Characterization of Yak Common Biofluids Metabolome by Means of Proton Nuclear Magnetic Resonance Spectroscopy


Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling of Biofluids
2.2. Metabolomics Analysis of Biofluids
2.3. Pathway Analysis
3. Results
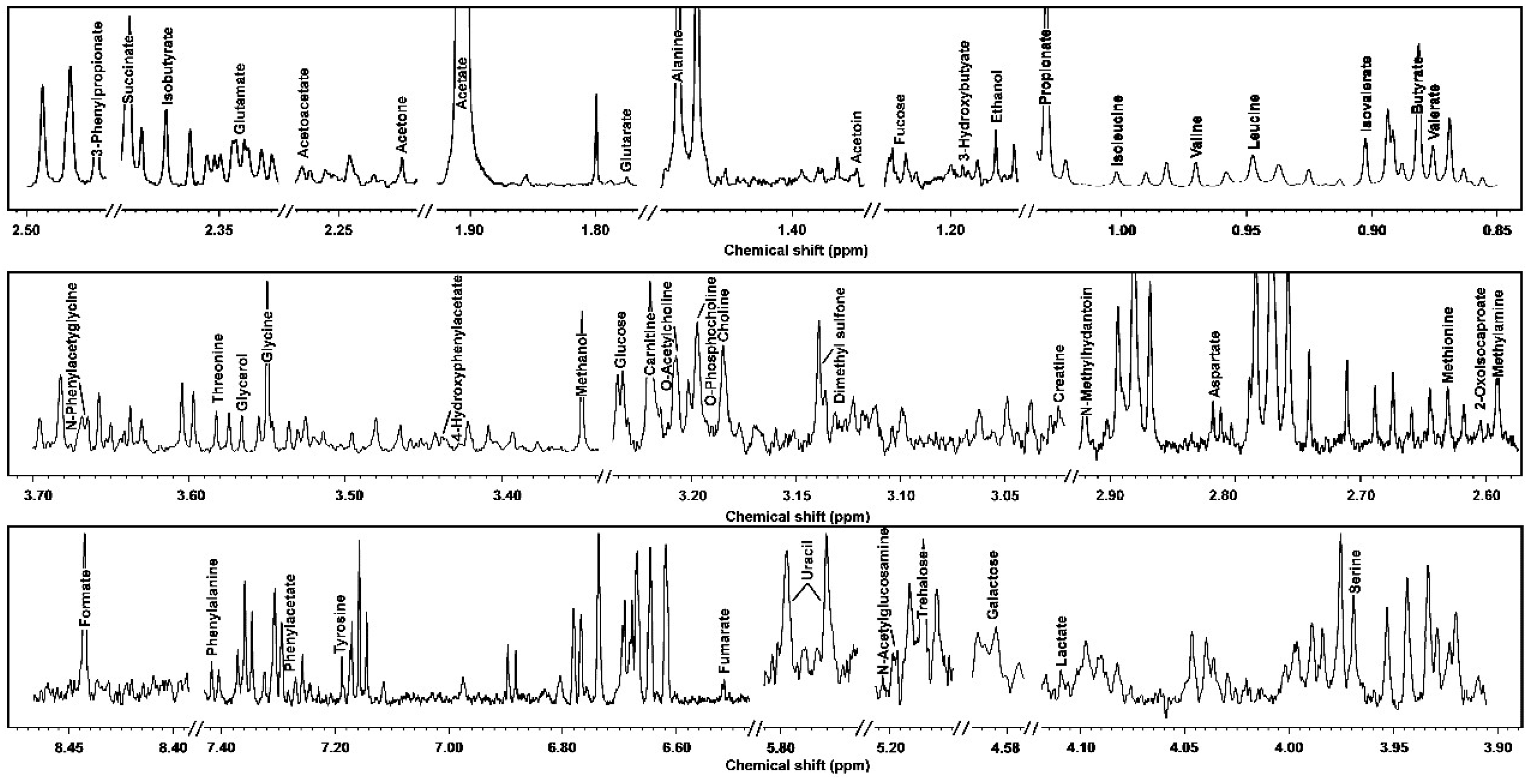
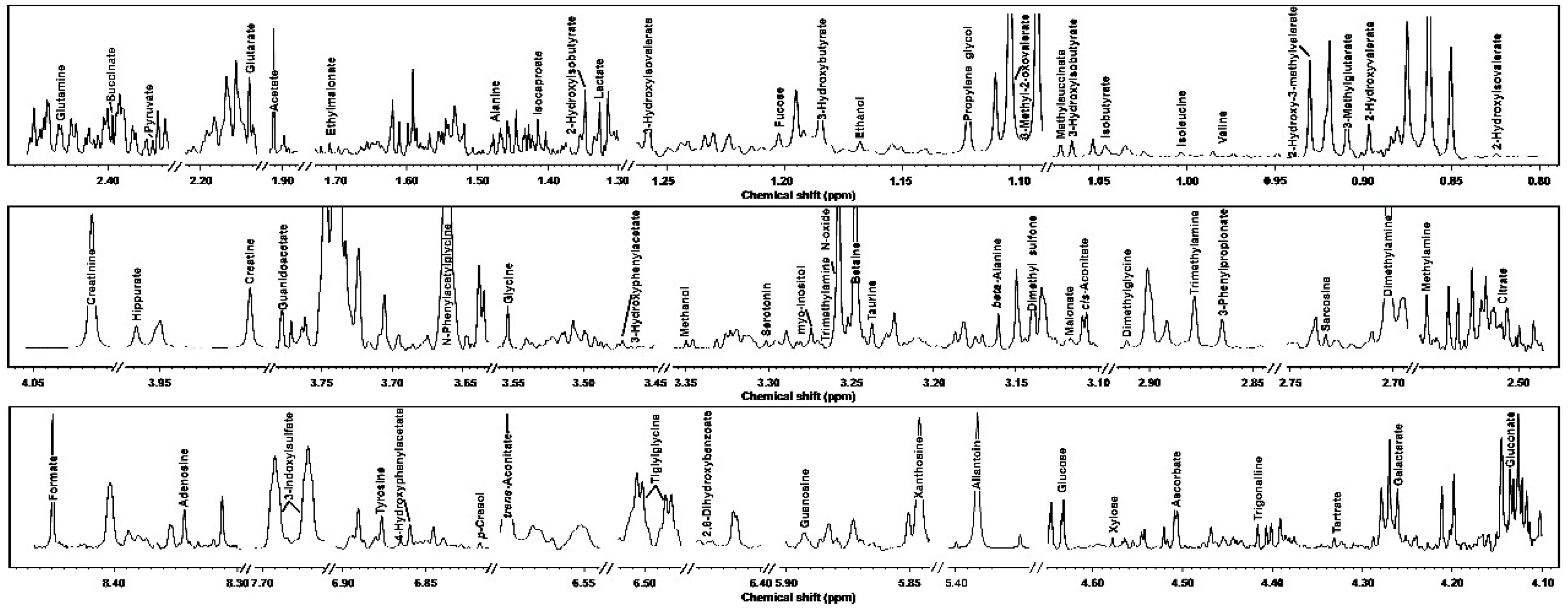
3.1. 1H-NMR Spectra of Yak Serum, Feces, and Urine
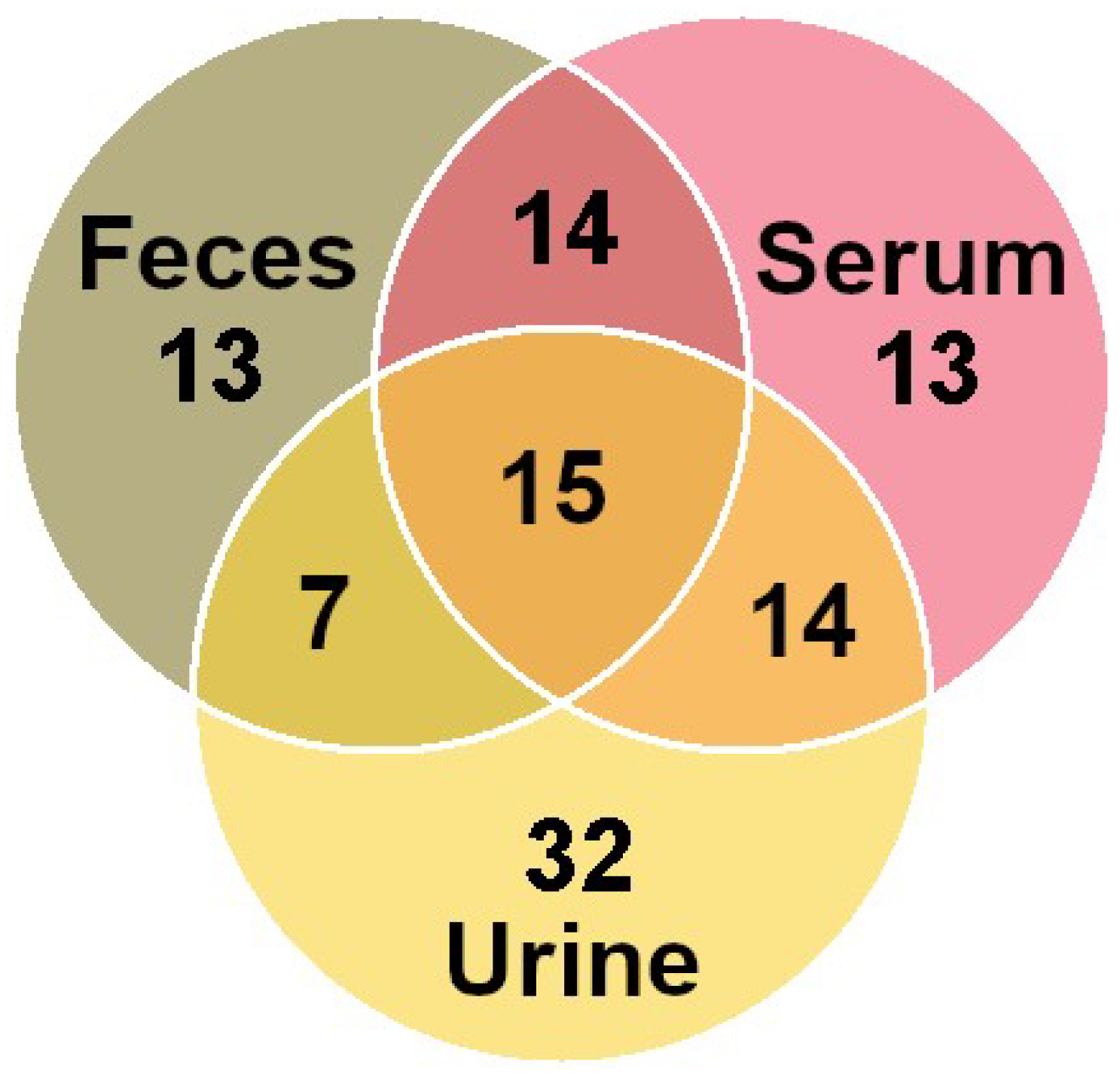
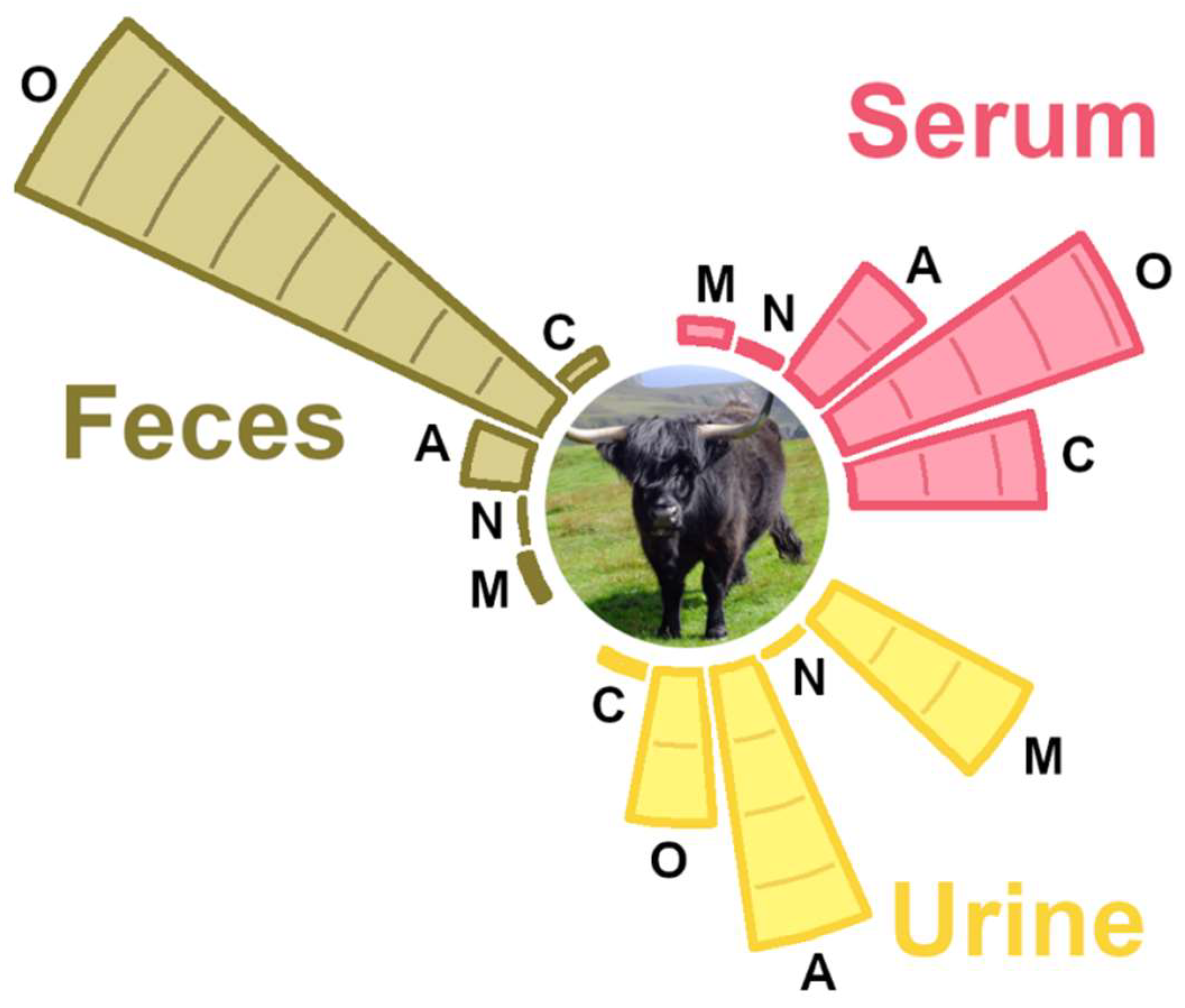
3.2. Molecule Distribution by Class
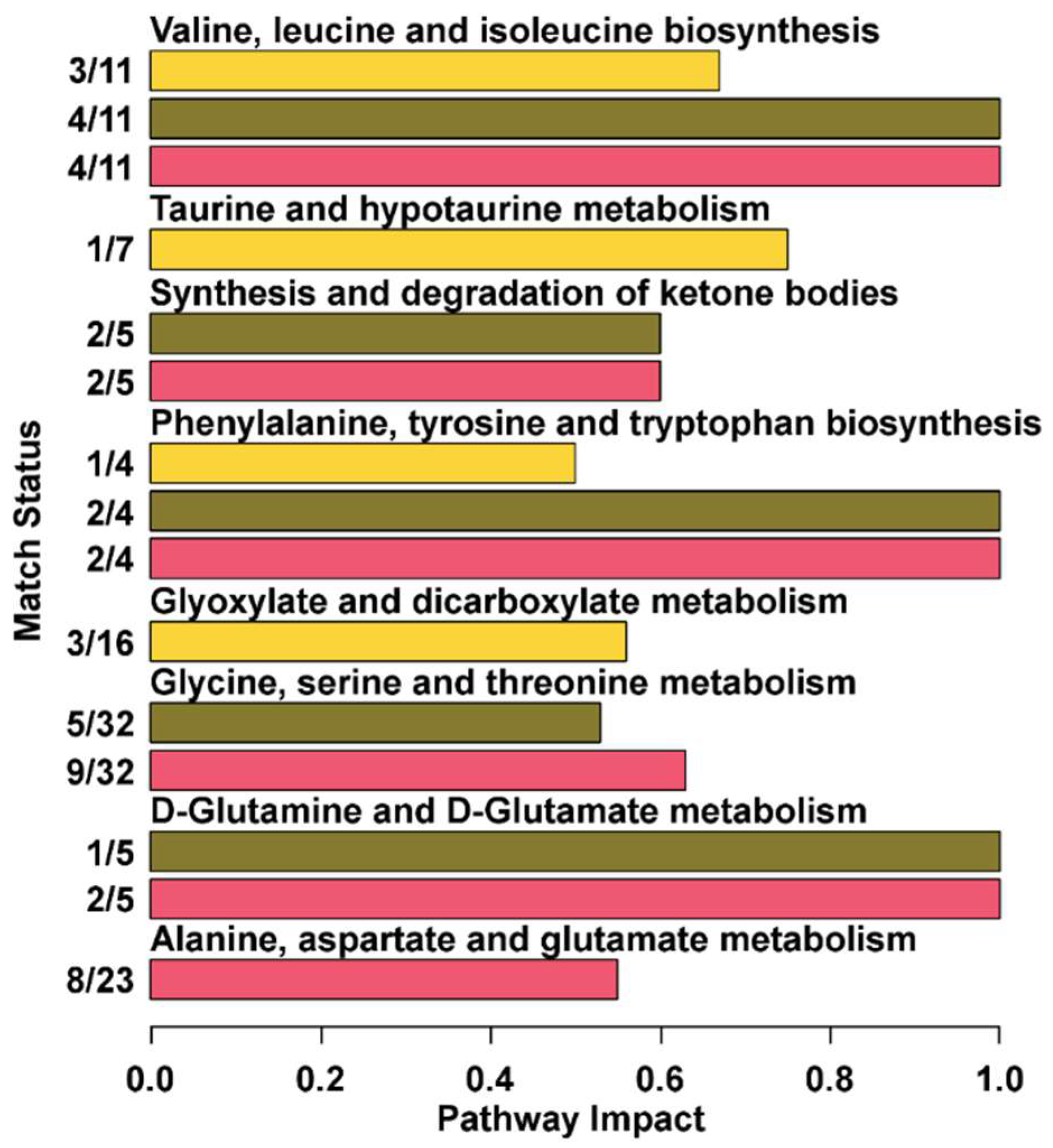
3.3. Pathway Analysis
4. Discussion
4.1. Yak Serum Metabolome
4.2. Yak Feces Metabolome
4.3. Yak Urine Metabolome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. “Metabonomics”: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Laghi, L.; Zhu, C.; Campagna, G.; Rossi, G.; Bazzano, M.; Laus, F. Probiotic Supplementation in Trained Trotter Horses: Effect on Blood Clinical Pathology Data and Urine Metabolomic Assessed in Field. J. Appl. Physiol. 2018, 125, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Faillace, V.; Laus, F.; Bazzano, M.; Laghi, L. Characterization of trotter horses urine metabolome by means of proton nuclear magnetic resonance spectroscopy. Metabolomics 2018, 14, 106. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, J.; Tu, L.; Xiong, X.; Hu, X.; Huang, J.; Xu, Z.; Zhang, X.; Hu, C.; Hu, X.; et al. 1H-NMR Spectroscopy Revealed Mycobacterium tuberculosis Caused Abnormal Serum Metabolic Profile of Cattle. PLoS One 2013, 8, e74507. