Mitochondrial Dysfunction in the Transition from NASH to HCC


{kind=link}
{kind=link}
Abstract
1. Introduction
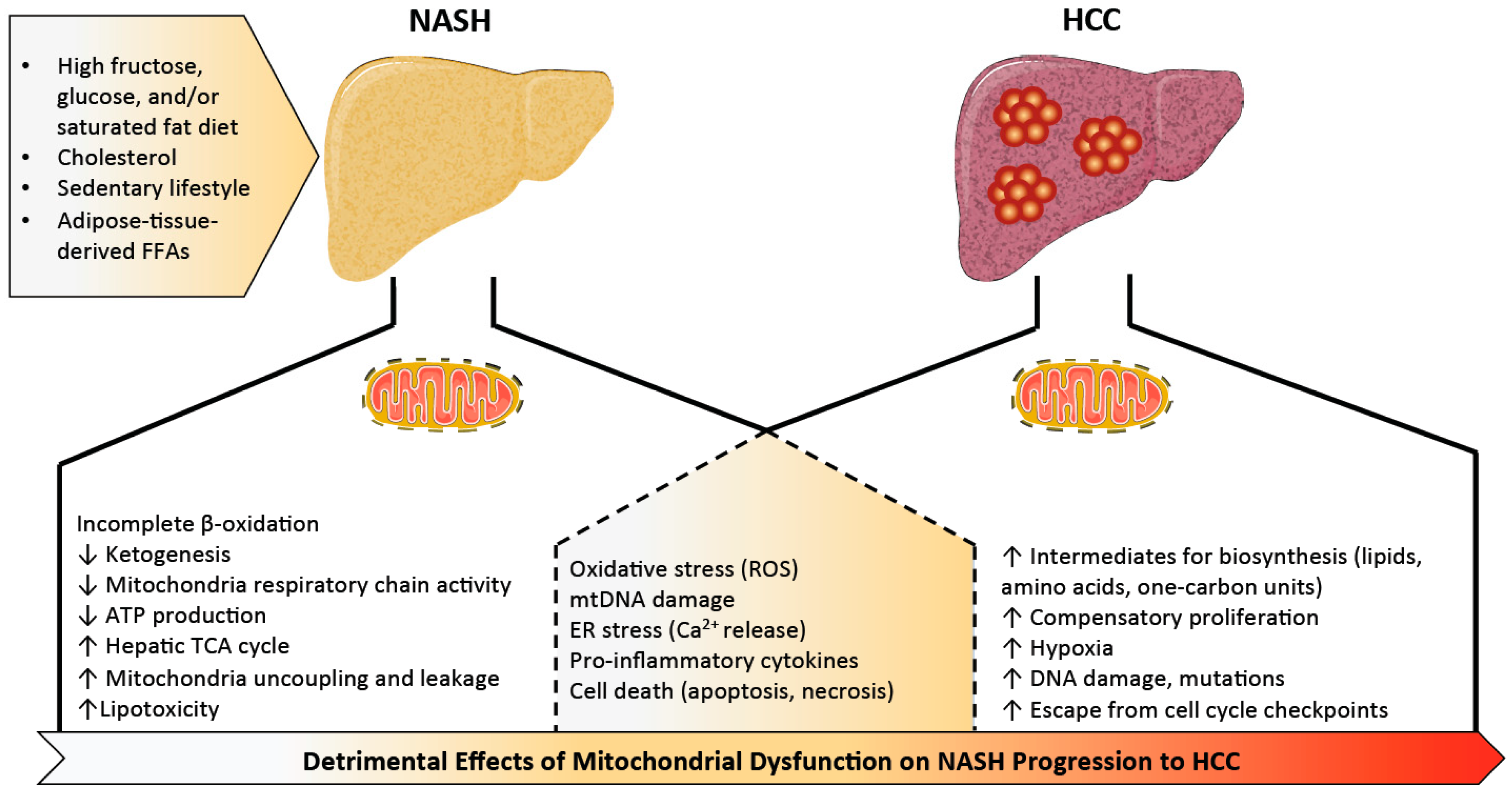
2. Mitochondrial Dysfunction in Non-Alcoholic Steatohepatitis (NASH)
2.1. Mitochondrial Adaptation and Flexibility
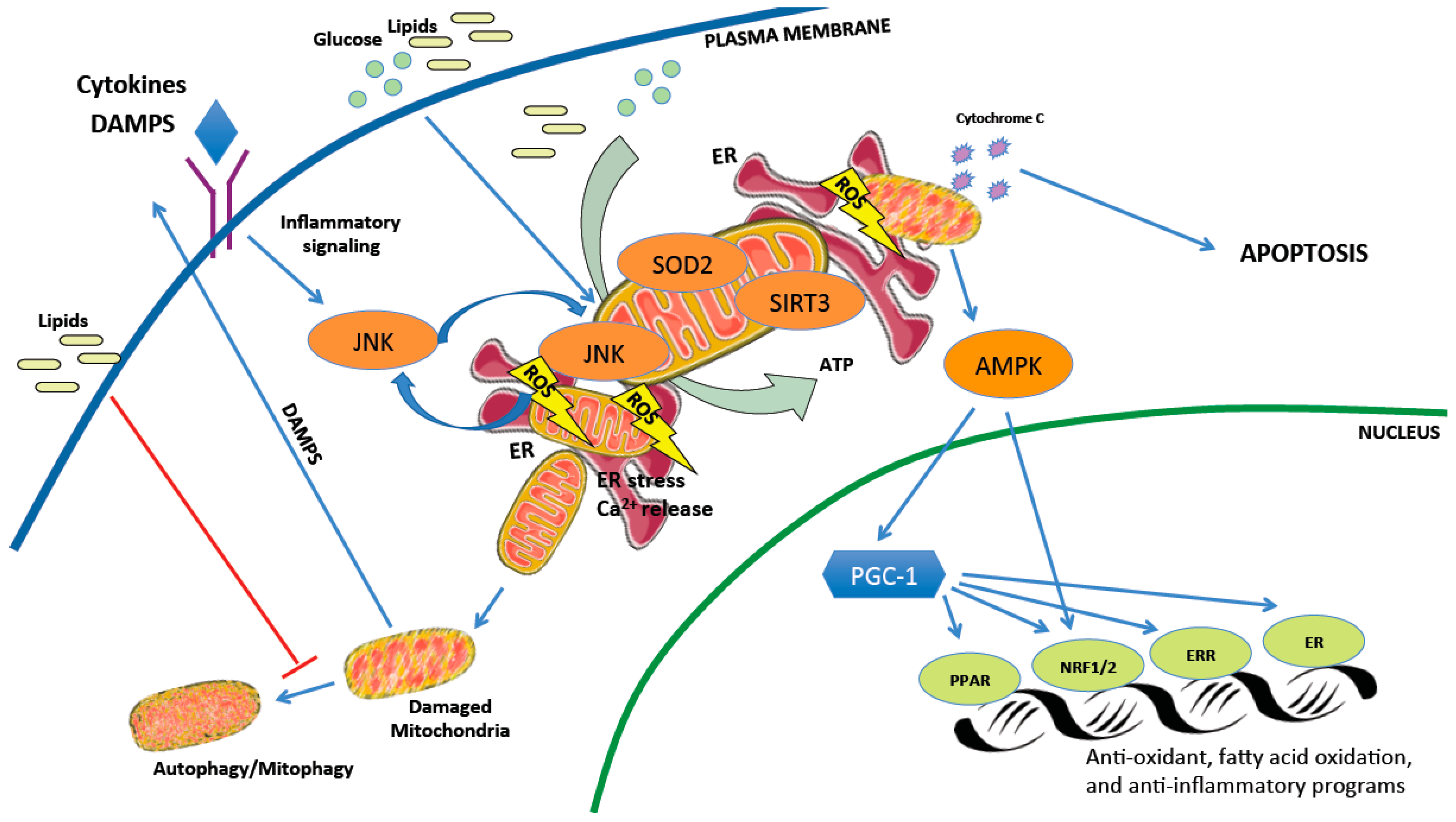
2.2. The Delicate Balance of Mitochondrial Reactive Oxygen Species
2.3. Mitochondrial Networks, Housekeeping, and Cell Death
2.4. We Are What We Eat
3. Mitochondrial Dysfunction in NASH-Associated HCC
3.1. From Cell Death to Proliferation
3.2. Cancer Cells: It is Not Only What You Burn, But When
3.3. Transcriptional Regulation of Mitochondrial Adaptation in HCC
4. Challenges in Targeting Mitochondria for NASH-Related HCC Treatment
4.1. Lifestyle Intervention
4.2. Antioxidants
4.3. Pioglitazone
4.4. NAD+ Precursors
4.5. Master Regulators of Metabolism
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yang, K.C.; Hung, H.-F.; Lu, C.-W.; Chang, H.-H.; Lee, L.-T.; Huang, K.-C. Association of Non-alcoholic Fatty Liver Disease with Metabolic Syndrome Independently of Central Obesity and Insulin Resistance. Sci. Rep. 2016, 6, 27034. [Google Scholar] [CrossRef]
- Asrih, M.; Jornayvaz, F.R. Metabolic syndrome and nonalcoholic fatty liver disease: Is insulin resistance the link? Mol. Cell. Endocrinol. 2015, 418, 55–65. [Google Scholar] [CrossRef]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease: Novel Mechanisms and Treatment Strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef]
- Portillo-Sanchez, P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High Prevalence of Nonalcoholic Fatty Liver Disease in Patients with Type 2 Diabetes Mellitus and Normal Plasma Aminotransferase Levels. J. Clin. Endocrinol. Metab. 2015, 100, 2231–2238. [Google Scholar] [CrossRef]
- Maximos, M.; Bril, F.; Sanchez, P.P.; Lomonaco, R.; Orsak, B.; Biernacki, D.; Suman, A.; Weber, M.; Cusi, K. The role of liver fat and insulin resistance as determinants of plasma aminotransferase elevation in nonalcoholic fatty liver disease. Hepatology 2015, 61, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.-J.; Fallon, M.B. Gender and racial differences in nonalcoholic fatty liver disease. World J. Hepatol. 2014, 6, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in NAFLD: State of the Art and Identification of Research Gaps. Hepatology 2019. [Google Scholar] [CrossRef]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306.e20. [Google Scholar] [CrossRef] [PubMed]
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 451–496. [Google Scholar] [CrossRef]
- Calzadilla Bertot, L.; Adams, L. The Natural Course of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 774. [Google Scholar] [CrossRef]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359.e2. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Otgonsuren, M.; Henry, L.; Venkatesan, C.; Mishra, A.; Erario, M.; Hunt, S. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015, 62, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Poklepovic, A.; Moyneur, E.; Barghout, V. Population-based risk factors and resource utilization for HCC: US perspective. Curr. Med. Res. Opin. 2010, 26, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Sood, G.K. Hepatocellular carcinoma review: Current treatment, and evidence-based medicine. World J. Gastroenterol. 2014, 20, 4115–4127. [Google Scholar] [CrossRef]
- Edeline, J.; Raoul, J.-L.; Vauleon, E.; Guillygomac’h, A.; Boudjema, K.; Boucher, E. Systemic chemotherapy for hepatocellular carcinoma in non-cirrhotic liver: A retrospective study. World J. Gastroenterol. 2009, 15, 713–716. [Google Scholar] [CrossRef]
- Medavaram, S.; Zhang, Y. Emerging therapies in advanced hepatocellular carcinoma. Exp. Hematol. Oncol. 2018, 7, 17. [Google Scholar] [CrossRef]
- Balogh, J.; Victor, D.; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P. Hepatocellular carcinoma: A review. J. Hepatocell. Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef]
- Cholankeril, G.; Patel, R.; Khurana, S.; Satapathy, S.K. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Current knowledge and implications for management. World J. Hepatol. 2017, 9, 533–543. [Google Scholar] [CrossRef]
- Kawada, N.; Imanaka, K.; Kawaguchi, T.; Tamai, C.; Ishihara, R.; Matsunaga, T.; Gotoh, K.; Yamada, T.; Tomita, Y. Hepatocellular carcinoma arising from non-cirrhotic nonalcoholic steatohepatitis. J. Gastroenterol. 2009, 44, 1190–1194. [Google Scholar] [CrossRef]
- Paradis, V.; Zalinski, S.; Chelbi, E.; Guedj, N.; Degos, F.; Vilgrain, V.; Bedossa, P.; Belghiti, J. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: A pathological analysis. Hepatology 2009, 49, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Said, A.; Ghufran, A. Epidemic of non-alcoholic fatty liver disease and hepatocellular carcinoma. World J. Clin. Oncol. 2017, 8, 429–436. [Google Scholar] [CrossRef]
- Shaker, M.; Tabbaa, A.; Albeldawi, M.; Alkhouri, N. Liver transplantation for nonalcoholic fatty liver disease: New challenges and new opportunities. World J. Gastroenterol. 2014, 20, 5320. [Google Scholar] [CrossRef] [PubMed]
- Degli Esposti, D.; Hamelin, J.; Bosselut, N.; Saffroy, R.; Sebagh, M.; Pommier, A.; Martel, C.; Lemoine, A. Mitochondrial roles and cytoprotection in chronic liver injury. Biochem. Res. Int. 2012, 2012, 387626. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Role of Mitochondria in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.-A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Hayata, Y.; Kawamura, S.; Yamada, T.; Fujiwara, N.; Koike, K. Lipid Metabolic Reprogramming in Hepatocellular Carcinoma. Cancers 2018, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Mello, T.; Simeone, I.; Galli, A. Mito-Nuclear Communication in Hepatocellular Carcinoma Metabolic Rewiring. Cells 2019, 8, 417. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.E.; Kalavalapalli, S.; Williams, C.M.; Nautiyal, M.; Mathew, J.T.; Martinez, J.; Reinhard, M.K.; McDougall, D.J.; Rocca, J.R.; Yost, R.A.; et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am. J. Physiol.-Endocrinol. Metab. 2016, 310, E484–E494. [Google Scholar] [CrossRef]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Méndez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Grieco, A.; Armuzzi, A.; Candelli, M.; Forgione, A.; Gasbarrini, A.; Gasbarrini, G. Hepatic mitochondrial beta-oxidation in patients with nonalcoholic steatohepatitis assessed by 13C-octanoate breath test. Am. J. Gastroenterol. 2003, 98, 2335–2336. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Satapati, S.; Fu, X.; He, T.; Mehdibeigi, R.; Spring-Robinson, C.; Duarte, J.; Potthoff, M.J.; Browning, J.D.; Burgess, S.C. Progressive adaptation of hepatic ketogenesis in mice fed a high-fat diet. Am. J. Physiol.-Endocrinol. Metab. 2010, 298, E1226–E1235. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.G.; Ercal, B.; Huang, X.; Leid, J.M.; d’Avignon, D.A.; Graham, M.J.; Dietzen, D.J.; Brunt, E.M.; Patti, G.J.; Crawford, P.A. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Investig. 2014, 124, 5175–5190. [Google Scholar] [CrossRef]
