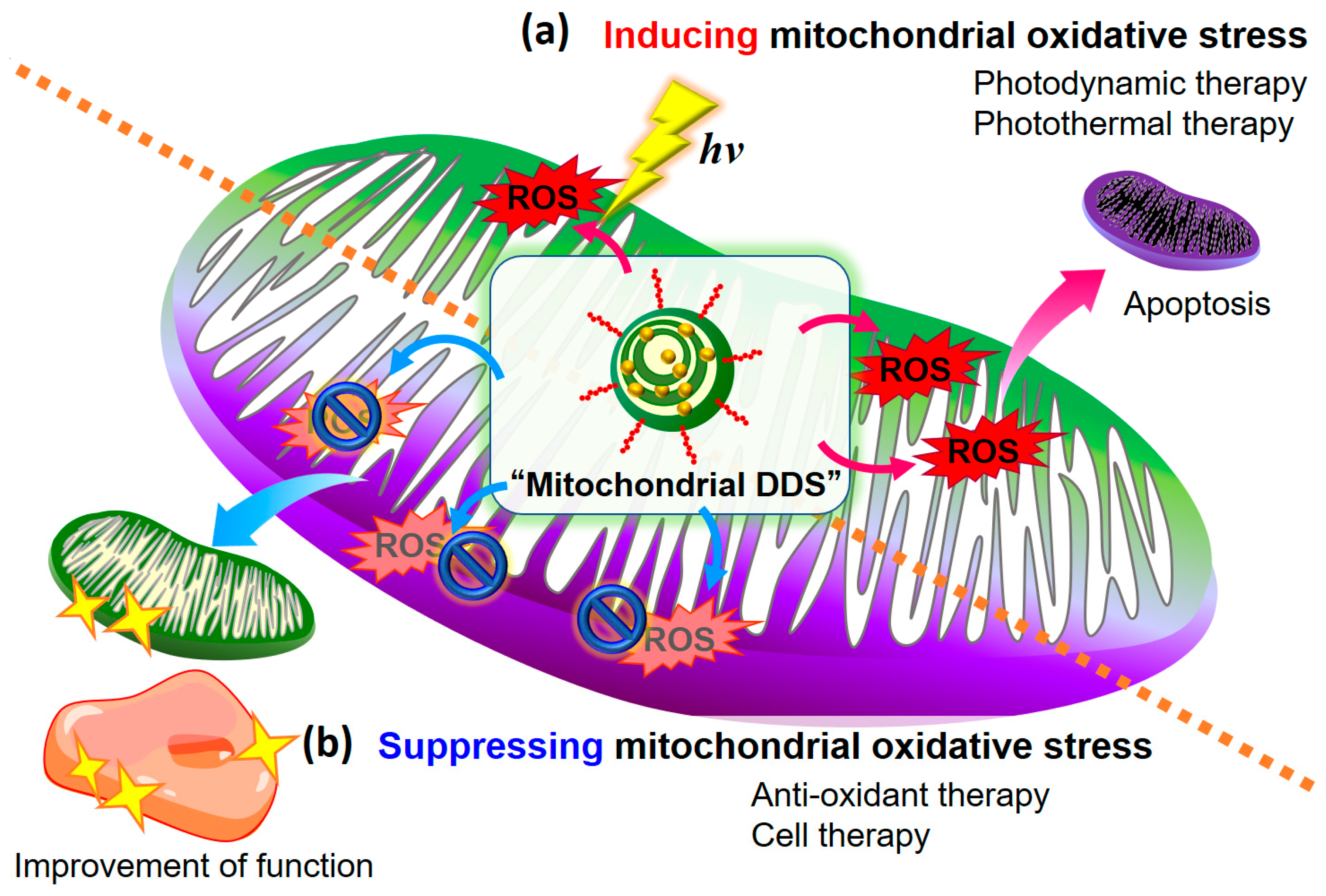
1. Introduction
2. Therapeutic Strategy to Induce Oxidative Stress
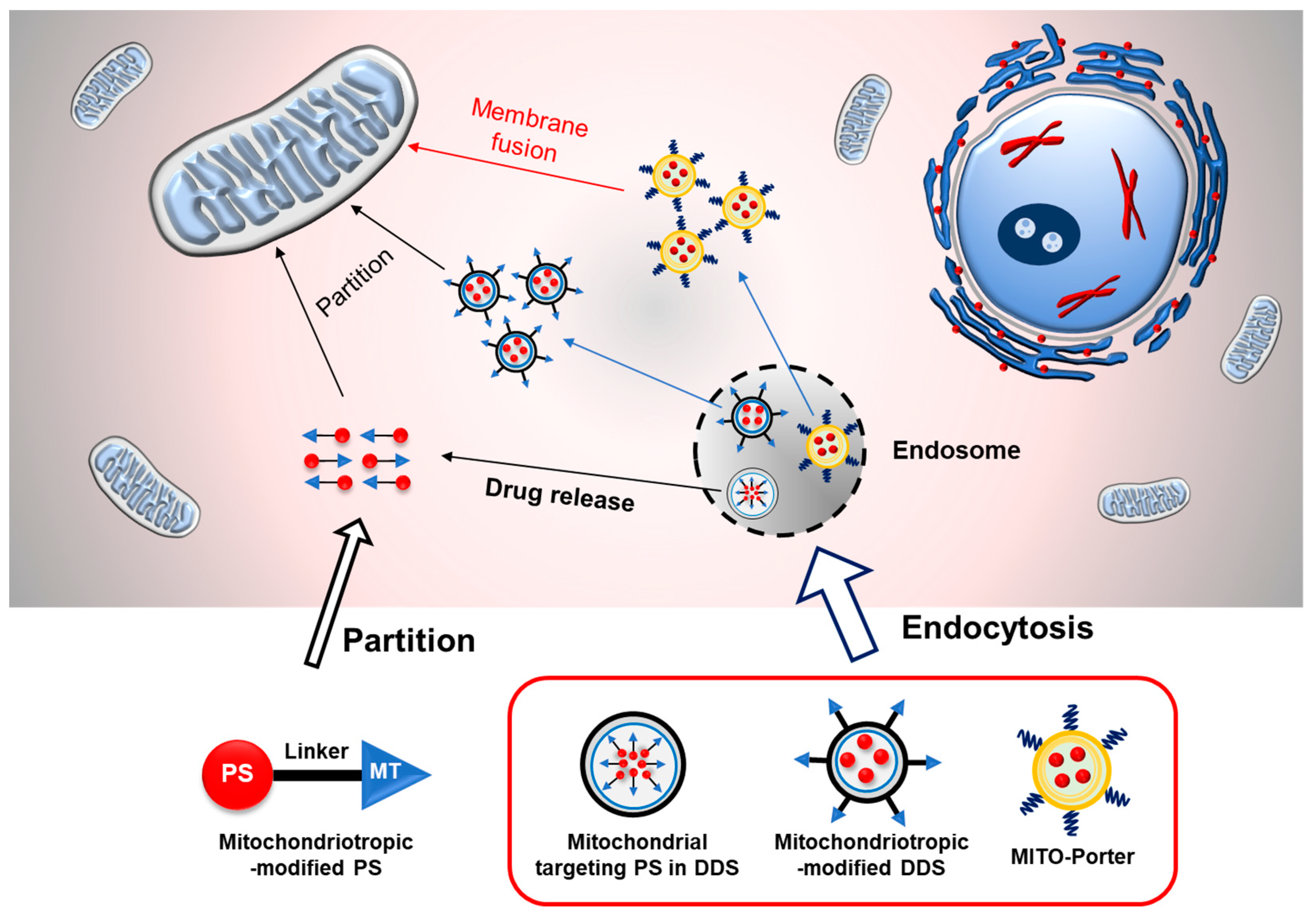
2.1. Mitochondrial Targeting PDT for Cancer Therapy
2.1.1. Chemical Conjugation of the PS with Mitochondrial Targeting Ligand
2.1.2. Mitochondrial Targeted PS in Drug Delivery System
2.1.3. Mitochondrial Targeting Ligand-Modified Drug Delivery System
2.1.4. Validation of Cancer Therapeutic Strategy Using MITO-Porter System
2.2. Other Phototherapies Targeting Mitochondria
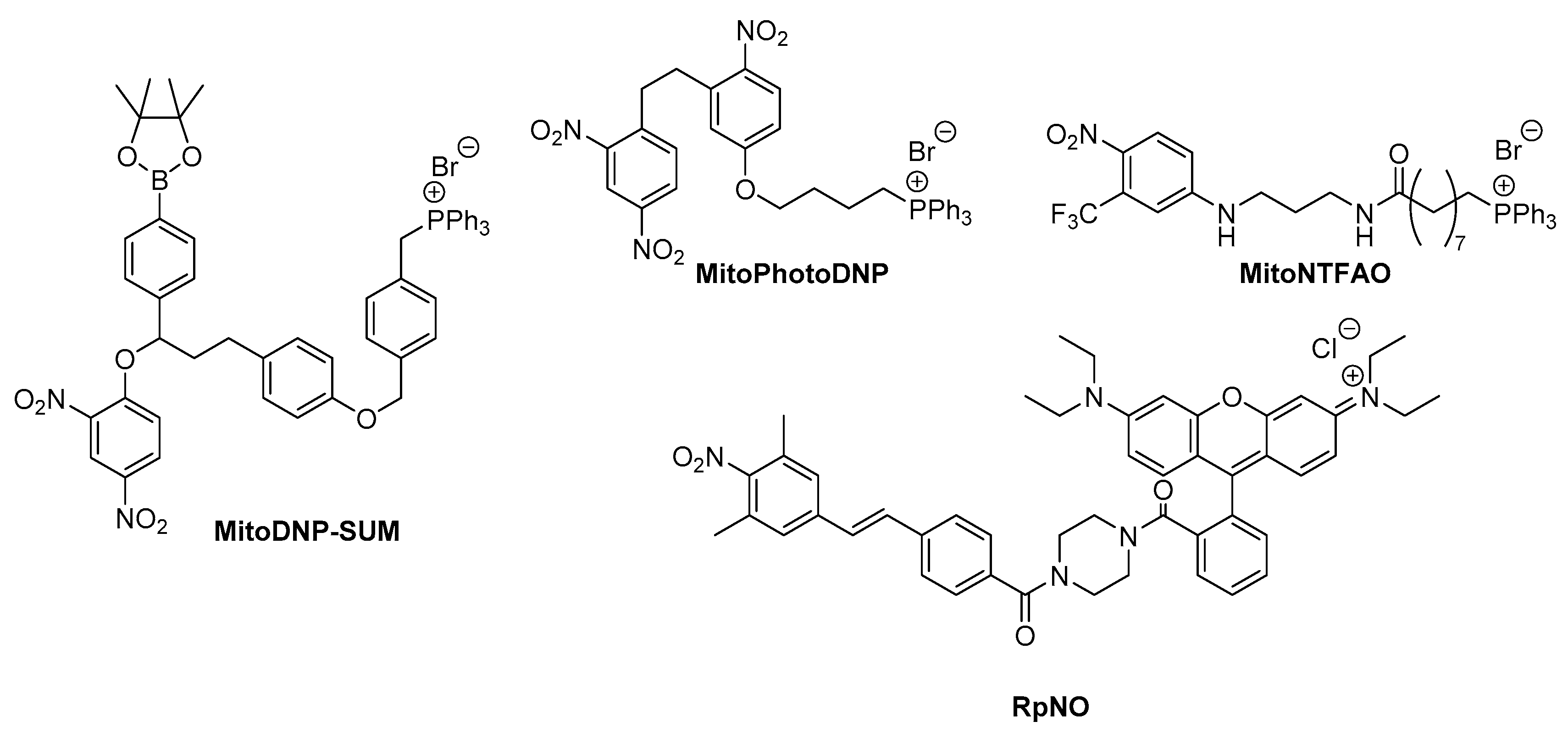
2.2.1. Mitochondria Targeted Photothermal Therapy
2.2.2. Photouncaging Strategy Targetting Mitochondria
2.2.3. Mitochondria Targeting Photoinduced Electron Transfer to Induce Redox Reactions
3. Therapeutic Strategy for Depressing Mitochondrial Oxidative Stress
3.1. Antioxidant Therapy Using Mitochondrial DDS
3.1.1. Mitochondria-Targeted Delivery of Antioxidants Using the TPP System
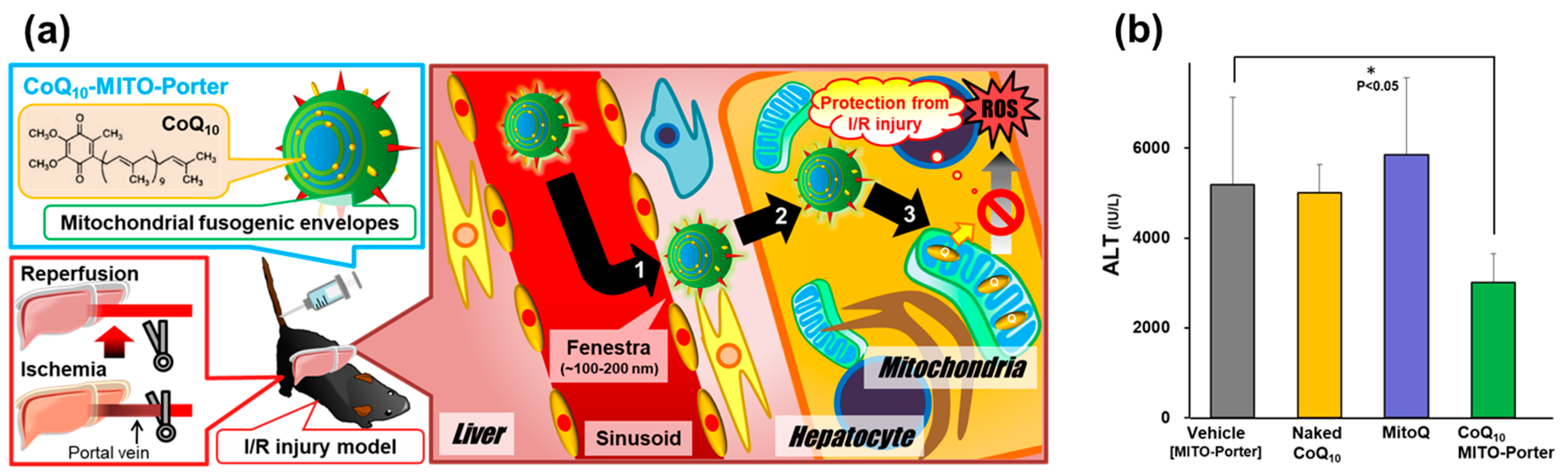
3.1.2. Validation of Anti-Oxidant Therapy by Mitochondrial Delivery of CoQ10 Using MITO-Porter System
3.2. Cell Therapy to Reduce Oxidative Stress in Mitochondria
3.2.1. Relationship between Mitochondrial Oxidative Stress and Cardiomyopathy
3.2.2. Cell Therapeutic Strategy for Treating Cardiomyopathy
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Indo, H.P.; Yen, H.C.; Nakanishi, I.; Matsumoto, K.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, Z. The pathophysiological role of mitochondrial oxidative stress in lung diseases. J. Transl. Med. 2017, 15, 207. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Wen, L.L.; Huang, Y.N.; Chen, Y.T.; Ku, M.C. Dual effects of antioxidants in neurodegeneration: Direct neuroprotection against oxidative stress and indirect protection via suppression of glia-mediated inflammation. Curr. Pharm. Des. 2006, 12, 3521–3533. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondrial dysfunction and oxidative damage in Alzheimer’s and Parkinson’s diseases and coenzyme Q10 as a potential treatment. J. Bioenerg. Biomembr. 2004, 36, 381–386. [Google Scholar] [CrossRef]
- Luo, K.; Yu, J.H.; Quan, Y.; Shin, Y.J.; Lee, K.E.; Kim, H.L.; Ko, E.J.; Chung, B.H.; Lim, S.W.; Yang, C.W. Therapeutic potential of coenzyme Q10 in mitochondrial dysfunction during tacrolimus-induced beta cell injury. Sci. Rep. 2019, 9, 7995. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Hawk, M.A.; McCallister, C.; Schafer, Z.T. Antioxidant Activity during Tumor Progression: A Necessity for the Survival of Cancer Cells? Cancers 2016, 8, 92. [Google Scholar] [CrossRef]
- Yoo, M.H.; Xu, X.M.; Carlson, B.A.; Gladyshev, V.N.; Hatfield, D.L. Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J. Biol. Chem. 2006, 281, 13005–13008. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef]
- Wang, J.; Luo, B.; Li, X.; Lu, W.; Yang, J.; Hu, Y.; Huang, P.; Wen, S. Inhibition of cancer growth in vitro and in vivo by a novel ROS-modulating agent with ability to eliminate stem-like cancer cells. Cell Death Dis. 2017, 8, e2887. [Google Scholar] [CrossRef]
- Pelicano, H.; Feng, L.; Zhou, Y.; Carew, J.S.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Inhibition of mitochondrial respiration: A novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J. Biol. Chem. 2003, 278, 37832–37839. [Google Scholar] [CrossRef]
- Yamada, Y.; Akita, H.; Kamiya, H.; Kogure, K.; Yamamoto, T.; Shinohara, Y.; Yamashita, K.; Kobayashi, H.; Kikuchi, H.; Harashima, H. MITO-Porter: A liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim. et Biophys. Acta 2008, 1778, 423–432. [Google Scholar] [CrossRef]
