Potent Antifungal Properties of Dimeric Acylphloroglucinols from Hypericum mexicanum and Mechanism of Action of a Highly Active 3′Prenyl Uliginosin B

 ,
,  , and
, and
Abstract
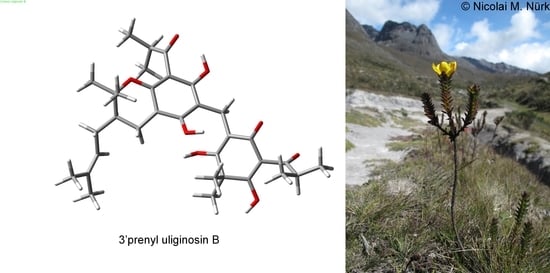
1. Introduction
2. Results and Discussion
2.1. Chemical Profile of Hypericum Mexicanum
2.2. Antifungal Activity of H. Mexicanum Extracts and Chemical Analysis of the Chloroformic Extract
2.3. Bioassay-Guided Isolation and Characterization of Antifungal Principles
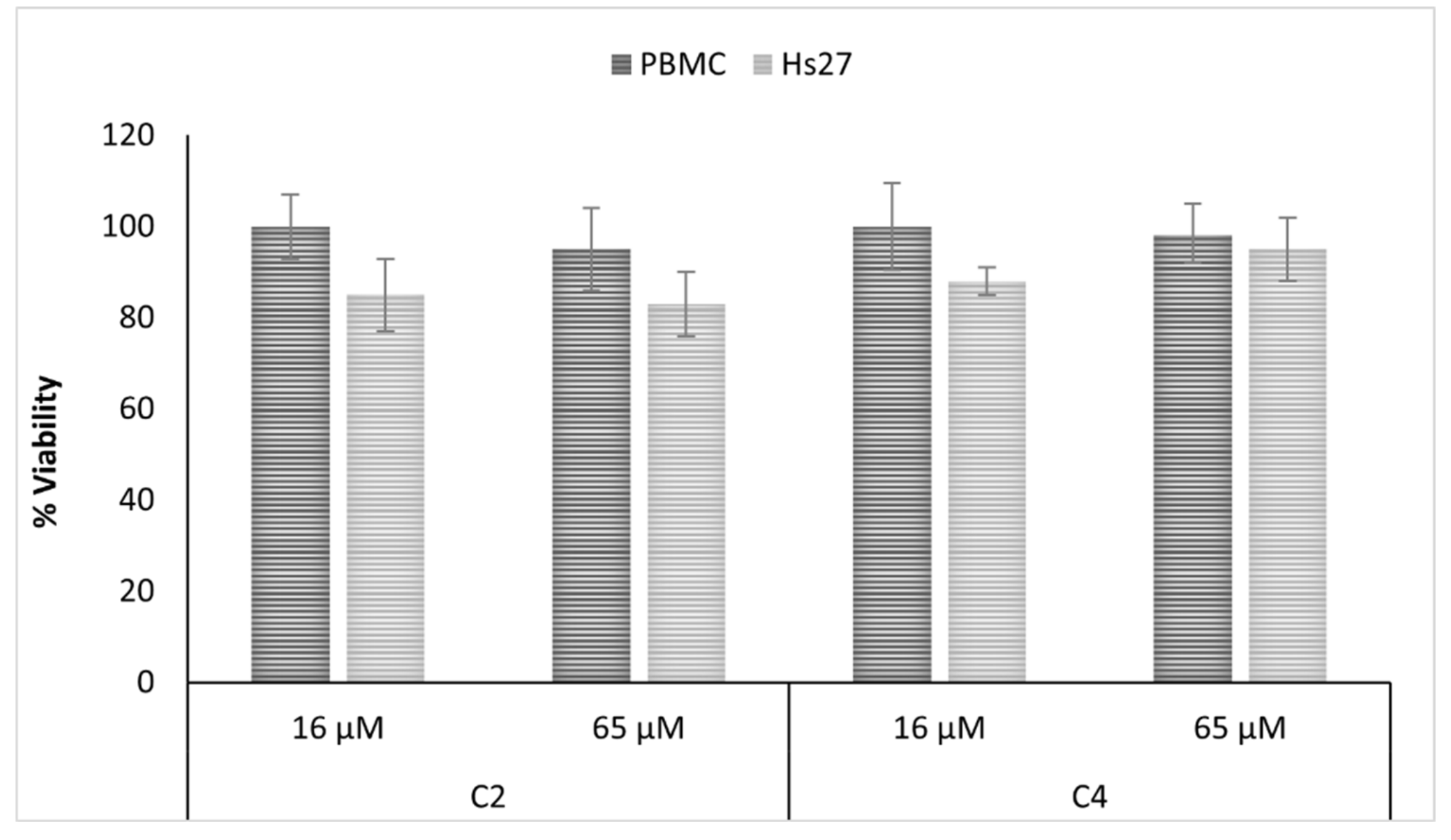
2.4. Antifungal and Cytotoxic Properties of Isolated Compunds
2.5. Mechanism of Action Analysis
3. Materials and Methods
3.1. Plant Material
3.2. Preparation of Plant Extracts
3.3. Chemicals
3.4. LC-MS Analysis
3.5. Compound Isolation and Mass Spectrometric Conditions
- Fraction I: Isocratic elution, 70% B and 30% A, flux: 0.9 mL/min;
- Fraction II: Gradient elution, starting with 75% B and reaching 85% B in 20 min.