- Männistö, V.T.; Simonen, M.; Hyysalo, J.; Soininen, P.; Kangas, A.J.; Kaminska, D.; Matte, A.K.; Venesmaa, S.; Käkelä, P.; Kärjä, V.; et al. Ketone body production is differentially altered in steatosis and non-alcoholic steatohepatitis in obese humans. Liver Int. 2015, 35, 1853–1861. [Google Scholar] [CrossRef]
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Chavin, K.D.; Yang, S.; Lin, H.Z.; Chatham, J.; Chacko, V.P.; Hoek, J.B.; Walajtys-Rode, E.; Rashid, A.; Chen, C.H.; Huang, C.C.; et al. Obesity induces expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. J. Biol. Chem. 1999, 274, 5692–5700. [Google Scholar] [CrossRef]
- Nair, S.; Chacko, V.P.; Arnold, C.; Diehl, A.M. Hepatic ATP reserve and efficiency of replenishing: Comparison between obese and nonobese normal individuals. Am. J. Gastroenterol. 2003, 98, 466–470. [Google Scholar]
- Cortez-Pinto, H.; Chatham, J.; Chacko, V.P.; Arnold, C.; Rashid, A.; Diehl, A.M. Alterations in Liver ATP Homeostasis in Human Nonalcoholic Steatohepatitis: A Pilot Study. JAMA 1999, 282, 1659–1664. [Google Scholar] [CrossRef]
- Schmid, A.I.; Szendroedi, J.; Chmelik, M.; Krššák, M.; Moser, E.; Roden, M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 2011, 34, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Szendroedi, J.; Chmelik, M.; Schmid, A.I.; Nowotny, P.; Brehm, A.; Krššák, M.; Moser, E.; Roden, M. Abnormal hepatic energy homeostasis in type 2 diabetes. Hepatology 2009, 50, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, M.; Koliaki, C.; Livingstone, R.; Phielix, E.; Bierwagen, A.; Meisinger, M.; Jelenik, T.; Strassburger, K.; Zimmermann, S.; Brockmann, K.; et al. Time course of postprandial hepatic phosphorus metabolites in lean, obese, and type 2 diabetes patients. Am. J. Clin. Nutr. 2015, 102, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Crespo, D.M.; Kang, H.S.; Al-Osaimi, A.M.S. Obesity and hepatocellular carcinoma. Gastroenterology 2004, 127, S97–S103. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef]
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2005, 100, 850–855. [Google Scholar] [CrossRef]
- Niederreiter, L.; Tilg, H. Cytokines and fatty liver diseases. Liver Res. 2018, 2, 14–20. [Google Scholar] [CrossRef]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef]
- Fromenty, B.; Robin, M.A.; Igoudjil, A.; Mansouri, A.; Pessayre, D. The ins and outs of mitochondrial dysfunction in NASH. Diabetes Metab. 2004, 30, 121–138. [Google Scholar] [CrossRef]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef]
- Leclercq, I.A.; Farrell, G.C.; Field, J.; Bell, D.R.; Gonzalez, F.J.; Robertson, G.R. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J. Clin. Investig. 2000, 105, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Besse-Patin, A.; Léveillé, M.; Oropeza, D.; Nguyen, B.N.; Prat, A.; Estall, J.L. Estrogen Signals Through Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α to Reduce Oxidative Damage Associated With Diet-Induced Fatty Liver Disease. Gastroenterology 2017, 152, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Satapati, S.; Kucejova, B.; Duarte, J.A.G.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.A.; Livingston, K.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Zhang, J.; Oo, C.; Min, R.W.M.; Kaplowitz, N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology 2018, 67, 2013–2024. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Min, R.W.M.; Aghajan, M.; Kaplowitz, N. JNK mediates mouse liver injury through a novel Sab (SH3BP5) dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 2016, 63, 1987–2003. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Le, B.H.A.; García-Ruiz, C.; Fernandez-Checa, J.C.; Kaplowitz, N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 2015, 62, 1367–1374. [Google Scholar] [CrossRef]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef]
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L.J. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell. Biol. 2014, 34, 3305–3320. [Google Scholar]
- Garcia, D.; Hellberg, K.; Chaix, A.; Wallace, M.; Herzig, S.; Badur, M.G.; Lin, T.; Shokhirev, M.N.; Pinto, A.F.M.; Ross, D.S.; et al. Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD. Cell Rep. 2019, 26, 192–208.e6. [Google Scholar] [CrossRef]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.-P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; Pierre, J.S.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARγ-coactivator 1α (PGC1α) and mitochondrial biogenesis in mice. Lab. Investig. 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Mello, T.; Materozzi, M.; Galli, A. PPARs and Mitochondrial Metabolism: From NAFLD to HCC. PPAR Res. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Casellas, A.; Amengual-Cladera, E.; Proenza, A.M.; Lladó, I.; Gianotti, M. Long-term high-fat-diet feeding impairs mitochondrial biogenesis in liver of male and female rats. Cell. Physiol. Biochem. 2010, 26, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kamat, A.; Pergola, P.; Swamy, A.; Tio, F.; Cusi, K. Metabolic factors in the development of hepatic steatosis and altered mitochondrial gene expression in vivo. Metabolism 2011, 60, 1090–1099. [Google Scholar] [CrossRef]
- Sczelecki, S.; Besse-Patin, A.; Abboud, A.; Kleiner, S.; Laznik-Bogoslavski, D.; Wrann, C.D.; Ruas, J.L.; Haibe-Kains, B.; Estall, J.L. Loss of Pgc-1α expression in aging mouse muscle potentiates glucose intolerance and systemic inflammation. Am. J. Physiol.-Endocrinol. Metab. 2013, 306, E157–E167. [Google Scholar] [CrossRef] [PubMed]
- Derbré, F.; Gomez-Cabrera, M.C.; Nascimento, A.L.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Tresguerres, J.A.F.; Fuentes, T.; Gratas-Delamarche, A.; Monsalve, M.; Viña, J. Age associated low mitochondrial biogenesis may be explained by lack of response of PGC-1α to exercise training. Age 2012, 34, 669–679. [Google Scholar] [CrossRef]
- Seo, A.Y.; Joseph, A.-M.; Dutta, D.; Hwang, J.C.Y.; Aris, J.P.; Leeuwenburgh, C. New insights into the role of mitochondria in aging: Mitochondrial dynamics and more. J. Cell Sci. 2010, 123, 2533–2542. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial Aging and Age-Related Dysfunction of Mitochondria. Available online: https://www.hindawi.com/journals/bmri/2014/238463/ (accessed on 25 July 2019).
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef]