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xu, C.; Li, C.; Xia, C.; Xu, C.; Wu, L.; Zhang, H. Characterization of the serum metabolic profile of dairy cows with milk fever using 1H-NMR spectroscopy. Vet. Q. 2014, 34, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, H.; Zhu, K.; Xu, C.; Shu, S.; Zheng, J.; Sun, L. Nuclear magnetic resonance-based serum metabolic profiling of dairy cows with footrot. J. Vet. Med. Sci. 2016, 78, 1421–1428. [Google Scholar] [CrossRef]
- Bazzano, M.; Laghi, L.; Zhu, C.; Magi, G.E.; Serri, E.; Spaterna, A.; Tesei, B.; Laus, F. Metabolomics of tracheal wash samples and exhaled breath condensates in healthy horses and horses affected by equine asthma. J. Breath Res. 2018, 12, 46015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, J.; Wang, X.; Liu, B.; Ma, B. Isolation of metallothionein genes and in silico structural characterization of their proteins Using molecular modeling from Yak (Bos grunniens). Biochem. Genet. 2012, 50, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Liang, J.B.; Long, R.; Wang, H.; Ding, L.; Guo, X. Comparison of Nitrogen Metabolism in Yak (Bos grunniens) and Indigenous Cattle (Bos taurus) on the Qinghai-Tibetan Plateau. Asian-Australasian J. Anim. Sci. 2011, 24, 766–773. [Google Scholar] [CrossRef]
- Zhou, J.W.; Liu, H.; Zhong, C.L.; Degen, A.A.; Yang, G.; Zhang, Y.; Qian, J.L.; Wang, W.W.; Hao, L.Z.; Qiu, Q.; et al. Apparent digestibility, rumen fermentation, digestive enzymes and urinary purine derivatives in yaks and Qaidam cattle offered forage-concentrate diets differing in nitrogen concentration. Livest. Sci. 2018, 208, 14–21. [Google Scholar] [CrossRef]