3.6. NMR Analysis
3.7. Microorganisms and Media
3.8. Antifungal Susceptibility Testings
3.9. Cytotoxicity Assay
3.10. Chemogenomics
3.11. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Ethical Statements
References
- Brown, G.D.; Denning, D.W.; Levitz, S.M. Tackling Human Fungal Infections. Science 2012, 336, 647. [Google Scholar]
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef]
- Ksiezopolska, E.; Gabaldón, T. Evolutionary emergence of drug resistance in candida opportunistic pathogens. Genes 2018, 9, 461. [Google Scholar]
- Rizzetto, L.; Weil, T.; Cavalieri, D. Systems Level Dissection of Candida Recognition by Dectins: A Matter of Fungal Morphology and Site of Infection. Pathogens 2015, 4, 639–661. [Google Scholar] [CrossRef] [PubMed]
- Odds, F.C.; Brown, A.J.P.; Gow, N.A.R. Antifungal agents: Mechanisms of action. Trends Microbiol. 2003, 11, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Arendrup, M.C. Update on antifungal resistance in Aspergillus and Candida. Clin. Microbiol. Infect. 2014, 20, 42–48. [Google Scholar]
- Roemer, T.; Krysan, D.J. Antifungal Drug Development: Challenges, Unmet Clinical Needs, and New Approaches. Cold Spring Harb. Perspect. Med. 2014, 4, a019703. [Google Scholar]
- Tocci, N.; Weil, T.; Perenzoni, D.; Narduzzi, L.; Madriñán, S.; Crockett, S.; Nürk, N.M.; Cavalieri, D.; Mattivi, F. Phenolic profile, chemical relationship and antifungal activity of Andean Hypericum species. Ind. Crops Prod. 2018, 112, 32–37. [Google Scholar] [CrossRef]
- Tocci, N.; Perenzoni, D.; Iamonico, D.; Fava, F.; Weil, T.; Mattivi, F. Extracts From Hypericum hircinum subsp. majus Exert Antifungal Activity Against a Panel of Sensitive and Drug-Resistant Clinical Strains. Front. Pharmacol. 2018, 9, 382. [Google Scholar] [CrossRef]
- Fenner, R.; Sortino, M.; Kuze Rates, S.M.; Dall’Agnol, R.; Ferraz, A.; Bernardi, A.P.; Albring, D.; Nör, C.; Von Poser, G.; Schapoval, E.; et al. Antifungal activity of some Brazilian Hypericum species. Phytomedicine 2005, 12, 236–240. [Google Scholar] [CrossRef]
- Meirelles, G.C.; Pippi, B.; Hatwig, C.; de Barros, F.M.C.; de Oliveira, L.F.S.; von Poser, G.L.; Fuentefria, A.M. Synergistic antifungal activity of the lipophilic fraction of Hypericum carinatum and fluconazole. Rev. Bras. Farmacogn. 2017, 27, 118–123. [Google Scholar] [CrossRef]
