Streamlining the Analysis of Dynamic 13C-Labeling Patterns for the Metabolic Engineering of Corynebacterium glutamicum as l-Histidine Production Host

 , , and
, , and 
Abstract
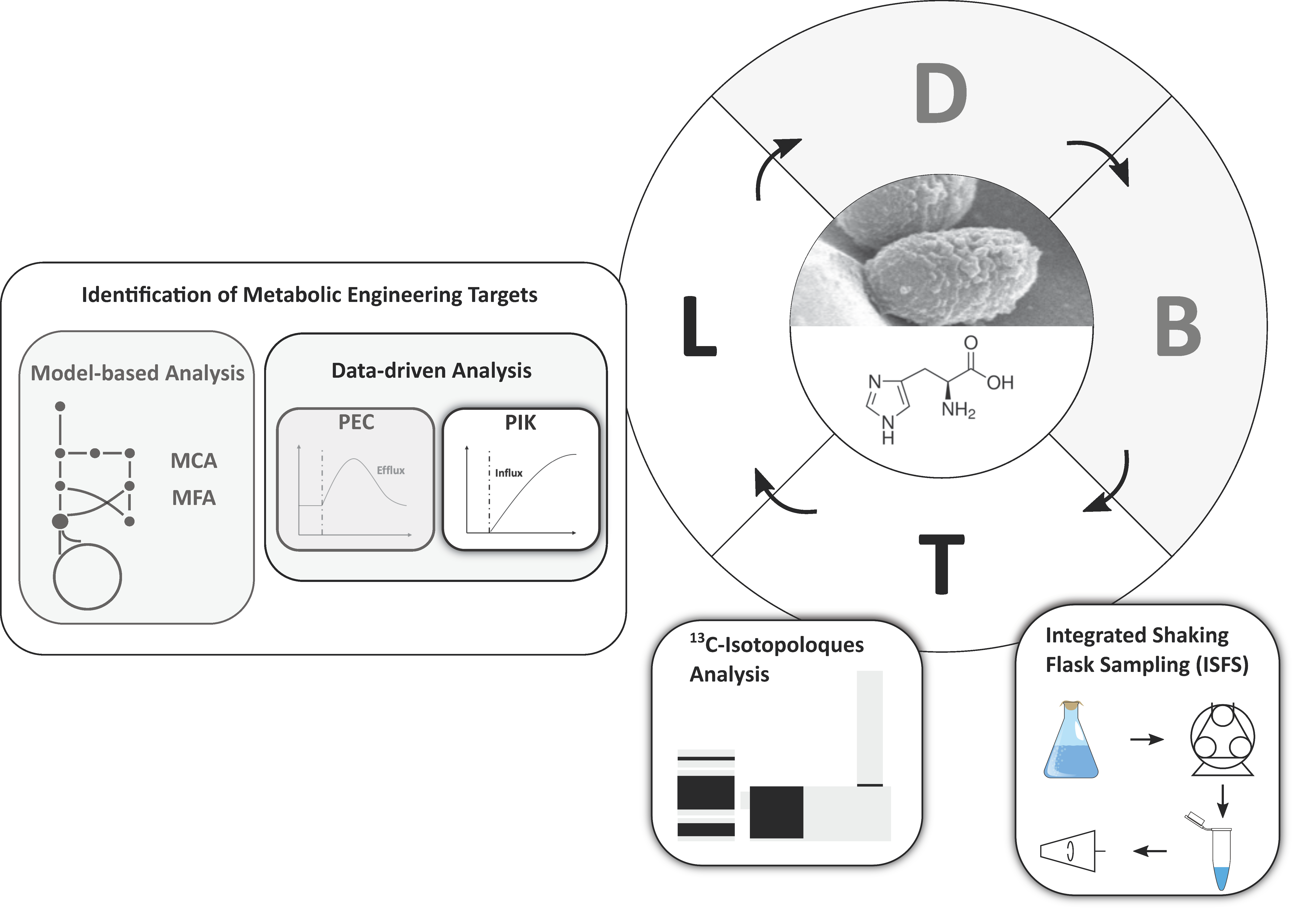
1. Introduction
2. Results
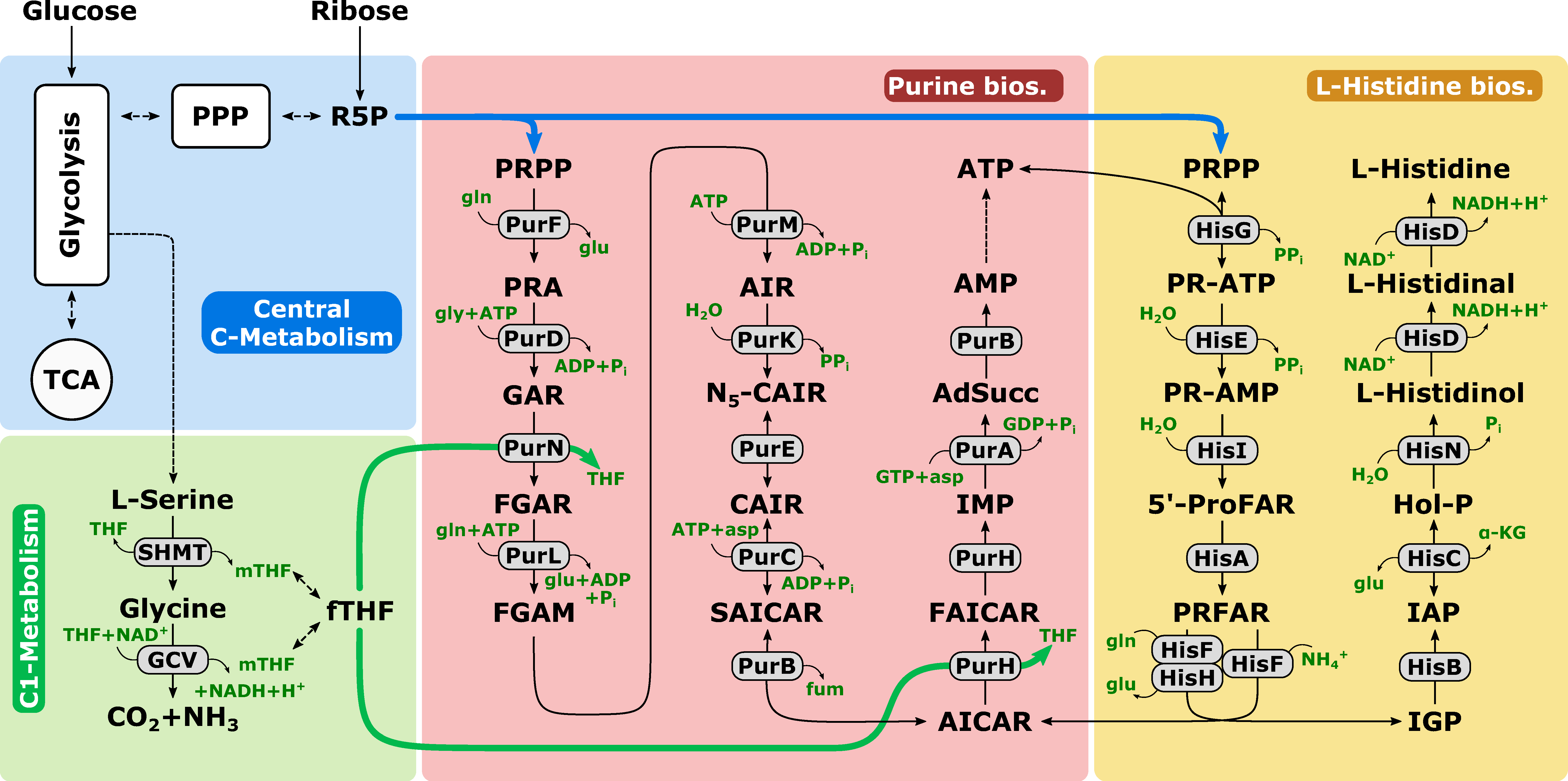
2.1. d-Ribose as a Tracer Molecule
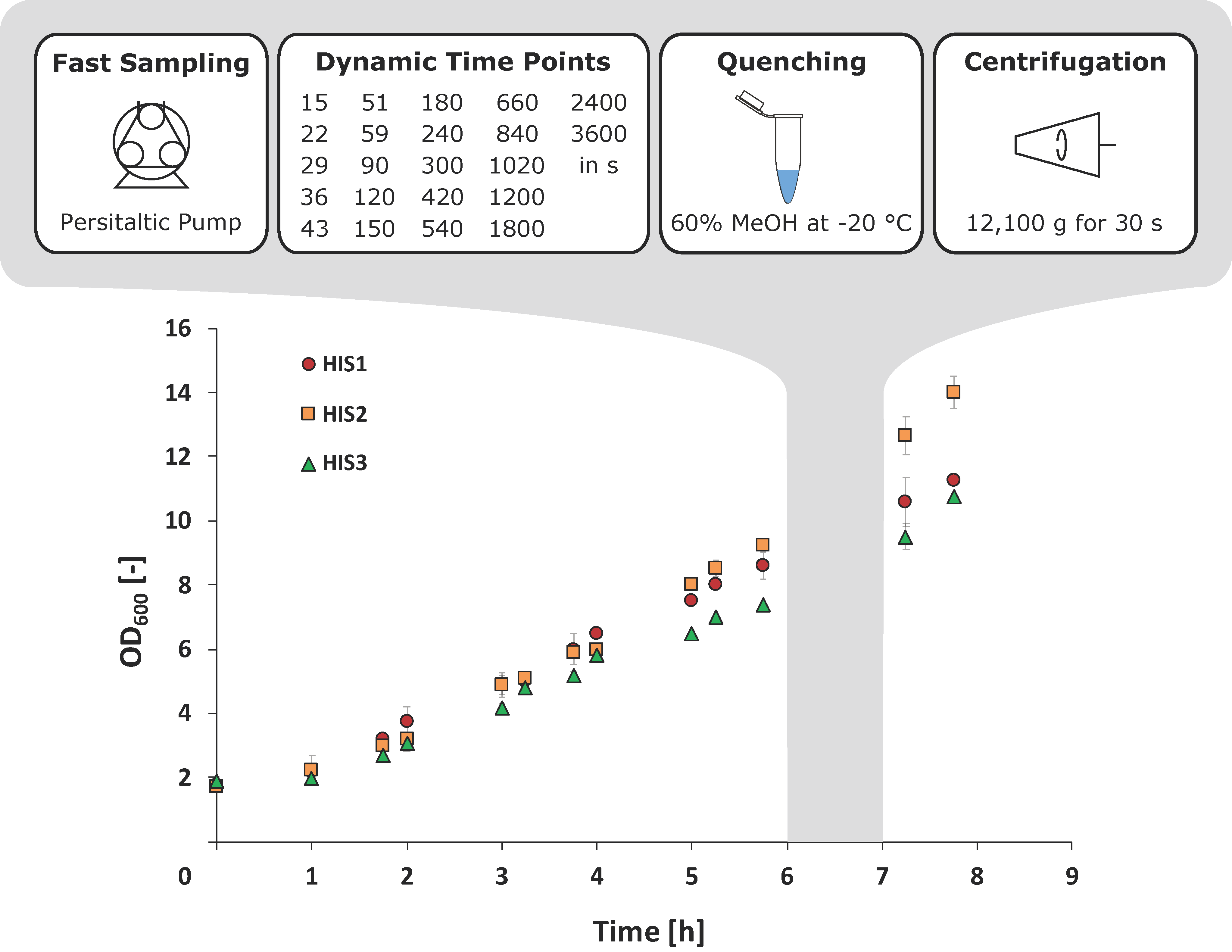
2.2. Integrated Shaking Flask Sampling (ISFS)
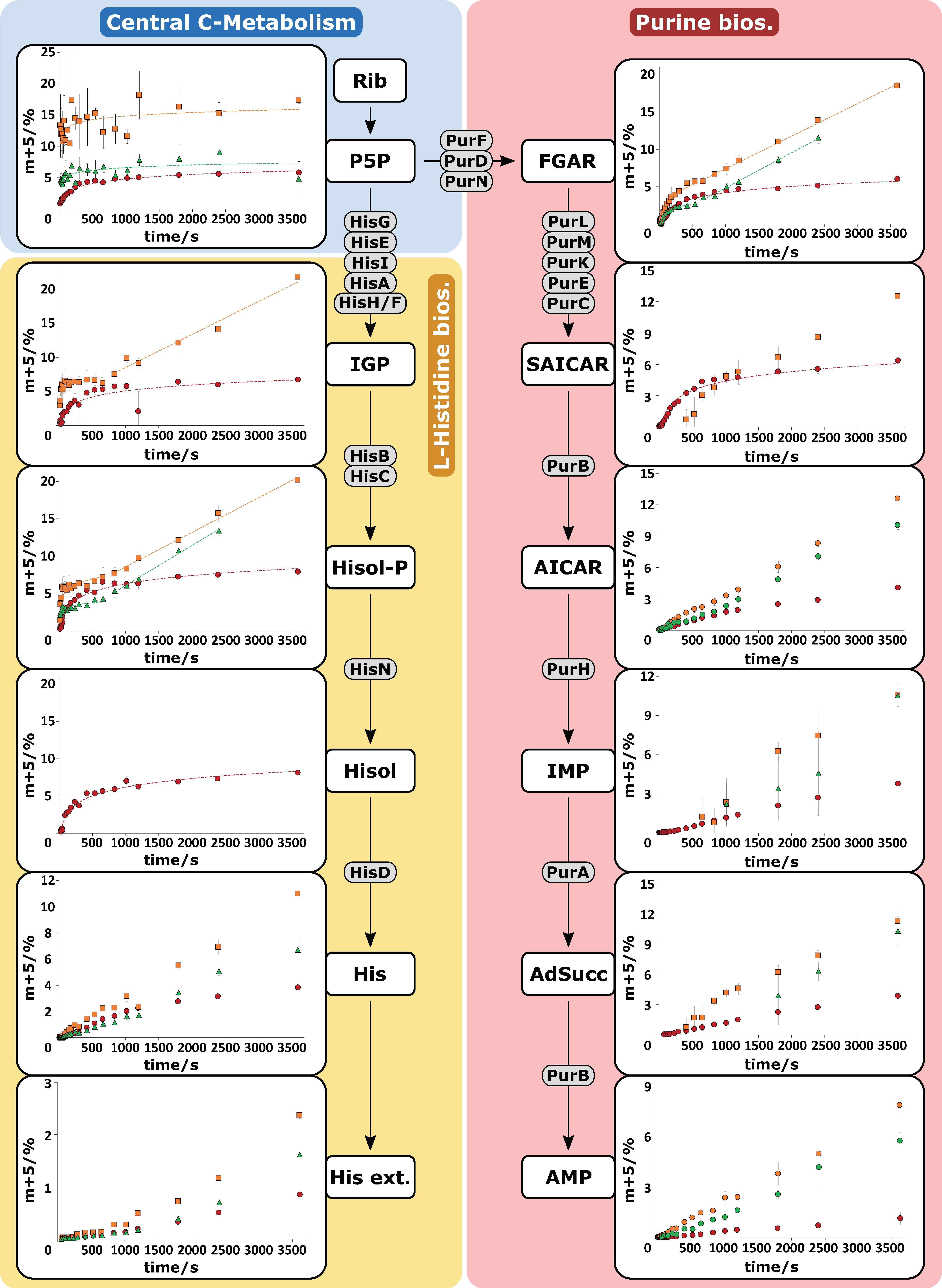
2.3. HILIC-QTOF-HRMS Analytics Enables Analysis of 13C-Isotopologues
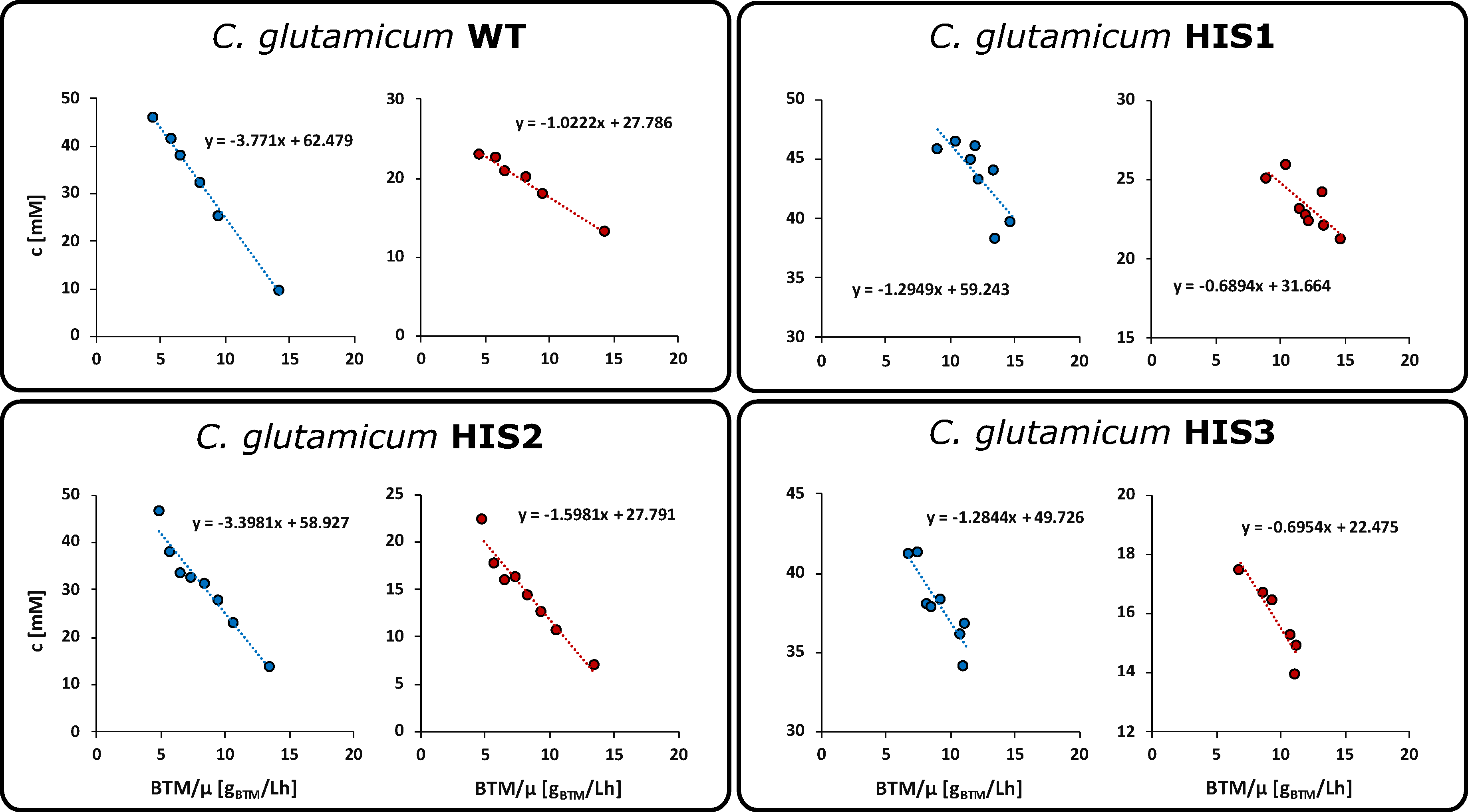
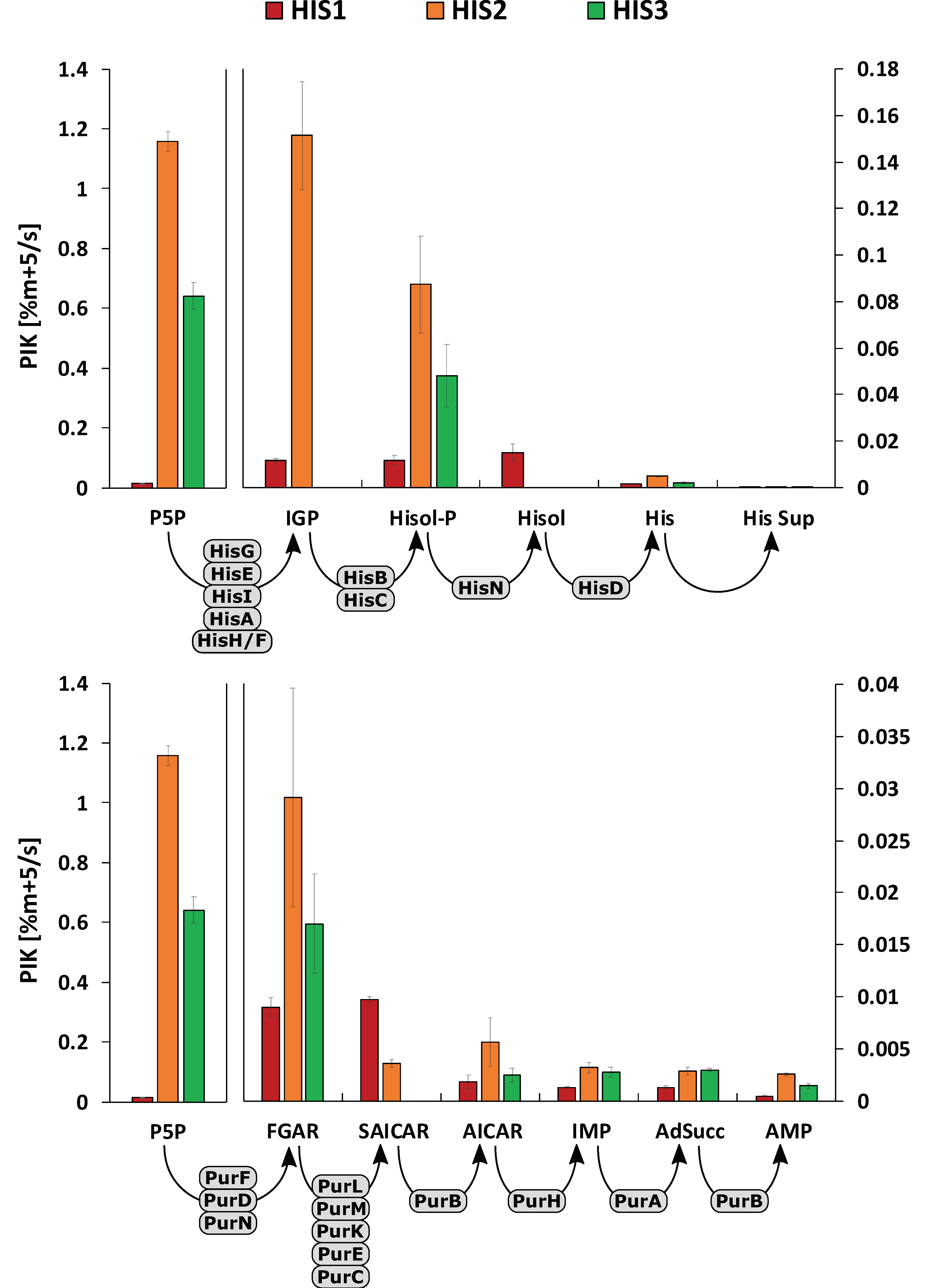
2.4. Pool Influx Kinetics as a Metabolic Engineering Tool
3. Discussion
4. Materials and Methods
4.1. Media and Cultivation Conditions
4.2. Chemicals
4.3. Fast Sampling Procedure and Methanol Quenching
4.4. Metabolite Extraction
4.5. LC-QTOF Analysis