- Yamada, Y.; Harashima, H. Mitochondrial drug delivery systems for macromolecule and their therapeutic application to mitochondrial diseases. Adv. Drug Deliv. Rev. 2008, 60, 1439–1462. [Google Scholar] [CrossRef]
- Yamada, Y.; Ishikawa, T.; Harashima, H. Validation of the use of an artificial mitochondrial reporter DNA vector containing a Cytomegalovirus promoter for mitochondrial transgene expression. Biomaterials 2017, 136, 56–66. [Google Scholar] [CrossRef]
- Ishikawa, T.; Somiya, K.; Munechika, R.; Harashima, H.; Yamada, Y. Mitochondrial transgene expression via an artificial mitochondrial DNA vector in cells from a patient with a mitochondrial disease. J. Control. Release 2018, 274, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Daikuhara, S.; Tamura, A.; Nishida, K.; Yui, N.; Harashima, H. Enhanced autophagy induction via the mitochondrial delivery of methylated beta-cyclodextrin-threaded polyrotaxanes using a MITO-Porter. Chem. Commun. 2019, 55, 7203–7206. [Google Scholar] [CrossRef] [PubMed]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Dysart, J.S.; Patterson, M.S. Characterization of Photofrin photobleaching for singlet oxygen dose estimation during photodynamic therapy of MLL cells in vitro. Phys. Med. Biol. 2005, 50, 2597–2616. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, S.M.; Ordaz, J.D.; Low, P.S. Targeting of a Photosensitizer to the Mitochondrion Enhances the Potency of Photodynamic Therapy. ACS Omega 2018, 3, 6066–6074. [Google Scholar] [CrossRef] [PubMed]
- Mitsunaga, M.; Ogawa, M.; Kosaka, N.; Rosenblum, L.T.; Choyke, P.L.; Kobayashi, H. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat. Med. 2011, 17, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, H.; Ito, H.; Inoue, M.; Tabata, K.; Sato, Y.; Yamagata, K.; Kizaka-Kondoh, S.; Kadonosono, T.; Yano, S.; Inoue, M.; et al. High resolution imaging of intracellular oxygen concentration by phosphorescence lifetime. Sci. Rep. 2015, 5, 10657. [Google Scholar] [CrossRef]
- Lv, W.; Zhang, Z.; Zhang, K.Y.; Yang, H.; Liu, S.; Xu, A.; Guo, S.; Zhao, Q.; Huang, W. A Mitochondria-Targeted Photosensitizer Showing Improved Photodynamic Therapy Effects Under Hypoxia. Angew. Chem. Int. Ed. 2016, 55, 9947–9951. [Google Scholar] [CrossRef]
- Qi, T.; Chen, B.; Wang, Z.; Du, H.; Liu, D.; Yin, Q.; Liu, B.; Zhang, Q.; Wang, Y. A pH-Activatable nanoparticle for dual-stage precisely mitochondria-targeted photodynamic anticancer therapy. Biomaterials 2019, 213, 119219. [Google Scholar] [CrossRef]
- Yu, Z.; Sun, Q.; Pan, W.; Li, N.; Tang, B. A Near-Infrared Triggered Nanophotosensitizer Inducing Domino Effect on Mitochondrial Reactive Oxygen Species Burst for Cancer Therapy. ACS Nano 2015, 9, 11064–11074. [Google Scholar] [CrossRef]
- Murphy, M.P. Targeting lipophilic cations to mitochondria. Biochim. et Biophys. Acta 2008, 1777, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef] [PubMed]
- Kirakci, K.; Zelenka, J.; Rumlova, M.; Cvacka, J.; Ruml, T.; Lang, K. Cationic octahedral molybdenum cluster complexes functionalized with mitochondria-targeting ligands: Photodynamic anticancer and antibacterial activities. Biomater. Sci. 2019, 7, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chan, P.S.; Li, H.; Wong, K.L.; Wong, R.N.; Mak, N.K.; Zhang, J.; Tam, H.L.; Wong, W.Y.; Kwong, D.W.; et al. Highly selective mitochondria-targeting amphiphilic silicon(IV) phthalocyanines with axially ligated rhodamine B for photodynamic therapy. Inorg. Chem. 2012, 51, 812–821. [Google Scholar] [CrossRef]