- Nürk, N.M.; Michling, F.; Linder, H.P. Are the radiations of temperate lineages in tropical alpine ecosystems pre-adapted? Glob. Ecol. Biogeogr. 2018, 27, 334–345. [Google Scholar] [CrossRef]
- Ccana-Ccapatinta, G.V.; de Barros, F.M.C.; Bridi, H.; von Poser, G.L. Dimeric acylphloroglucinols in Hypericum species from sections Brathys and Trigynobrathys. Phytochem. Rev. 2015, 14, 25–50. [Google Scholar] [CrossRef]
- Schmid, R.; Luteyn, J.L. Páramos: A Checklist of Plant Diversity, Geographical Distribution, and Botanical Literature. Taxon 1999, 48, 616. [Google Scholar] [CrossRef]
- Nürk, N.M.; Scheriau, C.; Madriñán, S. Explosive radiation in high Andean Hypericum—rates of diversification among New World lineages. Front. Genet. 2013, 4. [Google Scholar] [CrossRef]
- Pineda Forero, M. Estudio etnobotánico de seis (6) especies vegetales andinas en un área rural del Distrito Capital y realizar enriquecimiento y mantenimiento de la chagra experimental altoandina. In 3311303340318-Uso Sostenible de los Recursos Vegetales del Distrito Capital y la Región; Jardín Botánico José Celestino Mutis: Bogotá, Colombia, 2011. [Google Scholar]
- Corzo-Barragán, D.C.; Gaitán-Vaca, D.M. de Hypericum mexicanum L. E valuación de la efectividad de distintas Evaluation of the effectiveness of different formulations of soap with extract of Hypericum. Rev. Investig. Agrar. Ambient. 2017, 8, 131–138. [Google Scholar]
- Patiño-Bayona, W.R.; Plazas, E.; Bustos-cortes, J.J.; Prieto-Rodríguez, J.A.; Patiño-Ladino, O.J. Essential Oils of Three Hypericum Species from Colombia: Chemical Composition, Insecticidal and Repellent Activity Against Sitophilus zeamais Motsch. (Coleoptera: Curculionidae), Erika Plazas and Oscar J. Patiño-Ladino. Rec. Nat. Prod. 2020, 1–11. [Google Scholar] [CrossRef]
- Barros, F.M.C.; Ccana-Ccapatinta, G.V.; Meirelles, G.C.; Nunes, J.M.; Cargnin, S.T.; Sakamoto, S.; Bordignon, S.; del Carpio, C.; Crockett, S.L.; von Poser, G.L. Determination of phenolic compounds in flowers of Hypericum species native to South Brazil and Peruvian Páramos. Plant Syst. Evol. 2013, 299, 1865–1872. [Google Scholar] [CrossRef]
- Gonzalez, E.A. Tamizaje fitoquímico y actividad antibacteriana in vitro de extractos y fracciones de tres especies colombianas del género Hypericum. Rev. Cuba. Plantas Med. 2017, 22, 1–14. [Google Scholar]
- Bridi, H.; Meirelles, G.D.C.; Bordignon, S.A.D.L.; Rates, S.M.K.; Von Poser, G.L. Denudatin A, a Dimeric Acylphloroglucinol from Hypericum denudatum Presents an Antinociceptive Effect in Mice. Planta Med. 2017, 83, 1329–1334. [Google Scholar] [CrossRef]