4.5.1. Analysis of d-Glucose and d-Ribose in Supernatant Samples
4.5.2. Analysis of 13C-Labeled Intracellular Metabolite Extracts and Data Mining
4.6. Pool Influx Kinetics
Author Contributions
Funding
Conflicts of Interest
References
- Csörgő, B.; Nyerges, Á.; Pósfai, G.; Fehér, T. System-level genome editing in microbes. Curr. Opin. Microbiol. 2016, 33, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Storch, M.; Baldwin, G.S.; Ellis, T. Bricks and blueprints: Methods and standards for DNA assembly. Nat. Rev. Mol. Cell Biol. 2015, 16, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Dragosits, M.; Mattanovich, D. Adaptive laboratory evolution—principles and applications for biotechnology. Microb. Cell Fact. 2013, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef]
- Rogers, J.K.; Church, G.M. Multiplexed Engineering in Biology. Trends Biotechnol. 2016, 34, 198–206. [Google Scholar] [CrossRef]
- Reaves, M.L.; Rabinowitz, J.D. Metabolomics in systems microbiology. Curr. Opin. Biotechnol. 2011, 22, 17–25. [Google Scholar] [CrossRef]
- Carbonell, P.; Currin, A.; Jervis, A.J.; Rattray, N.J.W.; Swainston, N.; Yan, C.; Takano, E.; Breitling, R. Bioinformatics for the synthetic biology of natural products: Integrating across the Design–Build–Test cycle. Nat. Prod. Rep. 2016, 33, 925–932. [Google Scholar] [CrossRef]
- Petzold, C.J.; Chan, L.J.G.; Nhan, M.; Adams, P.D. Analytics for Metabolic Engineering. Front. Bioeng. Biotechnol. 2015, 3, 1–11. [Google Scholar] [CrossRef]
- Fiehn, O.; Weckwerth, W. Deciphering metabolic networks. Eur. J. Biochem. 2003, 270, 579–588. [Google Scholar] [CrossRef]