- Wen, Y.; He, Q.; Ding, J.; Wang, H.; Hou, Q.; Zheng, Y.; Li, C.; Ma, Y.; Zhang, H.; Kwok, L.Y. Cow, yak, and camel milk diets differentially modulated the systemic immunity and fecal microbiota of rats. Sci. Bull. 2017, 62, 405–414. [Google Scholar] [CrossRef]
- Yang, Y.; Zheng, N.; Zhao, X.; Zhang, Y.; Han, R.; Yang, J.; Zhao, S.; Li, S.; Guo, T.; Zang, C.; et al. Metabolomic biomarkers identify differences in milk produced by Holstein cows and other minor dairy animals. J. Proteomics 2016, 136, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Barrett-Wilt, G.; Fonseca, L.M.; Rankin, S.A. A profile of sphingolipids and related compounds tentatively identified in yak milk. J. Dairy Sci. 2016, 99, 5083–5092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Zhang, D.; Hasegawa, M.; Liu, W.; Zhang, J.; Ma, T.; Cao, C.; Lenstra, J.A.; Zang, X.; et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef]
- Zhang, Q.; Gong, J.; Wang, X.; Wu, X.; Li, Y.; Ma, Y.; Zhang, Y.; Zhao, X. Molecular cloning, bioinformatics analysis and expression of insulin-like growth factor 2 from tianzhu white yak,bos grunniens. Int. J. Mol. Sci. 2014, 15, 504–524. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.L.; Tong, Z.B.; Wei, Y.P.; Zhao, X.Q. Meat characteristics of Qinghai yak and semi-wild yak. Anim. Sci. J. 2006, 77, 230–234. [Google Scholar] [CrossRef]
- Wiener, G.; Han, J.; Long, R. The yak, 2nd ed.; FAO Regional Office for Asia and the, Pacific, Ed.; FAO Regional Office for Asia and the Pacific: Bangkok, 2003; ISBN 92-5-104965-3. [Google Scholar]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in1H NMR metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef] [PubMed]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar] [CrossRef]