- Iida, T.; Ichino, K.; Ito, K. Neolignans from Magnolia denudata. Phytochemistry 1980, 21, 2939–2941. [Google Scholar] [CrossRef]
- Chemical Abstracts Service: Columbus, OH. Available online: https://scifinder.cas.org (accessed on 12 November 2020).
- Giaever, G.; Flaherty, P.; Kumm, J.; Proctor, M.; Nislow, C.; Jaramillo, D.F.; Chu, A.M.; Jordan, M.I.; Arkin, A.P.; Davis, R.W. Chemogenomic profiling: Identifying the functional interactions of small molecules in yeast. Proc. Natl. Acad. Sci. USA 2004. [Google Scholar] [CrossRef] [PubMed]
- Dekker, C.; Roe, S.M.; Mccormack, A.; Beuron, F.; Pearl, L.H.; Willison, K.R. The crystal structure of yeast CCT reveals intrinsic asymmetry of eukaryotic cytosolic chaperonins. EMBO J. 2011, 30, 3078–3090. [Google Scholar] [CrossRef] [PubMed]
- Dekker, C.; Stirling, P.C.; Mccormack, E.A.; Paul, A.; Brost, R.L.; Costanzo, M.; Boone, C.; Leroux, M.R.; Willison, K.R. The interaction network of the chaperonin CCT. EMBO J. 2008, 27, 1827–1839. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Fleites, L.A.; Gabriel, D.W. A Small Wolbachia Protein Directly. mSphere 2017, 2, 1–12. [Google Scholar] [CrossRef]
- Willison, K.R. The substrate specificity of eukaryotic cytosolic chaperonin CCT. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170192. [Google Scholar] [CrossRef]
- Brackley, K.I.; Grantham, J. Activities of the chaperonin containing TCP-1 (CCT): Implications for cell cycle progression and cytoskeletal organisation. Cell Stress Chaperones 2009, 14, 23–31. [Google Scholar] [CrossRef]
- Rademacher, F.; Kehren, V.; Stoldt, V.R.; Ernst, J.F. A Candida albicans chaperonin subunit (CaCct8p) as a suppressor of morphogenesis and Ras phenotypes in C. albicans and Saccharomyces cerevisiae. Microbiology 1998, 144, 2951–2960. [Google Scholar] [CrossRef]
- Saville, S.P.; Lazzell, A.L.; Bryant, A.P.; Fretzen, A.; Monreal, A.; Solberg, E.O.; Monteagudo, C.; Lopez-Ribot, J.L.; Milne, G.T. Inhibition of Filamentation Can Be Used To Treat Disseminated Candidiasis. Antimicrob. Agents Chemother. 2006, 50, 3312–3316. [Google Scholar] [CrossRef]
- Robson, N.K.B. Studies in the genus Hypericum L. (Guttiferae) 8. Sections 29. Brathys (part 2) and 30. In Trigynobrathys*; Bulletin of the British Museum (Natural History); Botany Series Natural History Museum: London, UK, 1990; pp. 1–151. [Google Scholar]