- Suzuki, A.; Abdelmalek, M.F. Nonalcoholic fatty liver disease in women. Womens Health 2009, 5, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.M.; Meers, G.M.E.; Booth, F.W.; Fritsche, K.L.; Hardin, C.D.; Thyfault, J.P.; Ibdah, J.A. PGC-1α overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am. J. Physiol.-Gastrointest. Liver Physiol. 2012, 303, G979–G992. [Google Scholar] [CrossRef] [PubMed]
- Laye, M.J.; Rector, R.S.; Borengasser, S.J.; Naples, S.P.; Uptergrove, G.M.; Ibdah, J.A.; Booth, F.W.; Thyfault, J.P. Cessation of daily wheel running differentially alters fat oxidation capacity in liver, muscle, and adipose tissue. J. Appl. Physiol. 2009, 106, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Haase, T.N.; Ringholm, S.; Leick, L.; Biensø, R.S.; Kiilerich, K.; Johansen, S.; Nielsen, M.M.; Wojtaszewski, J.F.; Hidalgo, J.; Pedersen, P.A.; et al. Role of PGC-1α in exercise and fasting-induced adaptations in mouse liver. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1501–R1509. [Google Scholar] [CrossRef]
- Lezi, E.; Lu, J.; Selfridge, J.E.; Burns, J.M.; Swerdlow, R.H. Lactate administration reproduces specific brain and liver exercise-related changes. J. Neurochem. 2013, 127, 91–100. [Google Scholar]
- McCoin, C.S.; Von Schulze, A.; Allen, J.; Fuller, K.N.; Xia, Q.; Koestler, D.C.; Houchen, C.J.; Maurer, A.; Dorn, G.W., II; Shankar, K.; et al. Sex modulates hepatic mitochondrial adaptations to high fat diet and physical activity. Am. J. Physiol.-Endocrinol. Metab. 2019. [Google Scholar] [CrossRef] [PubMed]
- Buler, M.; Aatsinki, S.-M.; Skoumal, R.; Komka, Z.; Tóth, M.; Kerkelä, R.; Georgiadi, A.; Kersten, S.; Hakkola, J. Energy-sensing factors coactivator peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) and AMP-activated protein kinase control expression of inflammatory mediators in liver: Induction of interleukin 1 receptor antagonist. J. Biol. Chem. 2012, 287, 1847–1860. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Fuente-Martin, E.; Finan, B.; Kim, M.; Frank, A.; Garcia-Caceres, C.; Navas, C.R.; Gordillo, R.; Neinast, M.; Kalainayakan, S.P.; et al. Hypothalamic PGC-1α protects against high-fat diet exposure by regulating ERα. Cell Rep. 2014, 9, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Barroso, W.A.; Victorino, V.J.; Jeremias, I.C.; Petroni, R.C.; Ariga, S.K.K.; Salles, T.A.; Barbeiro, D.F.; de Lima, T.M.; de Souza, H.P. High-fat diet inhibits PGC-1α suppressive effect on NFκB signaling in hepatocytes. Eur. J. Nutr. 2018, 57, 1891–1900. [Google Scholar] [CrossRef]
- Piccinin, E.; Villani, G.; Moschetta, A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: The role of PGC1 coactivators. Nat. Rev. Gastroenterol Hepatol. 2018, 16, 160–174. [Google Scholar] [CrossRef]
- Ricci, C.; Pastukh, V.; Leonard, J.; Turrens, J.; Wilson, G.; Schaffer, D.; Schaffer, S.W. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am. J. Physiol.-Cell Physiol. 2008, 294, C413–C422. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.R.; Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. TEM 2012, 23, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-Sensitive Mitochondrial NAD+ Levels Dictate Cell Survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Liu, Y.; Fu, Y.; Liu, X.; Zhou, X. Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 2014, 44, 410–418. [Google Scholar] [PubMed]
- He, J.; Hu, B.; Shi, X.; Weidert, E.R.; Lu, P.; Xu, M.; Huang, M.; Kelley, E.E.; Xie, W. Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Mol. Cell. Biol. 2013, 33, 2047–2055. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Sirtuins and nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 10084–10092. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, L.; Wang, P.; Li, X.; Qiu, D.; Li, L.; Zhang, J.; Hou, X.; Han, L.; Ge, J.; et al. Sirt3-dependent deacetylation of SOD2 plays a protective role against oxidative stress in oocytes from diabetic mice. Cell Cycle 2017, 16, 1302–1308. [Google Scholar] [CrossRef]
- Bagul, P.K.; Katare, P.B.; Bugga, P.; Dinda, A.K.; Banerjee, S.K. SIRT-3 Modulation by Resveratrol Improves Mitochondrial Oxidative Phosphorylation in Diabetic Heart through Deacetylation of TFAM. Cells 2018, 7, 235. [Google Scholar] [CrossRef]
- Choudhury, M.; Jonscher, K.R.; Friedman, J.E. Reduced mitochondrial function in obesity-associated fatty liver: SIRT3 takes on the fat. Aging 2011, 3, 175–178. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef]
- Cho, E.-H. SIRT3 as a Regulator of Non-alcoholic Fatty Liver Disease. J. Lifestyle Med. 2014, 4, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Jeninga, E.H.; Canto, C.; Harach, T.; de Boer, V.C.J.; Andreux, P.; Moullan, N.; Pirinen, E.; Yamamoto, H.; Houten, S.M.; et al. Muscle or liver-specific Sirt3 deficiency induces hyperacetylation of mitochondrial proteins without affecting global metabolic homeostasis. Sci. Rep. 2012, 2, 425. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Quan, X.-B.; Zeng, W.-J.; Yang, X.-O.; Wang, M.-J. Mechanism of Hepatocyte Apoptosis. J. Cell Death 2016, 9, 19–29. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Kroemer, G. Mitochondrial control of apoptosis: An overview. Biochem. Soc. Symp. 1999, 66, 1–15. [Google Scholar] [CrossRef]
- Gupta, S.; Kass, G.E.; Szegezdi, E.; Joseph, B. The mitochondrial death pathway: A promising therapeutic target in diseases. J. Cell. Mol. Med. 2009, 13, 1004–1033. [Google Scholar] [CrossRef]
- Yin, X.; Zheng, F.; Pan, Q.; Zhang, S.; Yu, D.; Xu, Z.; Li, H. Glucose fluctuation increased hepatocyte apoptosis under lipotoxicity and the involvement of mitochondrial permeability transition opening. J. Mol. Endocrinol. 2015, 55, 169–181. [Google Scholar] [CrossRef]
- Schuster, S.; Johnson, C.D.; Hennebelle, M.; Holtmann, T.; Taha, A.Y.; Kirpich, I.A.; Eguchi, A.; Ramsden, C.E.; Papouchado, B.G.; McClain, C.J.; et al. Oxidized linoleic acid metabolites induce liver mitochondrial dysfunction, apoptosis, and NLRP3 activation in mice. J. Lipid Res. 2018, 59, 1597–1609. [Google Scholar] [CrossRef]
- Mishra, J.; Davani, A.J.; Stowe, D.F.; Kwok, W.-M.; Camara, A.K.S. Cyclosporin A: New Insights into its Potential Role in Mitochondrial Calcium Buffering. Biophys. J. 2018, 114, 659a. [Google Scholar] [CrossRef]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010, 24, 3052–3065. [Google Scholar] [CrossRef] [PubMed]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Sasai, M.; Linehan, M.M.; Iwasaki, A. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science 2010, 329, 1530–1534. [Google Scholar] [CrossRef]
- Zhang, N.-P.; Liu, X.-J.; Xie, L.; Shen, X.-Z.; Wu, J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Investig. 2019, 199, 749–763. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Pihán, P.; Hetz, C. The Unfolded Protein Response: At the Intersection between Endoplasmic Reticulum Function and Mitochondrial Bioenergetics. Front. Oncol. 2017, 7, 1. [Google Scholar] [CrossRef]