- Han, K.; Lei, Q.; Wang, S.B.; Hu, J.J.; Qiu, W.X.; Zhu, J.Y.; Yin, W.N.; Luo, X.; Zhang, X.Z. Dual-stage-light-guided tumor inhibition by mitochondria-targeted photodynamic therapy. Adv. Funct. Mater. 2015, 25, 2961–2971. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, L.; Ling, X.; Shao, P.; Wang, X.; Edwards, W.B.; Bai, M. Tumor mitochondria-targeted photodynamic therapy with a translocator protein (TSPO)-specific photosensitizer. Acta. Biomater. 2015, 28, 160–170. [Google Scholar] [CrossRef]
- Lucky, S.S.; Soo, K.C.; Zhang, Y. Nanoparticles in photodynamic therapy. Chem. Rev. 2015, 115, 1990–2042. [Google Scholar] [CrossRef]
- Thomas, A.P.; Palanikumar, L.; Jeena, M.T.; Kim, K.; Ryu, J.H. Cancer-mitochondria-targeted photodynamic therapy with supramolecular assembly of HA and a water soluble NIR cyanine dye. Chem. Sci. 2017, 8, 8351–8356. [Google Scholar] [CrossRef]
- Xu, J.; Zeng, F.; Wu, H.; Yu, C.; Wu, S. Dual-targeting nanosystem for enhancing photodynamic therapy efficiency. ACS Appl. Mater. Interfaces 2015, 7, 9287–9296. [Google Scholar] [CrossRef]
- Kang, J.H.; Ko, Y.T. Dual-selective photodynamic therapy with a mitochondria-targeted photosensitizer and fiber optic cannula for malignant brain tumors. Biomater. Sci. 2019, 7, 2812–2825. [Google Scholar] [CrossRef]
- Guo, F.; Yu, M.; Wang, J.; Tan, F.; Li, N. The mitochondria-targeted and IR780-regulated theranosomes for imaging and enhanced photodynamic/photothermal therapy. RSC Adv. 2016, 6, 11070–11076. [Google Scholar] [CrossRef]
- Gao, W.; Wang, Z.; Lv, L.; Yin, D.; Chen, D.; Han, Z.; Ma, Y.; Zhang, M.; Yang, M.; Gu, Y. Photodynamic Therapy Induced Enhancement of Tumor Vasculature Permeability Using an Upconversion Nanoconstruct for Improved Intratumoral Nanoparticle Delivery in Deep Tissues. Theranostics 2016, 6, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Zhou, J.; Zheng, K.; Bednarkiewicz, A.; Liu, X.; Jin, D. Advances in highly doped upconversion nanoparticles. Nat. Commun. 2018, 9, 2415. [Google Scholar] [CrossRef] [PubMed]
- Idris, N.M.; Gnanasammandhan, M.K.; Zhang, J.; Ho, P.C.; Mahendran, R.; Zhang, Y. In vivo photodynamic therapy using upconversion nanoparticles as remote-controlled nanotransducers. Nat. Med. 2012, 18, 1580–1585. [Google Scholar] [CrossRef]
- Zhang, D.; Wen, L.; Huang, R.; Wang, H.; Hu, X.; Xing, D. Mitochondrial specific photodynamic therapy by rare-earth nanoparticles mediated near-infrared graphene quantum dots. Biomaterials 2018, 153, 14–26. [Google Scholar] [CrossRef]
- Zhang, X.; Ai, F.; Sun, T.; Wang, F.; Zhu, G. Multimodal Upconversion Nanoplatform with a Mitochondria-Targeted Property for Improved Photodynamic Therapy of Cancer Cells. Inorg. Chem. 2016, 55, 3872–3880. [Google Scholar] [CrossRef]
- Abe, J.; Yamada, Y.; Harashima, H. Validation of a Strategy for Cancer Therapy: Delivering Aminoglycoside Drugs to Mitochondria in HeLa Cells. J. Pharm. Sci. 2016, 105, 734–740. [Google Scholar] [CrossRef]
- Yamada, Y.; Munechika, R.; Kawamura, E.; Sakurai, Y.; Sato, Y.; Harashima, H. Mitochondrial Delivery of Doxorubicin Using MITO-Porter Kills Drug-Resistant Renal Cancer Cells via Mitochondrial Toxicity. J. Pharm. Sci. 2017, 106, 2428–2437. [Google Scholar] [CrossRef]
- Satrialdi; Munechika, R.; Biju, V.; Takano, Y.; Harashima, H.; Yamada, Y. The optimization of cancer photodynamic therapy by utilization of a pi-extended porphyrin-type photosensitizer in combination with MITO-Porter. Chem. Commun. 2020, in press. [Google Scholar] [CrossRef]