- Vrhovsek, U.; Masuero, D.; Gasperotti, M.; Franceschi, P.; Caputi, L.; Viola, R.; Mattivi, F. A versatile targeted metabolomics method for the rapid quantification of multiple classes of phenolics in fruits and beverages. J. Agric. Food Chem. 2012, 60, 8831–8840. [Google Scholar] [CrossRef]
- Mattivi, F.; Reniero, F.; Korhammer, S. Isolation, Characterization, and Evolution in Red Wine Vinification of Resveratrol Monomers. J. Agric. Food Chem. 1995. [Google Scholar] [CrossRef]
- Arendrup, M.C.; Cuenca-Estrella, M.; Lass-Flörl, C.; Hope, W.; EUCAST-AFST. EUCAST technical note on the EUCAST definitive document EDef 7.2: Method for the determination of broth dilution minimum inhibitory concentrations of antifungal agents for yeasts EDef 7.2 (EUCAST-AFST). Clin. Microbiol. Infect. 2012, 18, E246–E247. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.G.; Chen, W.; Storey, J.D.; Gresham, D. Design and Analysis of Bar-seq Experiments. G3 Genes Genomes Genetics 2014, 4, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K.; Ritchie, M.; Thorne, N.; Wettenhall, J.; Shi, W.; Hu, Y. limma: Linear Models for Microarray Data User’s Guide. Software Manual. Available online: http://www.bioconductor.org (accessed on 2 October 2017).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Stem | Leaves Methanol Extracts * | Roots | Leaves Chloroform Extract * |
|---|---|---|---|---|
| Caffeic acid | nd | 0.16 ± 0.10 | nd | nd |
| 3,5-dihydroxy benzoic acid | 0.11 ± 0.05 | 2.02 ± 0.30 | 0.11 ± 0.05 | 0.13 ± 0.03 |
| Gallic acid | nd | nd | nd | nd |
| Kaempferol | nd | 1.78 ± 0.09 | 0.01 ± 0.01 | 0.40 ± 0.07 |
| Quercetin-3-glucuronide | nd | 0.09 ± 0.01 | nd | 0.21 ± 0.04 |
| Quercetin | nd | 97.07 ± 1.01 | 0.06 ± 0.01 | 2.87 ± 0.60 |
| Quercetin-3-glucoside | 0.05 ± 0.01 | 86.46 ± 3.03 | 0.04 ± 0.01 | nd |
| Kaempferol-3-glucuronide | nd | nd | nd | 0.01 ± 0.01 |
| Isorhamnetin | 0.02 ± 0.01 | 6.73 ± 0.02 | nd | 0.03 ± 0.01 |
| Syringetin-3-glucoside | nd | nd | nd | nd |
| Isorhamnetin-3-glucoside | 0.12 ± 0.01 | 96.58 ± 5.02 | nd | 0.13 ± 0.03 |
| Myricetin | nd | 1.80 ± 0.10 | nd | nd |
| Catechin | nd | nd | nd | nd |
| Epicatechin | 0.09 ± 0.03 | 36.17 ± 2.01 | nd | nd |
| Gallocatechin | nd | 0.12 ± 0.05 | nd | nd |
| Epigallocatechin | nd | nd | nd | nd |
| Procyanidin B1 | nd | 2.64 ± 0.30 | nd | 0.09 ± 0.03 |