- Van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta 2014, 1843, 2253–2262. [Google Scholar] [CrossRef]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.-A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef]
- Theurey, P.; Tubbs, E.; Vial, G.; Jacquemetton, J.; Bendridi, N.; Chauvin, M.-A.; Alam, M.R.; Le Romancer, M.; Vidal, H.; Rieusset, J. Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell Biol. 2016, 8, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Brumbaugh, D.E.; Friedman, J.E. Developmental origins of nonalcoholic fatty liver disease. Pediatric Res. 2014, 75, 140–147. [Google Scholar] [CrossRef]
- Peng, K.-Y.; Watt, M.J.; Rensen, S.; Greve, J.W.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J. Lipid Res. 2018, 59, 1977–1986. [Google Scholar] [CrossRef]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef]
- Rosqvist, F.; Kullberg, J.; Ståhlman, M.; Cedernaes, J.; Heurling, K.; Johansson, H.-E.; Iggman, D.; Wilking, H.; Larsson, A.; Eriksson, O.; et al. Overeating saturated fat promotes fatty liver and ceramides compared to polyunsaturated fat: A randomized trial. J. Clin. Endocrinol. Metab. 2019. [Google Scholar] [CrossRef]
- Mendez-Sanchez, N.; Cruz-Ramon, V.C.; Ramirez-Perez, O.L.; Hwang, J.P.; Barranco-Fragoso, B.; Cordova-Gallardo, J. New Aspects of Lipotoxicity in Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2018, 19, 2034. [Google Scholar] [CrossRef]
- García-Ruiz, I.; Solís-Muñoz, P.; Fernández-Moreira, D.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. In vitro treatment of HepG2 cells with saturated fatty acids reproduces mitochondrial dysfunction found in nonalcoholic steatohepatitis. Dis. Model. Mech. 2015, 8, 183–191. [Google Scholar] [CrossRef]
- Vanhorebeek, I.; De Vos, R.; Mesotten, D.; Wouters, P.J.; De Wolf-Peeters, C.; Van den Berghe, G. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet 2005, 365, 53–59. [Google Scholar] [CrossRef]
- Sun, L.; Xie, P.; Wada, J.; Kashihara, N.; Liu, F.; Zhao, Y.; Kumar, D.; Chugh, S.S.; Danesh, F.R.; Kanwar, Y.S. Rap1b GTPase Ameliorates Glucose-Induced Mitochondrial Dysfunction. J. Am. Soc. Nephrol. 2008, 19, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Dassanayaka, S.; Readnower, R.D.; Salabei, J.K.; Long, B.W.; Aird, A.L.; Zheng, Y.-T.; Muthusamy, S.; Facundo, H.T.; Hill, B.G.; Jones, S.P. High glucose induces mitochondrial dysfunction independently of protein O-GlcNAcylation. Biochem. J. 2015, 467, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Tien, T.; Zhang, J.; Muto, T.; Kim, D.; Sarthy, V.P.; Roy, S. High Glucose Induces Mitochondrial Dysfunction in Retinal Müller Cells: Implications for Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2915–2921. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.-L.; Zhu, C.; Zhao, Y.-P.; Chen, X.-H.; Ji, C.-B.; Zhang, C.-M.; Zhu, J.-G.; Xia, Z.-K.; Tong, M.-L.; Guo, X.-R. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 2010, 320, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.W.; Golovoy, D.; Vincent, A.M.; Mahendru, P.; Olzmann, J.A.; Mentzer, A.; Feldman, E.L. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 2002, 16, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, K.; Molina, A.J.A.; Roy, S. High glucose induces mitochondrial morphology and metabolic changes in retinal pericytes. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8657–8664. [Google Scholar] [CrossRef] [PubMed]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.-J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef]
- Cioffi, F.; Senese, R.; Lasala, P.; Ziello, A.; Mazzoli, A.; Crescenzo, R.; Liverini, G.; Lanni, A.; Goglia, F.; Iossa, S. Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients 2017, 9, 323. [Google Scholar] [CrossRef]
- García-Berumen, C.I.; Ortiz-Avila, O.; Vargas-Vargas, M.A.; del Rosario-Tamayo, B.A.; Guajardo-López, C.; Saavedra-Molina, A.; Rodríguez-Orozco, A.R.; Cortés-Rojo, C. The severity of rat liver injury by fructose and high fat depends on the degree of respiratory dysfunction and oxidative stress induced in mitochondria. Lipids Health Dis. 2019, 18, 78. [Google Scholar] [CrossRef]
- Ackerman, Z.; Oron-Herman, M.; Grozovski, M.; Rosenthal, T.; Pappo, O.; Link, G.; Sela, B.-A. Fructose-induced fatty liver disease: Hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension 2005, 45, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Collison, K.S.; Maqbool, Z.M.; Inglis, A.L.; Makhoul, N.J.; Saleh, S.M.; Bakheet, R.H.; Al-Johi, M.A.; Al-Rabiah, R.K.; Zaidi, M.Z.; Al-Mohanna, F.A.; et al. Effect of dietary monosodium glutamate on HFCS-induced hepatic steatosis: Expression profiles in the liver and visceral fat. Obesity 2010, 18, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Schuschke, D.A.; Zhou, Z.; Chen, T.; Shi, X.; Zhang, J.; Zhang, X.; Pierce, W.M.; Johnson, W.T.; Vos, M.B.; et al. Modest Fructose Beverage Intake Causes Liver Injury and Fat Accumulation in Marginal Copper Deficient Rats. Obesity 2013, 21, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.-K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.; Neil, D.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Hepatic adverse effects of fructose consumption independent of overweight/obesity. Int. J. Mol. Sci. 2013, 14, 21873–21886. [Google Scholar] [CrossRef] [PubMed]
- Bremer, A.A.; Stanhope, K.L.; Graham, J.L.; Cummings, B.P.; Wang, W.; Saville, B.R.; Havel, P.J. Fructose-fed rhesus monkeys: A nonhuman primate model of insulin resistance, metabolic syndrome, and type 2 diabetes. Clin. Transl. Sci. 2011, 4, 243–252. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef]
- Liang, J.Q.; Teoh, N.; Xu, L.; Pok, S.; Li, X.; Chu, E.S.H.; Chiu, J.; Dong, L.; Arfianti, E.; Haigh, W.G.; et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun. 2018, 9, 4490. [Google Scholar] [CrossRef]
- Domínguez-Pérez, M.; Simoni-Nieves, A.; Rosales, P.; Nuño-Lámbarri, N.; Rosas-Lemus, M.; Souza, V.; Miranda, R.U.; Bucio, L.; Uribe Carvajal, S.; Marquardt, J.U.; et al. Cholesterol burden in the liver induces mitochondrial dynamic changes and resistance to apoptosis. J. Cell. Physiol. 2019, 234, 7213–7223. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef]