- Oldiges, M.; Lütz, S.; Pflug, S.; Schroer, K.; Stein, N.; Wiendahl, C. Metabolomics: Current state and evolving methodologies and tools. Appl. Microbiol. Biotechnol. 2007, 76, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Fei, F.; Bowdish, D.M.E.; McCarry, B.E. Comprehensive and simultaneous coverage of lipid and polar metabolites for endogenous cellular metabolomics using HILIC-TOF-MS. Anal. Bioanal. Chem. 2014, 406, 3723–3733. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Combining Genomics, Metabolome Analysis, and Biochemical Modelling to Understand Metabolic Networks. Comp. Funct. Genom. 2001, 2, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.; Davey, H.M.; Broadhurst, D.; Heald, J.K.; Rowland, J.J.; Oliver, S.G.; Kell, D.B. High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nat. Biotechnol. 2003, 21, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Ellis, D.I. Metabolomics: Current analytical platforms and methodologies. TrAC Trends Anal. Chem. 2005, 24, 285–294. [Google Scholar] [CrossRef]
- Teleki, A.; Sánchez-Kopper, A.; Takors, R. Alkaline conditions in hydrophilic interaction liquid chromatography for intracellular metabolite quantification using tandem mass spectrometry. Anal. Biochem. 2015, 475, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Feith, A.; Teleki, A.; Graf, M.; Favilli, L.; Takors, R. HILIC-Enabled 13C Metabolomics Strategies: Comparing Quantitative Precision and Spectral Accuracy of QTOF High- and QQQ Low-Resolution Mass Spectrometry. Metabolites 2019, 9, 63. [Google Scholar] [CrossRef]
- Wiechert, W. 13C Metabolic Flux Analysis. Metab. Eng. 2001, 3, 195–206. [Google Scholar] [CrossRef]
- Zamboni, N.; Fendt, S.-M.; Rühl, M.; Sauer, U. 13C-based metabolic flux analysis. Nat. Protoc. 2009, 4, 878–892. [Google Scholar] [CrossRef]