| Procyanidin B2 | 0.24 ± 0.03 | 117.37 ± 4.02 | 2.72 ± 0.31 | 0.19 ± 0.08 |
| cis-piceid | nd | 0.11 ± 0.06 | nd | nd |
| Vanillic acid | nd | 0.47 ± 0.12 | 0.01 ± 0.01 | 0.08 ± 0.01 |
| Esculin | nd | 0.04 ± 0.01 | nd | 0.01 ± 0.01 |
| Neochlorogenic acid | nd | 6.98 ± 0.50 | 0.01 ± 0.01 | nd |
| Cryptochlorogenic acid | nd | 0.36 ± 0.02 | nd | nd |
| Chlorogenic acid | nd | nd | nd | nd |
| Luteolin | nd | 2.81 ± 0.81 | nd | 0.44 ± 0.09 |
| Luteolin-7- glucoside | nd | 0.42 ± 0.05 | nd | traces |
| Quercetin-3-arabinoside | nd | 2.39 ± 0.81 | nd | nd |
| 2,6-dihydroxy benzoic acid | nd | nd | nd | nd |
| p-hydroxybenzoic acid | nd | 1.31 ± 0.31 | 0.02 ± 0.01 | 0.04 ± 0.01 |
| Cinnamic acid | nd | 0.08 ± 0.01 | nd | 0.13 ± 0.01 |
| Naringenin-7-glucoside | nd | 0.12 ± 0.07 | nd | 1.74 ± 0.30 |
| Coniferyl alcohol | nd | nd | nd | nd |
| Kaempferol-3-glucoside | 0.01 ± 0.01 | 5.40 ± 1.01 | 0.01 ± 0.01 | nd |
| Phlorizin | 0.02 ± 0.01 | 4.51 ± 0.51 | 0.03 ± 0.01 | nd |
| Strain (N° Tested) | Chloroformic | Methanol |
|---|---|---|
| MIC50 µg/mL * | ||
| C. albicans (4) | 27 | 64 |
| C. parapsilosis (2) | 16 | 125 |
| C. glabrata (2) | 11 | 45 |
| C. lusitaniae (2) | 10 | 32 |
| C. tropicalis (1) | 23 | >125 |
| C. pararugosa (1) | 32 | >125 |
| C. deformans (1) | 32 | >125 |
| Strain | Fraction I | Fraction II | Fluconazole |
|---|---|---|---|
| MIC50 (µg/mL) | |||
| C. albicans MFB 051-1 | >50 | <32 | 64.0 ± 1.0 |
| C. albicans MFB032-1 | >50 | 32 ± 1.0 | 0.5 ± 0.1 |
| C. albicans MFB076-1 | >50 | 32 ± 0.5 | >64 |
| C. albicans MFB008 MM1 | >50 | <4 | >64 |
| C. albicans YMS 102-2 | >50 | <<4 | >64 |
| C. albicans YMS100-3 | >50 | 16 ± 0.5 | 0.5 ± 0.1 |
| C. lusitaniae MFB037 N1 | >50 | 8 ± 1.0 | 0.5 ± 0.2 |
| C. lusitaniae YMS 100-16 | >50 | <4 | 0.3 ± 0.1 |
| C. parapsilosis MFB014 CD7 | >50 | 4 ± 0.5 | >64 |
| C. parapsilosis MFB070 N1 | >50 | 125 ± 1 | >64 |
| C. parapsilosis YMS 100-1 | >50 | 4 ± 0.5 | 2 ± 0.5 |
| C. pararugosa MFB037 N3 | >50 | 8 ± 0.5 | 0.1 |
| Signal | 1 H-NMR Compound 2 | 1 H-NMR Compound 5 | 13 C-NMR Compound 2 | 13 C-NMR Compound 5 |
|---|---|---|---|---|
| 1 | 199.4 s | 199.4 s | ||
| 2 | 107.1 s | 107.1 s | ||
| 3 | 171.6 s | 171.6 s | ||
| 4 | 44.3 s | 44.3 s | ||
| 5 | 187.3 s | 187.3 s | ||
| 6 | 111.2 s | 111.4 s | ||
| 7 | 3.55 (2H; br d; 16.9) | 3.54 (2H; br d; 16.9) | 16.9 br t | 17.0 t |