- Kneen, M.A.; Annegarn, H.J. Algorithm for fitting XRF, SEM and PIXE X-ray spectra backgrounds. Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. with Mater. Atoms 1996, 109–110, 209–213. [Google Scholar] [CrossRef]
- Liland, K.H.; Almøy, T.; Mevik, B.H. Optimal choice of baseline correction for multivariate calibration of spectra. Appl. Spectrosc. 2010, 64, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Winning, H.; Larsen, F.H.; Bro, R.; Engelsen, S.B. Quantitative analysis of NMR spectra with chemometrics. J. Magn. Reson. 2008, 190, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Hoult, D.I. The principle of reciprocity. J. Magn. Reson. 2011, 213, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Soufan, O.; Chong, J.; Xia, J.; Bourque, G.; Li, S.; Caraus, I.; Wishart, D.S. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]
- Escalona, E.E.; Leng, J.; Dona, A.C.; Merrifield, C.A.; Holmes, E.; Proudman, C.J.; Swann, J.R. Dominant components of the Thoroughbred metabolome characterised by 1H-nuclear magnetic resonance spectroscopy: A metabolite atlas of common biofluids. Equine Vet. J. 2015, 47, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Bertram, H.C.; Yde, C.C.; Zhang, X.; Kristensen, N.B. Effect of dietary nitrogen content on the urine metabolite profile of dairy cows assessed by nuclear magnetic resonance (NMR)-based metabolomics. J. Agric. Food Chem. 2011, 59, 12499–12505. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S.; Valencia, A. MetPA: A web-based metabolomics tool for pathway analysis and visualization. In Bioinformatics; 2011; Volume 27, pp. 2342–2344. [Google Scholar]