- Jung, H.S.; Lee, J.H.; Kim, K.; Koo, S.; Verwilst, P.; Sessler, J.L.; Kang, C.; Kim, J.S. A Mitochondria-Targeted Cryptocyanine-Based Photothermogenic Photosensitizer. J. Am. Chem. Soc. 2017, 139, 9972–9978. [Google Scholar] [CrossRef]
- Ma, Z.; Han, K.; Dai, X.; Han, H. Precisely Striking Tumors without Adjacent Normal Tissue Damage via Mitochondria-Templated Accumulation. ACS Nano 2018, 6, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Marrache, S.; Dhar, S. The energy blocker inside the power house: Mitochondria targeted delivery of 3-bromopyruvate. Chem. Sci. 2015, 6, 1832–1845. [Google Scholar] [CrossRef] [PubMed]
- Jaque, D.; Martinez Maestro, L.; del Rosal, B.; Haro-Gonzalez, P.; Benayas, A.; Plaza, J.L.; Martin Rodriguez, E.; Garcia Sole, J. Nanoparticles for photothermal therapies. Nanoscale 2014, 6, 9494–9530. [Google Scholar] [CrossRef]
- Yuan, A.; Wu, J.; Tang, X.; Zhao, L.; Xu, F.; Hu, Y. Application of near-infrared dyes for tumor imaging, photothermal, and photodynamic therapies. J. Pharm. Sci. 2013, 102, 6–28. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.W.; Moseley, P.L.; Buettner, G.R. Increased flux of free radicals in cells subjected to hyperthermia: Detection by electron paramagnetic resonance spin trapping. FEBS Lett. 1998, 431, 285–286. [Google Scholar] [CrossRef]
- Zuo, L.; Christofi, F.L.; Wright, V.P.; Liu, C.Y.; Merola, A.J.; Berliner, L.J.; Clanton, T.L. Intra- and extracellular measurement of reactive oxygen species produced during heat stress in diaphragm muscle. Am. J. Physiol. Cell Physiol. 2000, 279, C1058–C1066. [Google Scholar] [CrossRef]
- Chung, H.; Dai, T.; Sharma, S.K.; Huang, Y.Y.; Carroll, J.D.; Hamblin, M.R. The nuts and bolts of low-level laser (light) therapy. Ann. Biomed. Eng. 2012, 40, 516–533. [Google Scholar] [CrossRef]
- Albuquerque-Pontes, G.M.; Vieira, R.P.; Tomazoni, S.S.; Caires, C.O.; Nemeth, V.; Vanin, A.A.; Santos, L.A.; Pinto, H.D.; Marcos, R.L.; Bjordal, J.M.; et al. Effect of pre-irradiation with different doses, wavelengths, and application intervals of low-level laser therapy on cytochrome c oxidase activity in intact skeletal muscle of rats. Lasers Med. Sci. 2015, 30, 59–66. [Google Scholar] [CrossRef]
- Zhou, F.; Xing, D.; Wu, B.; Wu, S.; Ou, Z.; Chen, W.R. New insights of transmembranal mechanism and subcellular localization of noncovalently modified single-walled carbon nanotubes. Nano Lett. 2010, 10, 1677–1681. [Google Scholar] [CrossRef]
- Zhou, F.; Wu, S.; Wu, B.; Chen, W.R.; Xing, D. Mitochondria-targeting single-walled carbon nanotubes for cancer photothermal therapy. Small 2011, 7, 2727–2735. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, N.; Hu, Y.; Jia, C.; Chou, T.; Du, H.; Wang, H. Gold nanoparticle-enhanced photodynamic therapy: Effects of surface charge and mitochondrial targeting. Ther. Deliv. 2015, 6, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.L. 3-Bromopyruvate (3BP) a fast acting, promising, powerful, specific, and effective “small molecule” anti-cancer agent taken from labside to bedside: Introduction to a special issue. J. Bioenerg. Biomembr. 2012, 44, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, H.; Lu, W.; Huang, P. Role of mitochondria-associated hexokinase II in cancer cell death induced by 3-bromopyruvate. Biochim. et Biophys. Acta 2009, 1787, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Zhu, Y.; Wen, L.; Yang, X.; Liu, X.; Meng, T.; Dai, S.; Ping, Y.; Yuan, H.; Hu, F. Mitochondria-Responsive Drug Release along with Heat Shock Mediated by Multifunctional Glycolipid Micelles for Precise Cancer Chemo-Phototherapy. Theranostics 2019, 9, 691–707. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.; Caldwell, S.T.; Quin, C.; Prime, T.A.; James, A.M.; Cairns, A.G.; Murphy, M.P.; McCarron, J.G.; Hartley, R.C. Selective uncoupling of individual mitochondria within a cell using a mitochondria-targeted photoactivated protonophore. J. Am. Chem. Soc. 2012, 134, 758–761. [Google Scholar] [CrossRef] [PubMed]