- Bellanti, F.; Mitarotonda, D.; Tamborra, R.; Blonda, M.; Iannelli, G.; Petrella, A.; Sanginario, V.; Iuliano, L.; Vendemiale, G.; Serviddio, G. OP2–6—Oxysterols induce mitochondrial impairment and hepatocellular toxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2014, 75, S16–S17. [Google Scholar] [CrossRef]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernández, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Tamborra, R.; Rollo, T.; Capitanio, N.; Romano, A.D.; Sastre, J.; Vendemiale, G.; Altomare, E. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut 2008, 57, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, C.; Mari, M.; Colell, A.; Morales, A.; Caballero, F.; Montero, J.; Terrones, O.; Basañez, G.; Fernández-Checa, J.C. Mitochondrial cholesterol in health and disease. Histol. Histopathol. 2009, 24, 117–132. [Google Scholar] [PubMed]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Enríquez-Cortina, C.; Bello-Monroy, O.; Rosales-Cruz, P.; Souza, V.; Miranda, R.U.; Toledo-Pérez, R.; Luna-López, A.; Simoni-Nieves, A.; Hernández-Pando, R.; Gutiérrez-Ruiz, M.C.; et al. Cholesterol overload in the liver aggravates oxidative stress-mediated DNA damage and accelerates hepatocarcinogenesis. Oncotarget 2017, 8, 104136–104148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. NAFLD Related-HCC: The Relationship with Metabolic Disorders. In Obesity, Fatty Liver and Liver Cancer; Yu, J., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2018; pp. 55–62. ISBN 978-981-10-8684-7. [Google Scholar]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondria, cholesterol and cancer cell metabolism. Clin. Transl. Med. 2016, 5, 22. [Google Scholar] [CrossRef]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular Pathogenesis of Nonalcoholic Steatohepatitis- (NASH-) Related Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef]
- Wang, W.; Xie, Q.; Zhou, X.; Yao, J.; Zhu, X.; Huang, P.; Zhang, L.; Wei, J.; Xie, H.; Zhou, L.; et al. Mitofusin-2 triggers mitochondria Ca2+ influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett. 2015, 358, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Mezale, D.; Strumfa, I.; Vanags, A.; Mezals, M.; Fridrihsone, I.; Strumfs, B.; Balodis, D. Non-Alcoholic Steatohepatitis, Liver Cirrhosis and Hepatocellular Carcinoma: The Molecular Pathways. Liver Cirrhosis-Update Curr. Chall. 2017. [Google Scholar] [CrossRef]
- Ryoo, H.D.; Bergmann, A. The Role of Apoptosis-Induced Proliferation for Regeneration and Cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008797. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R. Mitochondria and Cancer: A Growing Role in Apoptosis, Cancer Cell Metabolism and Dedifferentiation. In Advances in Mitochondrial Medicine; Scatena, R., Bottoni, P., Giardina, B., Eds.; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2012; pp. 287–308. ISBN 978-94-007-2869-1. [Google Scholar]
- Yu, C.; Wang, X.; Huang, L.; Tong, Y.; Chen, L.; Wu, H.; Xia, Q.; Kong, X. Deciphering the Spectrum of Mitochondrial DNA Mutations in Hepatocellular Carcinoma Using High-Throughput Sequencing. Gene Expr. J. Liver Res. 2018, 18, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-C.; Lee, H.-C.; Wei, Y.-H. Mitochondrial DNA alterations and mitochondrial dysfunction in the progression of hepatocellular carcinoma. World J. Gastroenterol. 2013, 19, 8880–8886. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.-Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef]
- Min, H.-K.; Mirshahi, F.; Verdianelli, A.; Pacana, T.; Patel, V.; Park, C.-G.; Choi, A.; Lee, J.-H.; Park, C.-B.; Ren, S.; et al. Activation of the GP130-STAT3 axis and its potential implications in nonalcoholic fatty liver disease. Am. J. Physiol.-Gastrointest. Liver Physiol. 2015, 308, G794–G803. [Google Scholar] [CrossRef]
- Jiang, N.; Sun, R.; Sun, Q. Leptin signaling molecular actions and drug target in hepatocellular carcinoma. Drug Des. Dev. Ther. 2014, 8, 2295–2302. [Google Scholar]
- Fernández-Real, J.M.; López-Bermejo, A.; Ricart, W. Cross-talk between iron metabolism and diabetes. Diabetes 2002, 51, 2348–2354. [Google Scholar] [CrossRef]
- Uysal, S.; Armutcu, F.; Aydogan, T.; Akin, K.; Ikizek, M.; Yigitoglu, M.R. Some inflammatory cytokine levels, iron metabolism and oxidan stress markers in subjects with nonalcoholic steatohepatitis. Clin. Biochem. 2011, 44, 1375–1379. [Google Scholar] [CrossRef]
- Pietrangelo, A. Iron in NASH, chronic liver diseases and HCC: How much iron is too much? J. Hepatol. 2009, 50, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.K.; Tennant, D.A.; McKeating, J.A. Hypoxia inducible factors in liver disease and hepatocellular carcinoma: Current understanding and future directions. J. Hepatol. 2014, 61, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Weljie, A.M.; Jirik, F.R. Hypoxia-induced metabolic shifts in cancer cells: Moving beyond the Warburg effect. Int. J. Biochem. Cell Biol. 2011, 43, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Foglia, B.; Sutti, S.; Morello, E.; Cannito, S.; Novo, E.; Bocca, C.; Maggiora, M.; Bruzzì, S.; Ramavath, N.N.; Rosso, C.; et al. NASH-related liver carcinogenesis is critically affected by hypoxia-inducible factor 2α. Dig. Liver Dis. 2019, 51, e8. [Google Scholar] [CrossRef]
- Ambade, A.; Satishchandran, A.; Saha, B.; Gyongyosi, B.; Lowe, P.; Kodys, K.; Catalano, D.; Szabo, G. Hepatocellular carcinoma is accelerated by NASH involving M2 macrophage polarization mediated by hif-1αinduced IL-10. Oncoimmunology 2016, 5, e1221557. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Smolková, K.; Plecitá-Hlavatá, L.; Bellance, N.; Benard, G.; Rossignol, R.; Ježek, P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int. J. Biochem. Cell Biol. 2011, 43, 950–968. [Google Scholar] [CrossRef]
- Kalhan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, A.J.; Hanson, R.W.; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism 2011, 60, 404–413. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Cabré, N.; Camps, J.; Joven, J. Inflammation, mitochondrial metabolism and nutrition: The multi-faceted progression of non-alcoholic fatty liver disease to hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2016, 5, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Dreval, K.; Kindrat, I.; Melnyk, S.; Jimenez, L.; de Conti, A.; Tryndyak, V.; Pogribna, M.; Ortega, J.F.; James, S.J.; et al. Epigenetically mediated inhibition of S-adenosylhomocysteine hydrolase and the associated dysregulation of 1-carbon metabolism in nonalcoholic steatohepatitis and hepatocellular carcinoma. FASEB J. 2018, 32, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Konno, M.; Koseki, J.; Nishida, N.; Kawamoto, K.; Yamada, D.; Asaoka, T.; Noda, T.; Wada, H.; Gotoh, K.; et al. The mitochondrial one-carbon metabolic pathway is associated with patient survival in pancreatic cancer. Oncol. Lett. 2018, 16, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Bjornson, E.; Zhang, C.; Klevstig, M.; Söderlund, S.; Ståhlman, M.; Adiels, M.; Hakkarainen, A.; Lundbom, N.; Kilicarslan, M.; et al. Personal model-assisted identification of NAD+ and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 2017, 13, 916. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef]