- De Buck, J.; Shaykhutdinov, R.; Barkema, H.W.; Vogel, H.J. Metabolomic profiling in cattle experimentally infected with mycobacterium avium subsp. paratuberculosis. PLoS One 2014, 9, e111872. [Google Scholar] [CrossRef] [PubMed]
- Trabi, M.; Keller, M.D.; Jonsson, N.N. NMR-based metabonomics of bovine blood: An investigation into the effects of long term storage on plasma samples. Metabolomics 2013, 9, 1041–1047. [Google Scholar] [CrossRef]
- Gold, A.; Johnson, T.; Costello, L. Effects of altitude stress on mitochondrial function. Am. J. Physiol. Content 1973, 224, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Messier, F.M.; Le Moyec, L.; Santi, C.; Gaston, A.-F.; Triba, M.N.; Roca, E.; Durand, F. The impact of moderate altitude on exercise metabolism in recreational sportsmen: a nuclear magnetic resonance metabolomic approach. Appl. Physiol. Nutr. Metab. 2017, 42, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Tissot van Patot, M.C.; Serkova, N.J.; Haschke, M.; Kominsky, D.J.; Roach, R.C.; Christians, U.; Henthorn, T.K.; Honigman, B. Enhanced leukocyte HIF-1α and HIF-1 DNA binding in humans after rapid ascent to 4300 m. Free Radic. Biol. Med. 2009, 46, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Firth, J.D.; Ebert, B.L.; Ratcliffe, P.J. Hypoxic regulation of lactate dehydrogenase A: Interaction between hypoxia-inducible factor 1 and cAMP response elements. J. Biol. Chem. 1995, 270, 21021–21027. [Google Scholar] [CrossRef] [PubMed]