- McQuaker, S.J.; Quinlan, C.L.; Caldwell, S.T.; Brand, M.D.; Hartley, R.C. A prototypical small-molecule modulator uncouples mitochondria in response to endogenous hydrogen peroxide production. Chembiochem 2013, 14, 993–1000. [Google Scholar] [CrossRef]
- Kerwin Jr, J.F.; Heller, M. The arginine-nitric oxide pathway: A target for new drugs. Med. Res. Rev. 1994, 14, 23–74. [Google Scholar] [CrossRef]
- Huang, Z.; Fu, J.; Zhang, Y. Nitric Oxide Donor-Based Cancer Therapy: Advances and Prospects. J. Med. Chem. 2017, 60, 7617–7635. [Google Scholar] [CrossRef]
- Sodano, F.; Rolando, B.; Spyrakis, F.; Failla, M.; Lazzarato, L.; Gazzano, E.; Riganti, C.; Fruttero, R.; Gasco, A.; Sortino, S. Tuning the Hydrophobicity of a Mitochondria-Targeted NO Photodonor. ChemMedChem 2018, 13, 1238–1245. [Google Scholar] [CrossRef]
- Horinouchi, T.; Nakagawa, H.; Suzuki, T.; Fukuhara, K.; Miyata, N. Photoinduced nitric oxide release from a nitrobenzene derivative in mitochondria. Chemistry 2011, 17, 4809–4813. [Google Scholar] [CrossRef]
- Balzani, V. Principles, Theories, Methods, and Techniques; WILEY-VCH Verlag GmbH: Weinheim, Germany, 2001; Volume 1. [Google Scholar]
- Takano, Y.; Hanai, E.; Imahori, H. Photoinduced electron transfer reaction in mitochondria for spatiotemporal selective photo-oxidation of lipids by donor/acceptor linked molecules. Nanoscale 2017, 9, 17909–17913. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Munechika, R.; Biju, V.; Harashima, H.; Imahori, H.; Yamada, Y. Optical control of mitochondrial reductive reactions in living cells using an electron donor-acceptor linked molecule. Nanoscale 2017, 9, 18690–18698. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, C.; Yamasoba, T. Mitochondria-Targeted Antioxidants for Treatment of Hearing Loss: A Systematic Review. Antioxidants 2019, 8, 109. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef]
- Jelinek, A.; Heyder, L.; Daude, M.; Plessner, M.; Krippner, S.; Grosse, R.; Diederich, W.E.; Culmsee, C. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 2018, 117, 45–57. [Google Scholar] [CrossRef]
- Du, K.; Farhood, A.; Jaeschke, H. Mitochondria-targeted antioxidant Mito-Tempo protects against acetaminophen hepatotoxicity. Arch. Toxicol. 2017, 91, 761–773. [Google Scholar] [CrossRef]
- Finichiu, P.G.; Larsen, D.S.; Evans, C.; Larsen, L.; Bright, T.P.; Robb, E.L.; Trnka, J.; Prime, T.A.; James, A.M.; Smith, R.A.; et al. A mitochondria-targeted derivative of ascorbate: MitoC. Free Radic. Biol. Med. 2015, 89, 668–678. [Google Scholar] [CrossRef]
- Silachev, D.N.; Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Balakireva, A.V.; Gulyaev, M.V.; Pirogov, Y.A.; Skulachev, V.P.; Zorov, D.B. Neuroprotective Effects of Mitochondria-Targeted Plastoquinone in a Rat Model of Neonatal Hypoxic(-)Ischemic Brain Injury. Molecules 2018, 23, 1871. [Google Scholar] [CrossRef]
- Brenza, T.M.; Ghaisas, S.; Ramirez, J.E.V.; Harischandra, D.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G.; Narasimhan, B. Neuronal protection against oxidative insult by polyanhydride nanoparticle-based mitochondria-targeted antioxidant therapy. Nanomedicine 2017, 13, 809–820. [Google Scholar] [CrossRef]