- Gaggini, M.; Carli, F.; Rosso, C.; Buzzigoli, E.; Marietti, M.; Latta, V.D.; Ciociaro, D.; Abate, M.L.; Gambino, R.; Cassader, M.; et al. Altered amino acid concentrations in NAFLD: Impact of obesity and insulin resistance. Hepatology 2018, 67, 145–158. [Google Scholar] [CrossRef]
- Yamakado, M.; Tanaka, T.; Nagao, K.; Imaizumi, A.; Komatsu, M.; Daimon, T.; Miyano, H.; Tani, M.; Toda, A.; Yamamoto, H.; et al. Plasma amino acid profile associated with fatty liver disease and co-occurrence of metabolic risk factors. Sci. Rep. 2017, 7, 14485. [Google Scholar] [CrossRef]
- Gao, R.; Cheng, J.; Fan, C.; Shi, X.; Cao, Y.; Sun, B.; Ding, H.; Hu, C.; Dong, F.; Yan, X. Serum Metabolomics to Identify the Liver Disease-Specific Biomarkers for the Progression of Hepatitis to Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 18175. [Google Scholar] [CrossRef]
- Stepien, M.; Duarte-Salles, T.; Fedirko, V.; Floegel, A.; Barupal, D.K.; Rinaldi, S.; Achaintre, D.; Assi, N.; Tjønneland, A.; Overvad, K.; et al. Alteration of amino acid and biogenic amine metabolism in hepatobiliary cancers: Findings from a prospective cohort study. Int. J. Cancer 2016, 138, 348–360. [Google Scholar] [CrossRef]
- Ji, L.; Tang, Y.; Pang, X.; Zhang, Y. Increased Expression of Serine Hydroxymethyltransferase 2 (SHMT2) is a Negative Prognostic Marker in Patients with Hepatocellular Carcinoma and is Associated with Proliferation of HepG2 Cells. Med. Sci. Monit. 2019, 25, 5823–5832. [Google Scholar] [CrossRef]
- Woo, C.C.; Chen, W.C.; Teo, X.Q.; Radda, G.K.; Lee, P.T.H. Downregulating serine hydroxymethyltransferase 2 (SHMT2) suppresses tumorigenesis in human hepatocellular carcinoma. Oncotarget 2016, 7, 53005–53017. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial one-carbon metabolism maintains redox balance during hypoxia. Cancer Discov. 2014, 4, 1371–1373. [Google Scholar] [CrossRef]
- Rubio-Aliaga, I.; Roos, B.D.; Sailer, M.; McLoughlin, G.A.; Boekschoten, M.; van Erk, M.J.; Bachmair, E.M.; van Schothorst, E.M.; Keijer, J.; Coort, S.L.M.; et al. Alterations in hepatic one-carbon metabolism and related pathways following a high-fat dietary intervention. Physiol. Genom. 2011, 43, 408–416. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Salomao, M.; Remotti, H.; Vaughan, R.; Siegel, A.B.; Lefkowitch, J.H.; Moreira, R.K. The steatohepatitic variant of hepatocellular carcinoma and its association with underlying steatohepatitis. Hum. Pathol. 2012, 43, 737–746. [Google Scholar] [CrossRef]
- Lin, M.; Lv, D.; Zheng, Y.; Wu, M.; Xu, C.; Zhang, Q.; Wu, L. Downregulation of CPT2 promotes tumorigenesis and chemoresistance to cisplatin in hepatocellular carcinoma. OncoTargets Ther. 2018, 11, 3101–3110. [Google Scholar] [CrossRef]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef]
- Kudo, Y.; Tanaka, Y.; Tateishi, K.; Yamamoto, K.; Yamamoto, S.; Mohri, D.; Isomura, Y.; Seto, M.; Nakagawa, H.; Asaoka, Y.; et al. Altered composition of fatty acids exacerbates hepatotumorigenesis during activation of the phosphatidylinositol 3-kinase pathway. J. Hepatol. 2011, 55, 1400–1408. [Google Scholar] [CrossRef]
- Wang, B.; Hsu, S.-H.; Frankel, W.; Ghoshal, K.; Jacob, S.T. Stat3-mediated activation of microRNA-23a suppresses gluconeogenesis in hepatocellular carcinoma by down-regulating glucose-6-phosphatase and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha. Hepatology 2012, 56, 186–197. [Google Scholar] [CrossRef]
- Martínez-Jiménez, C.P.; Gómez-Lechón, M.J.; Castell, J.V.; Jover, R. Underexpressed coactivators PGC1alpha and SRC1 impair hepatocyte nuclear factor 4 alpha function and promote dedifferentiation in human hepatoma cells. J. Biol. Chem. 2006, 281, 29840–29849. [Google Scholar] [CrossRef]
- Lee, H.-J.; Su, Y.; Yin, P.-H.; Lee, H.-C.; Chi, C.-W. PPARγ/PGC-1α Pathway in E-Cadherin Expression and Motility of HepG2 Cells. Anticancer Res. 2009, 29, 5057–5063. [Google Scholar]
- Sen, N.; Satija, Y.K.; Das, S. PGC-1α, a Key Modulator of p53, Promotes Cell Survival upon Metabolic Stress. Mol. Cell 2011, 44, 621–634. [Google Scholar] [CrossRef]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Ou, L.; Twaddel, W.; Fang, H.; Vafai, S.B.; Vazquez, F.; Puigserver, P.; Boros, L.; et al. PGC1α promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011, 71, 6888–6898. [Google Scholar] [CrossRef]
- Piccinin, E.; Peres, C.; Bellafante, E.; Ducheix, S.; Pinto, C.; Villani, G.; Moschetta, A. Hepatic peroxisome proliferator-activated receptor γ coactivator 1β drives mitochondrial and anabolic signatures that contribute to hepatocellular carcinoma progression in mice. Hepatology 2018, 67, 884–898. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef]
- Chambers, K.T.; Chen, Z.; Lai, L.; Leone, T.C.; Towle, H.C.; Kralli, A.; Crawford, P.A.; Finck, B.N. PGC-1β and ChREBP partner to cooperatively regulate hepatic lipogenesis in a glucose concentration-dependent manner. Mol. Metab. 2013, 2, 194–204. [Google Scholar] [CrossRef]
- Zhang, X.; Han, K.; Yuan, D.-H.; Meng, C.-Y. Overexpression of NAD(P)H: Quinone Oxidoreductase 1 Inhibits Hepatocellular Carcinoma Cell Proliferation and Induced Apoptosis by Activating AMPK/PGC-1α Pathway. DNA Cell Biol. 2017, 36, 256–263. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Li, J.; Zheng, L.; Feng, M.; Wang, X.; Han, K.; Pi, H.; Li, M.; Huang, X.; et al. SIRT1 facilitates hepatocellular carcinoma metastasis by promoting PGC-1α-mediated mitochondrial biogenesis. Oncotarget 2016, 7, 29255–29274. [Google Scholar]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a New Target of PGC-1α, Plays an Important Role in the Suppression of ROS and Mitochondrial Biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.-L.; Cheng, W.; Yin, X.-M.; Jiang, B. The expression of SIRT3 in primary hepatocellular carcinoma and the mechanism of its tumor suppressing effects. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 978–998. [Google Scholar]
- Zeng, X.; Wang, N.; Zhai, H.; Wang, R.; Wu, J.; Pu, W. SIRT3 functions as a tumor suppressor in hepatocellular carcinoma. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Liu, L.; Cai, M.; Pan, Y.; Fu, J.; Cao, Y.; Yun, J. Low SIRT3 Expression Correlates with Poor Differentiation and Unfavorable Prognosis in Primary Hepatocellular Carcinoma. PLoS ONE 2012, 7, e51703. [Google Scholar] [CrossRef]
- Daher, S.; Massarwa, M.; Benson, A.A.; Khoury, T. Current and Future Treatment of Hepatocellular Carcinoma: An Updated Comprehensive Review. J. Clin. Transl. Hepatol. 2018, 6, 69–78. [Google Scholar] [CrossRef]
- Hannah, W.N.; Harrison, S.A. Lifestyle and Dietary Interventions in the Management of Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1365–1374. [Google Scholar] [CrossRef]