| 8 | 1.45 (3H; s) | 1.45 (3H; s) | 24.2 brq | 19.1 brq |
| 9 | 1.53 (3H; s) | 1.53 (3H; s) | 25.4 brq | 19.5 brq |
| 10 | 4.21 (1H; septet; 6.8) | 4.21 (1H; septet; 6.8) | 210.9 s | 210.8 s |
| 11 | 36.6 d | 36.6 d | ||
| 12 | 1.18 (3H, s) | 1.18 (3H, s) | 19.3 brq | 19.3 brq |
| 13 | 1.18 (3H, s) | 1.18 (3H, s) | 16.7 brq | 20.5 brq |
| 2′ | 78.1 s | 79.5 s | ||
| 2′-Me | 1.48 s | 1.47 s | 27.7 brq | 27.5 brq |
| 1.48 s | 1.23 | 28.1 brq | 20.5 brq | |
| 3′ | 5.44 (d, 10.1) | 1.71 (1H, m) | 124.6 d | 40.8 d |
| 4′ | 6.70 (d,10.1) | 2.83 (1H; dd,5.4,17.1) | 117.3 d | 22.7 t |
| 2.17 (1H; dd 10.2,17.1) | ||||
| 5′ | 155.4 s | 155.3 s | ||
| 6′ | 108.1 s | 107.1 s | ||
| 7′ | 162.2 s | 161.6 s | ||
| 8′ | 104.2 s | 104.7 s | ||
| 9′ | 159.3 s | 161.4 s | ||
| 10′ | 103.6 s | 102.4 s | ||
| 11′ | 210.8 s | 210.7 s | ||
| 12′ | 3.81(1H; septet; 6.8) | 3.89(1H; septet; 6.8) | 45.7 d | 38.9 d |
| 13′ | 1.18 (3H; s) | 1.18 (3H; s) | 19.1 q | 19.1 q |
| 14′ | 1.42/1.88 (2H; m) | 1.42/1.88 (2H; m) | 26.7 t | 27.5 t |
| 15′ | 0.94 (3H; t; 6.7) | 0.94 (3H; t; 6.7) | 11.9 q | 11.9 q |
| 1′’ | 2.32/1.81 (2H; m) | 29.4 t | ||
| 2′’ | 5.19 (1H, br t, 6.8) | 122.1 d | ||
| 3′’ | 133.1 s | |||
| 4′’ | 1.73 (3H; brs) | 25.8 q | ||
| 5′’ | 1.62 (3H. brs) | 17.2 q |
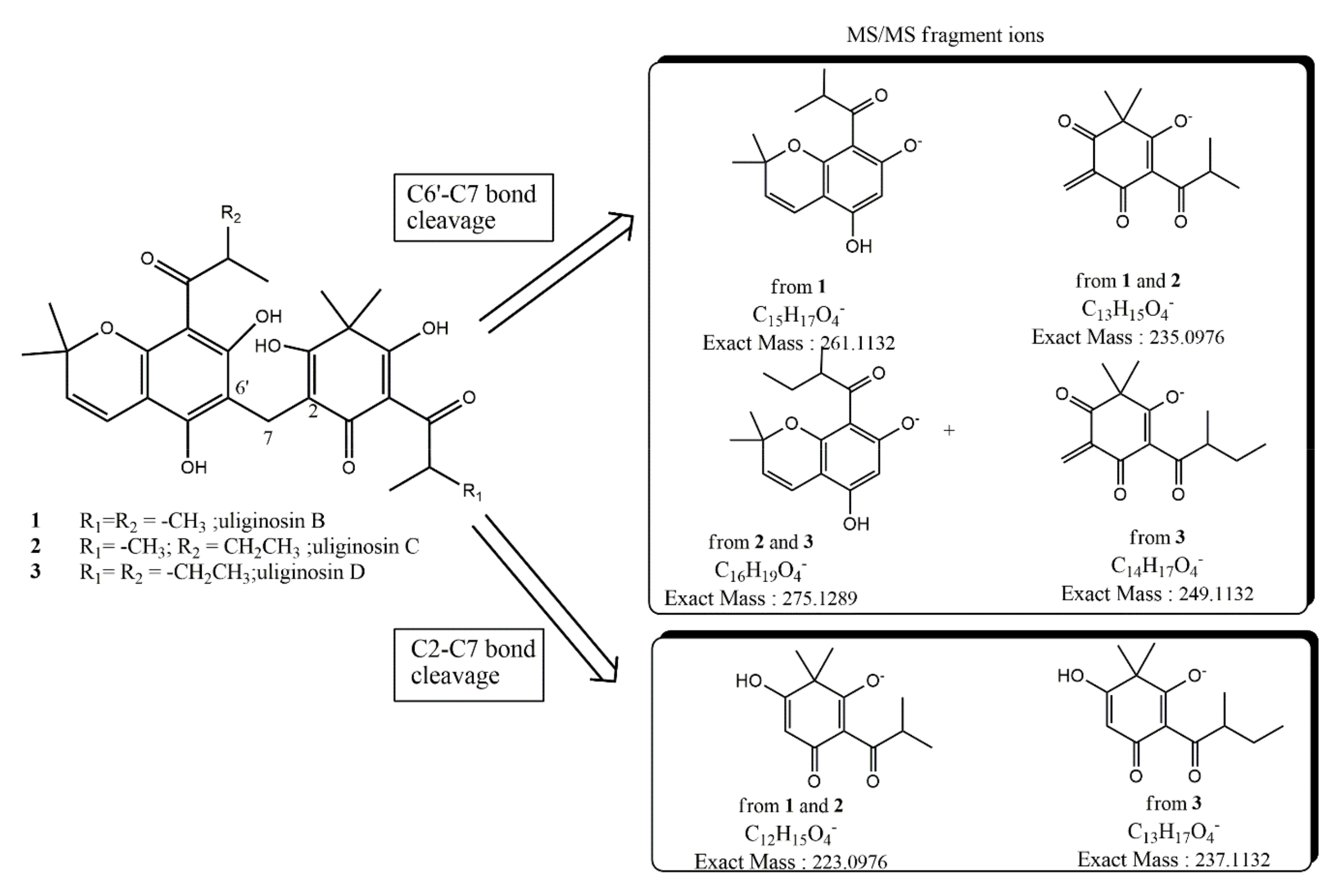
| Compound | ESI (−) Ion Type | Molecular Formula | Calculated Exact Mass (m/z) | High Resolution Mass Measurement (±0.0010) |
|---|---|---|---|---|
| 2 | [M − H]− | C29H35O8 | 511.2337 | 511.2315 |
| Fragment-ion C2-C7 | C12H15O4 | 223.0976 | 223.0978 | |
| Fragment-ion C6′-C7 | C13H15O4 | 235.0976 | 235.0978 | |
| Fragment-ion C6′-C7 | C16H19O4 | 275.1289 | ||
| 3 | [M − H]− | C30H37O8 | 525.2493 | 525.2483 |
| Fragment-ion C2-C7 | C13H17O4 | 237.1132 | 237.1133 | |
| Fragment-ion C6′-C7 | C14H17O4 | 249.1132 | ||
| Fragment-ion C6′-C7 | C16H19O4 | 275.1289 | 275.1293 | |
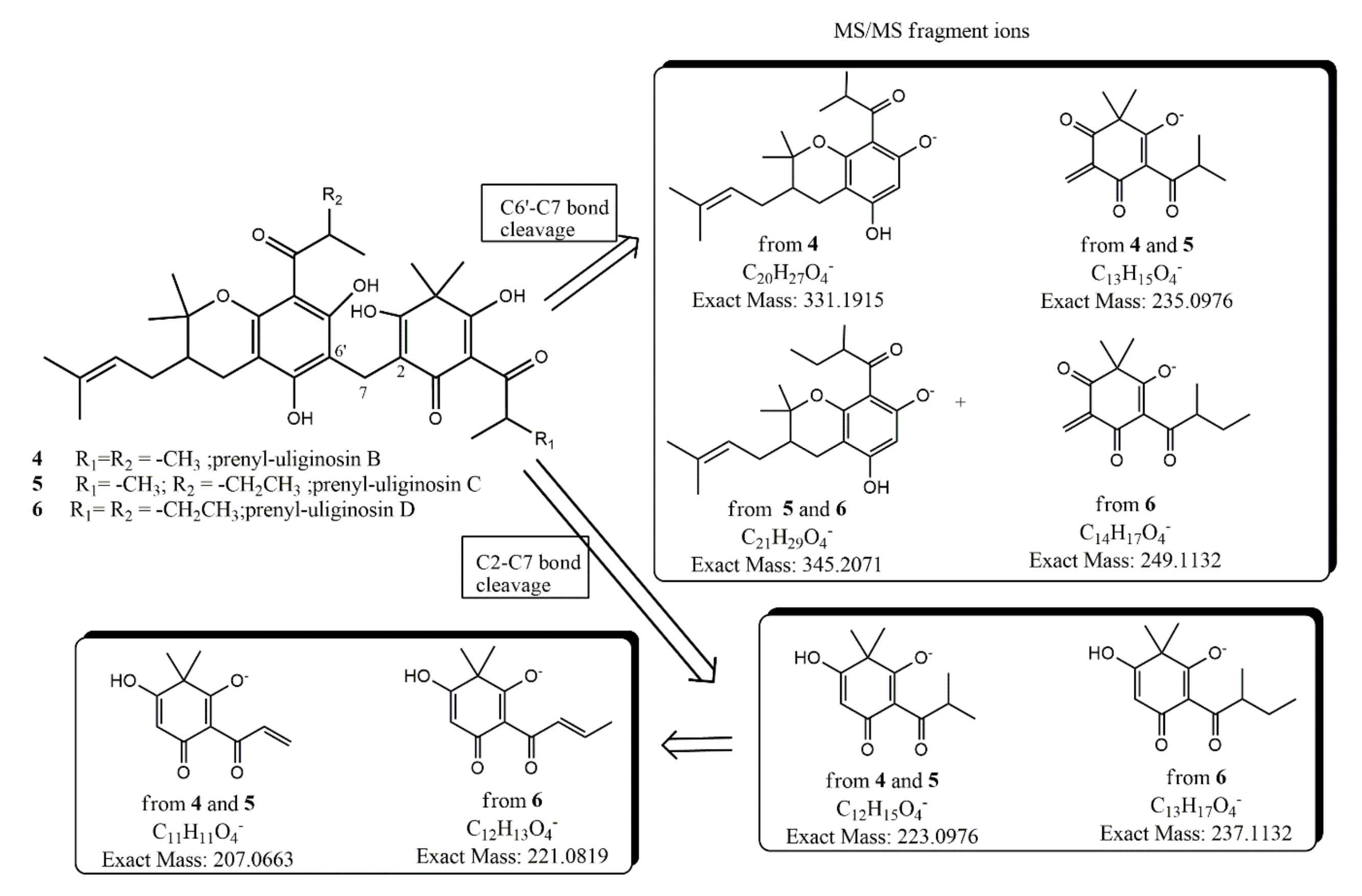
| 4 | [M − H]− | C33H43O8 | 567.2943 | 567.2941 |
| Fragment-ion C2-C7 | C12H15O4 | 223.0976 | 223.0978 | |
| Fragment-ion C6′-C7 | C13H15O4 | 235.0976 | 235.0978 | |
| Fragment-ion C6′-C7 | C20H27O4 | 331.1915 | 331.1918 | |
| 5 | [M − H]− | C34H45O8 | 581.3120 | 581.3111 |
| Fragment-ion C2-C7 | C12H15O4 | 223.0976 | 223.0978 | |
| Fragment-ion C6′-C7 | C13H15O4 | 235.0976 | 235.0978 | |
| Fragment-ion C6′-C7 | C21H29O4 | 345.2071 | ||
| 6 | [M − H]− | C35H47O8 | 595.3276 | 595.3267 |