- Banchero, N. Cardiovascular Responses to Chronic Hypoxia. Annu. Rev. Physiol. 1987, 204, 3133–3139. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, D.M.; Deltimple, N.; van Velzen, E.; van Dorsten, F.A.; Bingham, M.; Vaughan, E.E.; van Duynhoven, J. 1H NMR metabolite profiling of feces as a tool to assess the impact of nutrition on the human microbiome. NMR Biomed. 2008, 21, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Qiu, Q.; Wang, W.; Wang, L.; Zhang, Z.; Zhao, F.; Zhou, J.; Long, R.; Shi, P.; Xu, D.; et al. Convergent Evolution of Rumen Microbiomes in High-Altitude Mammals. Curr. Biol. 2016, 26, 1873–1879. [Google Scholar] [CrossRef]
- Liang, G.; Chen, Y.; Guan, L.L.; Kong, R.S.G.; Stothard, P. Transcriptome profiling of the rumen epithelium of beef cattle differing in residual feed intake. BMC Genomics 2016, 17, 592. [Google Scholar] [CrossRef]
- Graf, G.C.; Petersen, W.E. Changes in Respiration and Heart Rates, Body Temperatures, Plasma Lactic Acid Levels and Plasma Creatinine Levels Caused by Stress in Dairy Cattle. J. Dairy Sci. 1953, 36, 1036–1048. [Google Scholar] [CrossRef]
- Kaur, T.; Mazumder, A.; Gandhi, S.; Khushu, S.; Koundal, S. “Omics” of High Altitude Biology: A Urinary Metabolomics Biomarker Study of Rats Under Hypobaric Hypoxia. Omi. A J. Integr. Biol. 2015, 19, 757–765. [Google Scholar] [CrossRef]
- Vercoe, J.E. Urinary allantoin excretion and digestible dry-matter intake in cattle and buffalo. J. Agric. Sci. 1976, 86, 613–615. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | ppm * | Functional Group | Multiplicity ** | Source [25] *** |