- Wu, M.; Liao, L.; Jiang, L.; Zhang, C.; Gao, H.; Qiao, L.; Liu, S.; Shi, D. Liver-targeted Nano-MitoPBN normalizes glucose metabolism by improving mitochondrial redox balance. Biomaterials 2019, 222, 119457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, J.; Hu, S.; Wu, F.; Zhang, X. Triphenylphosphonium and D-alpha-tocopheryl polyethylene glycol 1000 succinate-modified, tanshinone IIA-loaded lipid-polymeric nanocarriers for the targeted therapy of myocardial infarction. Int. J. Nanomed. 2018, 13, 4045–4057. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Liaw, K.; Sharma, R.; Zhang, Z.; Kannan, S.; Kannan, R.M. Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia using Dendrimer-Based Therapeutics. Theranostics 2018, 8, 5529–5547. [Google Scholar] [CrossRef] [PubMed]
- Velichkovska, M.; Surnar, B.; Nair, M.; Dhar, S.; Toborek, M. Targeted Mitochondrial COQ10 Delivery Attenuates Antiretroviral-Drug-Induced Senescence of Neural Progenitor Cells. Mol. Pharm. 2019, 16, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Nakamura, K.; Abe, J.; Hyodo, M.; Haga, S.; Ozaki, M.; Harashima, H. Mitochondrial delivery of Coenzyme Q10 via systemic administration using a MITO-Porter prevents ischemia/reperfusion injury in the mouse liver. J. Control. Release 2015, 213, 86–95. [Google Scholar] [CrossRef]
- Yamada, Y.; Burger, L.; Kawamura, E.; Harashima, H. Packaging of the Coenzyme Q(10) into a Liposome for Mitochondrial Delivery and the Intracellular Observation in Patient Derived Mitochondrial Disease Cells. Biol. Pharm. Bull. 2017, 40, 2183–2190. [Google Scholar] [CrossRef]
- Hibino, M.; Yamada, Y.; Fujishita, N.; Sato, Y.; Maeki, M.; Tokeshi, M.; Harashima, H. The Use of a Microfluidic Device to Encapsulate a Poorly Water-Soluble Drug CoQ10 in Lipid Nanoparticles and an Attempt to Regulate Intracellular Trafficking to Reach Mitochondria. J. Pharm. Sci. 2019, 108, 2668–2676. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Burwell, L.S.; Nadtochiy, S.M.; Brookes, P.S. Cardioprotection by metabolic shut-down and gradual wake-up. J. Mol. Cell. Cardiol. 2009, 46, 804–810. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Singal, P.K.; Iliskovic, N. Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 1998, 339, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, S.; Ohtsuki, S.; Tarui, S.; Ousaka, D.; Eitoku, T.; Kondo, M.; Okuyama, M.; Kobayashi, J.; Baba, K.; Arai, S.; et al. Intracoronary autologous cardiac progenitor cell transfer in patients with hypoplastic left heart syndrome: The TICAP prospective phase 1 controlled trial. Circ. Res. 2015, 116, 653–664. [Google Scholar] [CrossRef]
- Heusch, G. Cardioprotection: Chances and challenges of its translation to the clinic. Lancet 2013, 381, 166–175. [Google Scholar] [CrossRef]
- De Angelis, A.; Piegari, E.; Cappetta, D.; Marino, L.; Filippelli, A.; Berrino, L.; Ferreira-Martins, J.; Zheng, H.; Hosoda, T.; Rota, M.; et al. Anthracycline cardiomyopathy is mediated by depletion of the cardiac stem cell pool and is rescued by restoration of progenitor cell function. Circulation 2010, 121, 276–292. [Google Scholar] [CrossRef]
- Takehara, N.; Tsutsumi, Y.; Tateishi, K.; Ogata, T.; Tanaka, H.; Ueyama, T.; Takahashi, T.; Takamatsu, T.; Fukushima, M.; Komeda, M.; et al. Controlled delivery of basic fibroblast growth factor promotes human cardiosphere-derived cell engraftment to enhance cardiac repair for chronic myocardial infarction. J. Am. Coll. Cardiol. 2008, 52, 1858–1865. [Google Scholar] [CrossRef]