| Fragment-ion C2-C7 | C13H17O4 | 237.1132 | 237.1141 | |
| Fragment-ion C6′-C7 | C14H17O4 | 249.1132 | 249.1120 | |
| Fragment-ion C6′-C7 | C21H29O4 | 345.2071 | 345.2075 |
| Candida Strain | Uliginosin C | 3′ Prenyl Uliginosin B | Fluconazole |
|---|---|---|---|
| MIC50 (µM) | |||
| C. albicans MFB 076N1 | 16 ± 0.5 | 15 ± 1 | 208 ± 2 |
| C. albicans MFB 008 MM1 | 6 ± 0.2 | 3 ± 0.2 | 208 ± 2 |
| C. albicans MFB YMS 100-3 | >32 | >30 | 1.6 ± 0.1 |
| C. albicans MFB YMS 102-2 | 8 ± 0.7 | 4 ± 0.3 | >208 |
| C. parapsilosis MFB YMS 100-1 | 32 ± 1 | 6 ± 0.6 | 6 ± 0.5 |
| C. parapsilosis MFB 014 CD7 | 32 ± 1 | 30 ± 2 | >208 |
| C. parapsilosis MFB 070 N1 | >32 | >30 | >208 |
| C. lusitaniae MFB YMS 100-16 | >32 | 30 ± 2 | 0.8 ± 0.2 |
| C. lusitaniae MFB 037 N1 | 8 ± 0.2 | 30 ± 1 | 1.6 ± 0.5 |
| C. pararugosa MFB 037 N3 | 8 ± 0.7 | 15 ± 2 | 0.4 ± 0.0 |
| C. glabrata MFB004 | 16 ± 1 | 4 ± 0.1 | 0.13 ± 0.0 |
| C. glabrata MFB005FS4 | 8 ± 0.4 | 6 ± 0.1 | 0.13 ± 0.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tocci, N.; Weil, T.; Perenzoni, D.; Moretto, M.; Nürk, N.; Madriñán, S.; Ferrazza, R.; Guella, G.; Mattivi, F. Potent Antifungal Properties of Dimeric Acylphloroglucinols from Hypericum mexicanum and Mechanism of Action of a Highly Active 3′Prenyl Uliginosin B. Metabolites 2020, 10, 459. https://doi.org/10.3390/metabo10110459
Tocci N, Weil T, Perenzoni D, Moretto M, Nürk N, Madriñán S, Ferrazza R, Guella G, Mattivi F. Potent Antifungal Properties of Dimeric Acylphloroglucinols from Hypericum mexicanum and Mechanism of Action of a Highly Active 3′Prenyl Uliginosin B. Metabolites. 2020; 10(11):459. https://doi.org/10.3390/metabo10110459
Chicago/Turabian StyleTocci, Noemi, Tobias Weil, Daniele Perenzoni, Marco Moretto, Nicolai Nürk, Santiago Madriñán, Ruggero Ferrazza, Graziano Guella, and Fulvio Mattivi. 2020. "Potent Antifungal Properties of Dimeric Acylphloroglucinols from Hypericum mexicanum and Mechanism of Action of a Highly Active 3′Prenyl Uliginosin B" Metabolites 10, no. 11: 459. https://doi.org/10.3390/metabo10110459
APA StyleTocci, N., Weil, T., Perenzoni, D., Moretto, M., Nürk, N., Madriñán, S., Ferrazza, R., Guella, G., & Mattivi, F. (2020). Potent Antifungal Properties of Dimeric Acylphloroglucinols from Hypericum mexicanum and Mechanism of Action of a Highly Active 3′Prenyl Uliginosin B. Metabolites, 10(11), 459. https://doi.org/10.3390/metabo10110459