|---|---|---|---|---|
| 3-Hydroxybutyrate | 1.1863 | CH3 | d | E |
| Acetate | 1.9071 | CH3 | s | P |
| Alanine | 1.4675 | CH3 | d | P |
| Creatine | 3.0222 | CH2 | s | P |
| Dimethyl sulfone | 3.1391 | CH3 | s | D, M |
| Ethanol | 1.1699 | CH3 | t | E, M |
| Formate | 8.4446 | CH | s | E |
| Glucose | 3.2233 | CH-2 | dd | D, E |
| Glycine | 3.5533 | CH2 | s | P |
| Isoleucine | 1.0020 | CH3-9 | d | P |
| Lactate | 4.1059 | CH | dd | E |
| Methanol | 3.3481 | CH3 | s | E |
| Succinate | 2.3933 | CH2 | s | P, E |
| Tyrosine | 7.1776 | CH-3 | d | P |
| Valine | 1.0206 | CH3-7 | d | P |
| Molecules | Serum (mmol/L) | Feces (mmol/g) | Urine (mmol/L) |
|---|---|---|---|
| 3-Hydroxybutyrate | 1.40 × 10−1 (3.56 × 10−2) | 1.52 × 10−5 (1.30 × 10−5) | 1.28 × 10−3 (7.54 × 10−4) |
| Acetate | 1.40 × 10−1 (1.59 × 10−1) | 3.66 × 10−2 (1.06 × 10−2) | 5.66 × 10−4 (4.03 × 10−4) |
| Alanine | 2.78 × 10−1 (1.27 × 10−2) | 5.16 × 10−4 (3.25 × 10−4) | 2.14 × 10−4 (4.23 × 10−5) |
| Creatine | 2.01 × 10−1 (2.12 × 10−1) | 1.91 × 10−5 (1.88 × 10−5) | 4.00 × 10−2 (1.80 × 10−2) |
| Dimethyl sulfone | 1.23 × 10−2 (4.15 × 10−3) | 1.29 × 10−5 (1.07 × 10−5) | 6.15 × 10−4 (1.40 × 10−4) |
| Ethanol | 4.69 × 10−3 (2.74 × 10−3) | 7.15 × 10−5 (2.98 × 10−5) | 2.62 × 10−4 (3.81 × 10−5) |
| Formate | 1.78 × 10−2 (5.69 × 10−3) | 1.17 × 10−4 (2.43 × 10−5) | 2.50 × 10−4 (1.70 × 10−4) |
| Glucose | 1.37 (5.97 × 10−1) | 3.51 × 10−4 (5.47 × 10−5) | 8.33 × 10−4 (4.09 × 10−4) |