- Aonuma, T.; Takehara, N.; Maruyama, K.; Kabara, M.; Matsuki, M.; Yamauchi, A.; Kawabe, J.; Hasebe, N. Apoptosis-Resistant Cardiac Progenitor Cells Modified with Apurinic/Apyrimidinic Endonuclease/Redox Factor 1 Gene Overexpression Regulate Cardiac Repair After Myocardial Infarction. Stem. Cells Transl. Med. 2016, 5, 1067–1078. [Google Scholar] [CrossRef]
- Abe, J.; Yamada, Y.; Takeda, A.; Harashima, H. Cardiac progenitor cells activated by mitochondrial delivery of resveratrol enhance the survival of a doxorubicin-induced cardiomyopathy mouse model via the mitochondrial activation of a damaged myocardium. J. Control. Release 2018, 269, 177–188. [Google Scholar] [CrossRef]
- Abe, J.; Yamada, Y.; Harashima, H. The use of cardiac progenitor cells for transplantation in congenital heart disease and an innovative strategy for activating mitochondrial function in such cells. J. Thorac. Dis 2018, 10, 2119–2121. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System Name | Irradiation Conditions | Evaluation | In Vitro EC50 | Refs. |
|---|---|---|---|---|
| Mitochondriotropic-Modified PS | ||||
| Verteporfin-TPP TPP-modified verteporfin PS | Laser 690 nm | KB cells | n.a. | [25] |
| Ir-P(ph)3 Iridium (III) complex as singlet oxygen PS conjugated with TPP | Xenon lamp 475 nm (39.6 J/cm2) | HeLa cells | n.a. | [28] |
| Cationic octahedral molybdenum complex Octahedral molybdenum complex PS conjugated with TPP | LED 460 nm (18 J/cm2) | HeLa cells | 0.10 ± 0.02 μM | [33] |
| Rh-SiPc Si(IV)-phthalocyanine PS axially ligated with two rhodamine B molecules as mitochondriotropic | Tungsten lamp 500 nm (1–4 J/cm2) | HK-1 | n.a. | [34] |
| PpIX-PEG-(KLAKLAK)2 Conjugation of protoporphyrin IX (PpIX) PS with mitochondria-targeted (KLAKLAK)2 peptide | 400–700 nm (4.75 J/cm2) | HeLa cells | 30 mg/L | [35] |
| IR700DX-6T Chemical conjugation of IR700 PS with mitochondrial membrane TSPO ligand | LED 690 ± 20 nm (54 J/cm2) | MDA-MB-231 & MCF-7 | n.a. | [36] |
| Mitochondrial Targeted PS in DDS | ||||
| M-TPPa pH-responsive mPEG-b-PDPA labeled with Cy7.5 as a nanocarrier system for TPP-conjugated pyropheophorbide-a | Laser 660 nm (15 J/cm2) | HO8910 | n.a. | [29] |
| HA-IR-Pyr Micellar aggregate of mitochondrial targeting PS, IR-Pyr, and Hyaluronic acid as cancer selective delivery agent | Laser 808 nm (36 J/cm2) | HeLa cells & MDA-MB-231 | 5–7 μM | [38] |
| NGO-PEG-FA/MitoTPP Mitochondrial targeting PS (MitoTPP) incorporated into PEGylated-nanographene (NGO) functionalized with tumor targeting folic acid | LED 650 nm (18 J/cm2) | HeLa cells, L929, & A549 | n.a. | [39] |
| PS@chol-BSA NPs TPP-modified pheophorbide-a as the mitochondria selective PS encapsulated with folate-cholesteryl bovine serum albumin as the tumor-selective nanocarrier | Laser 671 ± 1 nm (1.5 J/cm2) | U87MG | 0.81 μg/mL | [40] |
| Mitochondriotropic-Modified DDS | ||||
| TPP-IR780/Ce6-TNS Chlorin e6 PS and IR780 incorporated into TPP-modified lipid nanoparticles. IR780 acts as PTT agent and to control PDT process | Laser 808 nm (IR780) followed by 660 nm (Chlorin e6) | HeLa cells | n.a. | [41] |
| UCNPs@TiO2-TPP TPP-modified TiO2-coated UCNPs. UCNPs harvest NIR light and emit UV light to activate TiO2 for ROS production | Laser 980 nm (90-450 J/cm2) | MCF-7 | n.a. | [30] |
| UCNP-GQD/TRITC Hybrid nanoparticles of UCNP and 1O2 generator of graphene quantum dot (GQD) with TRITC surface modification as mitochondriotropic | Laser 980 nm (720 J/cm2) | 4T1 cells | n.a. | [45] |
| TAT-Ppa-UCNPs PEGylated polymer-coated UCNP carrying pyropheophorbide a (Ppa) PS with TAT surface modification | Laser 808 nm (1800 J/cm2) | HeLa cells | n.a. | [46] |