- Dethlefsen, M.M.; Kristensen, C.M.; Tøndering, A.S.; Lassen, S.B.; Ringholm, S.; Pilegaard, H. Impact of liver PGC-1α on exercise and exercise training-induced regulation of hepatic autophagy and mitophagy in mice on HFF. Physiol. Rep. 2018, 6, e13731. [Google Scholar] [CrossRef]
- Santos-Alves, E.; Marques-Aleixo, I.; Rizo-Roca, D.; Torrella, J.R.; Oliveira, P.J.; Magalhães, J.; Ascensão, A. Exercise modulates liver cellular and mitochondrial proteins related to quality control signaling. Life Sci. 2015, 135, 124–130. [Google Scholar] [CrossRef]
- Yun, Y.H.; Lim, M.K.; Won, Y.-J.; Park, S.M.; Chang, Y.J.; Oh, S.W.; Shin, S.A. Dietary preference, physical activity, and cancer risk in men: National health insurance corporation study. BMC Cancer 2008, 8, 366. [Google Scholar] [CrossRef]
- Tsai, J.-H.; Ferrell, L.D.; Tan, V.; Yeh, M.M.; Sarkar, M.; Gill, R.M. Aggressive non-alcoholic steatohepatitis following rapid weight loss and/or malnutrition. Mod. Pathol. 2017, 30, 834–842. [Google Scholar] [CrossRef]
- Besse-Patin, A.; Estall, J.L. An Intimate Relationship between ROS and Insulin Signalling: Implications for Antioxidant Treatment of Fatty Liver Disease. Int. J. Cell Biol. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Baumgardner, J.N.; Shankar, K.; Hennings, L.; Albano, E.; Badger, T.M.; Ronis, M.J.J. N-acetylcysteine attenuates progression of liver pathology in a rat model of nonalcoholic steatohepatitis. J. Nutr. 2008, 138, 1872–1879. [Google Scholar] [CrossRef]
- De Oliveira, C.P.M.S.; de Lima, V.M.R.; Simplicio, F.I.; Soriano, F.G.; de Mello, E.S.; de Souza, H.P.; Alves, V.A.F.; Laurindo, F.R.M.; Carrilho, F.J.; de Oliveira, M.G. Prevention and reversion of nonalcoholic steatohepatitis in OB/OB mice by S-nitroso-N-acetylcysteine treatment. J. Am. Coll. Nutr. 2008, 27, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Kretzmann, N.A.; Chiela, E.; Matte, U.; Marroni, N.; Marroni, C.A. N-acetylcysteine improves antitumoural response of Interferon alpha by NF-kB downregulation in liver cancer cells. Comp. Hepatol. 2012, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liu, X.; Yu, J.; Hua, F.; Hu, Z. Antioxidant N-acetylcysteine attenuates hepatocarcinogenesis by inhibiting ROS/ER stress in TLR2 deficient mouse. PLoS ONE 2013, 8, e74130. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Atkinson, J. Vitamin E, Antioxidant and Nothing More. Free Radic. Biol. Med. 2007, 43, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Draeger, C.L.; Naves, A.; Marques, N.; Baptistella, A.B.; Carnauba, R.A.; Paschoal, V.; Nicastro, H. Controversies of antioxidant vitamins supplementation in exercise: Ergogenic or ergolytic effects in humans? J. Int. Soc. Sports Nutr. 2014, 11, 4. [Google Scholar] [CrossRef]
- Erhardt, A.; Stahl, W.; Sies, H.; Lirussi, F.; Donner, A.; Häussinger, D. Plasma levels of vitamin E and carotenoids are decreased in patients with nonalcoholic steatohepatitis (NASH). Eur. J. Med. Res. 2011, 16, 76–78. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The TONIC randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Hironaka, K.; Factor, V.M.; Thorgeirsson, S.S. Vitamin E down-modulates iNOS and NADPH oxidase in c-Myc/TGF-alpha transgenic mouse model of liver cancer. J. Hepatol. 2004, 41, 815–822. [Google Scholar] [CrossRef]
- Fantappiè, O.; Lodovici, M.; Fabrizio, P.; Marchetti, S.; Fabbroni, V.; Solazzo, M.; Lasagna, N.; Pantaleo, P.; Mazzanti, R. Vitamin E Protects DNA from Oxidative Damage in Human Hepatocellular Carcinoma Cell Lines. Free Radic. Res. 2004, 38, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shu, X.-O.; Li, H.; Yang, G.; Cai, H.; Ji, B.-T.; Gao, J.; Gao, Y.-T.; Zheng, W.; Xiang, Y.-B. Vitamin intake and liver cancer risk: A report from two cohort studies in China. J. Natl. Cancer Inst. 2012, 104, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Tajima, K.; Nakamura, A.; Shirakawa, J.; Togashi, Y.; Orime, K.; Sato, K.; Inoue, H.; Kaji, M.; Sakamoto, E.; Ito, Y.; et al. Metformin prevents liver tumorigenesis induced by high-fat diet in C57Bl/6 mice. Am. J. Physiol.-Endocrinol. Metab. 2013, 305, E987–E998. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, S.; Houseright, R.A.; Graves, A.L.; Golenberg, N.; Korte, B.G.; Miskolci, V.; Huttenlocher, A. Metformin modulates innate immune-mediated inflammation and early progression of NAFLD-associated hepatocellular carcinoma in zebrafish. J. Hepatol. 2019, 70, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases: Hepatology, Vol. XX, No. X, 2017. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients with Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef]
- Aithal, G.P.; Thomas, J.A.; Kaye, P.V.; Lawson, A.; Ryder, S.D.; Spendlove, I.; Austin, A.S.; Freeman, J.G.; Morgan, L.; Webber, J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology 2008, 135, 1176–1184. [Google Scholar] [CrossRef]
- Kalavalapalli, S.; Bril, F.; Koelmel, J.P.; Abdo, K.; Guingab, J.; Andrews, P.; Li, W.-Y.; Jose, D.; Yost, R.A.; Frye, R.F.; et al. Pioglitazone improves hepatic mitochondrial function in a mouse model of nonalcoholic steatohepatitis. Am. J. Physiol.-Endocrinol. Metab. 2018, 315, E163–E173. [Google Scholar] [CrossRef]
- Li, S.; Ghoshal, S.; Sojoodi, M.; Arora, G.; Masia, R.; Erstad, D.J.; Lanuti, M.; Hoshida, Y.; Baumert, T.F.; Tanabe, K.K.; et al. Pioglitazone Reduces Hepatocellular Carcinoma Development in Two Rodent Models of Cirrhosis. J. Gastrointest. Surg. 2019, 23, 101–111. [Google Scholar] [CrossRef]
- Sumie, S.; Kawaguchi, T.; Kawaguchi, A.; Kuromatsu, R.; Nakano, M.; Satani, M.; Yamada, S.; Okamura, S.; Yonezawa, Y.; Kakuma, T.; et al. Effect of pioglitazone on outcome following curative treatment for hepatocellular carcinoma in patients with hepatitis C virus infection: A prospective study. Mol. Clin. Oncol. 2015, 3, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Lin, J.-W.; Wu, L.-C.; Lai, M.-S.; Chuang, L.-M.; Chan, K.A. Association of thiazolidinediones with liver cancer and colorectal cancer in type 2 diabetes mellitus. Hepatology 2012, 55, 1462–1472. [Google Scholar] [CrossRef] [PubMed]
- Desquiret-Dumas, V.; Gueguen, N.; Leman, G.; Baron, S.; Nivet-Antoine, V.; Chupin, S.; Chevrollier, A.; Vessières, E.; Ayer, A.; Ferré, M.; et al. Resveratrol Induces a Mitochondrial Complex I-dependent Increase in NADH Oxidation Responsible for Sirtuin Activation in Liver Cells. J. Biol. Chem. 2013, 288, 36662–36675. [Google Scholar] [CrossRef] [PubMed]
- Gusdon, A.M.; Song, K.-X.; Qu, S. Nonalcoholic Fatty liver disease: Pathogenesis and therapeutics from a mitochondria-centric perspective. Oxid. Med. Cell. Longev. 2014, 2014, 637027. [Google Scholar] [CrossRef] [PubMed]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef]