| Glycine | 5.11 × 10−1 (3.91 × 10−1) | 1.77 × 10−4 (7.46 × 10−6) | 8.52 × 10−4 (5.64 × 10−4) |
| Isoleucine | 4.35 × 10−2 (1.62 × 10−2) | 6.20 × 10−5 (1.39 × 10−4) | 1.33 × 10−4 (7.24 × 10−5) |
| Lactate | 7.25 (5.46 × 10−1) | 5.57 × 10−5 (5.13 × 10−5) | 9.37 × 10−4 (1.87 × 10−4) |
| Methanol | 7.14 × 10−3 (1.78 × 10−3) | 9.12 × 10−5 (4.36 × 10−5) | 3.95 × 10−5 (2.32 × 10−5) |
| Succinate | 2.01 × 10−1 (4.09 × 10−2) | 1.12 × 10−4 (4.47 × 10−5) | 9.15 × 10−5 (7.46 × 10−5) |
| Tyrosine | 2.53 × 10−2 (1.22 × 10−2) | 1.16 × 10−4 (6.81 × 10−5) | 1.23 × 10−3 (2.26 × 10−5) |
| Valine | 1.48 × 10−1 (1.14 × 10−2) | 1.91 × 10−4 (5.42 × 10−5) | 1.22 × 10−4 (3.45 × 10−5) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, C.; Li, C.; Wang, Y.; Laghi, L. Characterization of Yak Common Biofluids Metabolome by Means of Proton Nuclear Magnetic Resonance Spectroscopy. Metabolites 2019, 9, 41. https://doi.org/10.3390/metabo9030041
Zhu C, Li C, Wang Y, Laghi L. Characterization of Yak Common Biofluids Metabolome by Means of Proton Nuclear Magnetic Resonance Spectroscopy. Metabolites. 2019; 9(3):41. https://doi.org/10.3390/metabo9030041
Chicago/Turabian StyleZhu, Chenglin, Cheng Li, Yaning Wang, and Luca Laghi. 2019. "Characterization of Yak Common Biofluids Metabolome by Means of Proton Nuclear Magnetic Resonance Spectroscopy" Metabolites 9, no. 3: 41. https://doi.org/10.3390/metabo9030041
APA StyleZhu, C., Li, C., Wang, Y., & Laghi, L. (2019). Characterization of Yak Common Biofluids Metabolome by Means of Proton Nuclear Magnetic Resonance Spectroscopy. Metabolites, 9(3), 41. https://doi.org/10.3390/metabo9030041