| MITO-Porter System | ||||
| rTPA-MITO-Porter An NIR PS, rTPA, delivered by MITO-Porter system via macropinocytosis followed by electrostatic interaction and fusion with mitochondrial membrane | Xenon lamp 700 ± 6 nm (12.8 J/cm2 & 20.6 J/cm2) | HeLa cells & SAS cells | 0.16 – 0.41 μM (0.26–0.64 μg/mL) | [49] |
| PTT Reagents | ||||
| Mito-CCy Cryptocyanine-based PTT reagent with mitochondrial targeting moiety TPP | Laser 730 nm (1.4 kJ/cm2) | HeLa cells | n.a. | [50] |
| TPP-Au Gold nanoparticles tethered with mitochondrial targeting moiety TPP | Laser 808 nm (1.4 kJ/cm2) | HeLa cells & COS7 cells | 7.0–8.5 μg/mL | [51] |
| T-3-BP-AuNP Gold nanoparticles tethered with mitochondrial targeting moiety TPP and 3-bromopyruvate which is a decoupling reagent of the respiration | Laser 660 nm (1.2 J) | PC3 cells | 10 μg/mL | [52] |
| Name | Cargo | Model/Administration Route | Nanoparticle | Outcomes | Refs. |
|---|---|---|---|---|---|
| MitoQ | Coenzyme Q10 | Neuronal HT22 cells and Mouse embryonic fibroblasts | — | Decreasing oxidative stress | Jelinek A. et al. (2018) [77] |
| Mito-TEMPO | 2,2,6,6-tetramethylpiperidine 1-oxyl | APAP-induced liver injury mouse/Intraperitoneal injection | — | Attenuating the mitochondrial oxidant stress and preventing peroxynitrite formation and the subsequent mitochondrial dysfunction | Du K. et al. (2017) [78] |
| MitoC | Ascorbate | Rat liver mitochondria | — | Preventing mitochondrial oxidative damage | Finichiu PG. et al. (2015) [79] |
| Mito-Apo | Apocynin | Immortalized rat mesencephalic cells (N27), LUHMES cells | Brain targeting nanoparticle (CPH:SA = 20/80) with FA | Protection against oxidative stress-induced mitochondrial dysfunction and neuronal damage in a dopaminergic neuronal cells. | Brenza TM. et al. (2017) [81] |
| MitoPBN | PBN | L02 cells and 293T cells Diabetic mouse model/Intraperitoneal injection | Liver targeting nanoparticle (Cholesterol:lecothin = 1/2) | Alleviating ROS-induced mitochondrial dysfunction | Wu M. et al. (2019) [82] |
| Disease Model | Type of Cell Source | Cell Modification | Method of Transplant | Main Outcome | Refs. |
|---|---|---|---|---|---|
| IRI, pig | Human CDC | None | Cell sheet | Reduced infarct size and improved EF | Takehara N. et al. (2008) [99] |
| IRI, mouse | Mouse CPC | Overexpression of APE1/REF1 gene | Intramyocardial injection | Attenuation of fibrosis and improved EF | Aonuma T. et al. (2016) [100] |
| DOX-CM, mouse | Mouse CPC | Delivery of resveratrol into mitochondria | Intramyocardial injection | Reduced oxidative stress of myocardium and longer survival time | Abe J. et al. (2018) [101] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamada, Y.; Takano, Y.; Satrialdi; Abe, J.; Hibino, M.; Harashima, H. Therapeutic Strategies for Regulating Mitochondrial Oxidative Stress. Biomolecules 2020, 10, 83. https://doi.org/10.3390/biom10010083
Yamada Y, Takano Y, Satrialdi, Abe J, Hibino M, Harashima H. Therapeutic Strategies for Regulating Mitochondrial Oxidative Stress. Biomolecules. 2020; 10(1):83. https://doi.org/10.3390/biom10010083
Chicago/Turabian StyleYamada, Yuma, Yuta Takano, Satrialdi, Jiro Abe, Mitsue Hibino, and Hideyoshi Harashima. 2020. "Therapeutic Strategies for Regulating Mitochondrial Oxidative Stress" Biomolecules 10, no. 1: 83. https://doi.org/10.3390/biom10010083
APA StyleYamada, Y., Takano, Y., Satrialdi, Abe, J., Hibino, M., & Harashima, H. (2020). Therapeutic Strategies for Regulating Mitochondrial Oxidative Stress. Biomolecules, 10(1), 83. https://doi.org/10.3390/biom10010083