- De Oliveira, M.R.; Nabavi, S.F.; Manayi, A.; Daglia, M.; Hajheydari, Z.; Nabavi, S.M. Resveratrol and the mitochondria: From triggering the intrinsic apoptotic pathway to inducing mitochondrial biogenesis, a mechanistic view. Biochim. Biophys. Acta BBA-Gen. Subj. 2016, 1860, 727–745. [Google Scholar] [CrossRef]
- Singh, C.K.; Ndiaye, M.A.; Ahmad, N. Resveratrol and cancer: Challenges for clinical translation. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2015, 1852, 1178–1185. [Google Scholar] [CrossRef]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis. Oncol. 2017, 1, 35. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The in Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Okabe, K.; Yaku, K.; Tobe, K.; Nakagawa, T. Implications of altered NAD metabolism in metabolic disorders. J. Biomed. Sci. 2019, 26, 34. [Google Scholar] [CrossRef]
- Barbosa, M.T.P.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Du, M.; Tan, X.; Yang, L.; Li, X.; Jiang, Y.; Wang, C.; Zhang, F.; Zhu, F.; Cheng, M.; et al. PARP1-mediated PPARα poly(ADP-ribosyl) action suppresses fatty acid oxidation in non-alcoholic fatty liver disease. J. Hepatol. 2017, 66, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Gariani, K.; Ryu, D.; Menzies, K.J.; Yi, H.-S.; Stein, S.; Zhang, H.; Perino, A.; Lemos, V.; Katsyuba, E.; Jha, P.; et al. Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease. J. Hepatol. 2017, 66, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Horváth, B.; Rajesh, M.; Varga, Z.V.; Gariani, K.; Ryu, D.; Cao, Z.; Holovac, E.; Park, O.; Zhou, Z.; et al. PARP inhibition protects against alcoholic and non-alcoholic steatohepatitis. J. Hepatol. 2017, 66, 589–600. [Google Scholar] [CrossRef]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef]
- Pham, T.X.; Bae, M.; Kim, M.-B.; Lee, Y.; Hu, S.; Kang, H.; Park, Y.-K.; Lee, J.-Y. Nicotinamide riboside, an NAD+ precursor, attenuates the development of liver fibrosis in a diet-induced mouse model of liver fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2451–2463. [Google Scholar] [CrossRef]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD (+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.J.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef]
- Airhart, S.E.; Shireman, L.M.; Risler, L.J.; Anderson, G.D.; Nagana Gowda, G.A.; Raftery, D.; Tian, R.; Shen, D.D.; O’Brien, K.D. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE 2017, 12, e0186459. [Google Scholar] [CrossRef]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Djouder, N. Boosting NAD+ for the prevention and treatment of liver cancer. Mol. Cell. Oncol. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Tummala, K.S.; Gomes, A.L.; Yilmaz, M.; Graña, O.; Bakiri, L.; Ruppen, I.; Ximénez-Embún, P.; Sheshappanavar, V.; Rodriguez-Justo, M.; Pisano, D.G.; et al. Inhibition of De Novo NAD + Synthesis by Oncogenic URI Causes Liver Tumorigenesis through DNA Damage. Cancer Cell 2014, 26, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.S.; Haydar, G.; Taniane, C.; Farrell, G.; Arias, I.M.; Lippincott-Schwartz, J.; Fu, D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLoS ONE 2016, 11, e0165638. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xiong, R.; Xiong, D.; Zhao, W.; Zhang, S.; Yin, T.; Zhang, X.; Jiang, G.; Yin, Z. The Adenosine Monophosphate (AMP) Analog, 5-Aminoimidazole-4-Carboxamide Ribonucleotide (AICAR) Inhibits Hepatosteatosis and Liver Tumorigenesis in a High-Fat Diet Murine Model Treated with Diethylnitrosamine (DEN). Med. Sci. Monit. 2018, 24, 8533–8543. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Huang, T.; Li, Y.; Guo, Y.; Zhu, Y.; Wang, Q.; Tan, X.; Chen, W.; Zhang, Y.; Cheng, W.; et al. AMP-Activated Protein Kinase Suppresses the in Vitro and in Vivo Proliferation of Hepatocellular Carcinoma. PLoS ONE 2014, 9, e93256. [Google Scholar] [CrossRef]
- Yang, C.-C.; Chang, S.-F.; Chao, J.-K.; Lai, Y.-L.; Chang, W.-E.; Hsu, W.-H.; Kuo, W.-H. Activation of AMP-activated protein kinase attenuates hepatocellular carcinoma cell adhesion stimulated by adipokine resistin. BMC Cancer 2014, 14, 112. [Google Scholar] [CrossRef]
- Pavlov, T.S.; Levchenko, V.; Ilatovskaya, D.V.; Li, H.; Palygin, O.; Pastor-Soler, N.M.; Hallows, K.R.; Staruschenko, A. Lack of Effects of Metformin and AICAR Chronic Infusion on the Development of Hypertension in Dahl Salt-Sensitive Rats. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Morishita, M.; Kawamoto, T.; Hara, H.; Onishi, Y.; Ueha, T.; Minoda, M.; Katayama, E.; Takemori, T.; Fukase, N.; Kurosaka, M.; et al. AICAR induces mitochondrial apoptosis in human osteosarcoma cells through an AMPK-dependent pathway. Int. J. Oncol. 2016, 50, 23–30. [Google Scholar] [CrossRef]
- Jose, C.; Hébert-Chatelain, E.; Bellance, N.; Larendra, A.; Su, M.; Nouette-Gaulain, K.; Rossignol, R. AICAR inhibits cancer cell growth and triggers cell-type distinct effects on OXPHOS biogenesis, oxidative stress and Akt activation. Biochim. Biophys. Acta 2011, 1807, 707–718. [Google Scholar] [CrossRef]
- Suwa, M.; Egashira, T.; Nakano, H.; Sasaki, H.; Kumagai, S. Metformin increases the PGC-1alpha protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J. Appl. Physiol. 2006, 101, 1685–1692. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Khang, R.; Ham, S.; Jeong, G.R.; Kim, H.; Jo, M.; Lee, B.D.; Lee, Y.I.; Jo, A.; Park, C.; et al. Activation of the ATF2/CREB-PGC-1α pathway by metformin leads to dopaminergic neuroprotection. Oncotarget 2017, 8, 48603–48618. [Google Scholar] [PubMed]
- Higashida, K.; Kim, S.H.; Jung, S.R.; Asaka, M.; Holloszy, J.O.; Han, D.-H. Effects of Resveratrol and SIRT1 on PGC-1α Activity and Mitochondrial Biogenesis: A Reevaluation. PLoS Biol. 2013, 11, e1001603. [Google Scholar] [CrossRef] [PubMed]
- Aatsinki, S.-M.; Buler, M.; Salomäki, H.; Koulu, M.; Pavek, P.; Hakkola, J. Metformin induces PGC-1α expression and selectively affects hepatic PGC-1α functions. Br. J. Pharmacol. 2014, 171, 2351–2363. [Google Scholar] [CrossRef]
- Uchida, D.; Takaki, A.; Adachi, T.; Okada, H. Beneficial and Paradoxical Roles of Anti-Oxidative Nutritional Support for Non-Alcoholic Fatty Liver Disease. Nutrients 2018, 10, 977. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Léveillé, M.; Estall, J.L. Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites 2019, 9, 233. https://doi.org/10.3390/metabo9100233
Léveillé M, Estall JL. Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites. 2019; 9(10):233. https://doi.org/10.3390/metabo9100233
Chicago/Turabian StyleLéveillé, Mélissa, and Jennifer L. Estall. 2019. "Mitochondrial Dysfunction in the Transition from NASH to HCC" Metabolites 9, no. 10: 233. https://doi.org/10.3390/metabo9100233
APA StyleLéveillé, M., & Estall, J. L. (2019). Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites, 9(10), 233. https://doi.org/10.3390/metabo9100233