- Visser, D.; Heijnen, J.J. The mathematics of Metabolic Control Analysis revisited. Metab. Eng. 2002, 4, 114–123. [Google Scholar] [CrossRef]
- Thomas, S.; Fell, D.A. Metabolic Control Analysis: Sensitivity of Control Coefficients to Experimentally Determined Variables. J. Theor. Biol. 1994, 167, 175–200. [Google Scholar] [CrossRef]
- de Noronha Pissara, P.; Nielsen, J.; Bazin, M.J. Pathway kinetics and metabolic control analysis of a high-yielding strain ofPenicillium chrysogenum during fed batch cultivations. Biotechnol. Bioeng. 1996, 51, 168–176. [Google Scholar] [CrossRef]
- van der Meer, R.; Westerhoff, H.V.; Van Dam, K. Linear relation between rate and thermodynamic force in enzyme-catalyzed reactions. Biochim. Biophys. Acta Bioenerg. 1980, 591, 488–493. [Google Scholar] [CrossRef]
- Visser, D.; Heijnen, J.J. Dynamic simulation and metabolic re-design of a branched pathway using linlog kinetics. Metab. Eng. 2003, 5, 164–176. [Google Scholar] [CrossRef]
- Nikerel, I.E.; van Winden, W.A.; Verheijen, P.J.T.; Heijnen, J.J. Model reduction and a priori kinetic parameter identifiability analysis using metabolome time series for metabolic reaction networks with linlog kinetics. Metab. Eng. 2009, 11, 20–30. [Google Scholar] [CrossRef]
- Magnus, J.B.; Oldiges, M.; Takors, R. The identification of enzyme targets for the optimization of a valine producing Corynebacterium glutamicum strain using a kinetic model. Biotechnol. Prog. 2009, 25, 754–762. [Google Scholar] [CrossRef]
- Teleki, A.; Rahnert, M.; Bungart, O.; Gann, B.; Ochrombel, I.; Takors, R. Robust identification of metabolic control for microbial l -methionine production following an easy-to-use puristic approach. Metab. Eng. 2017, 41, 159–172. [Google Scholar] [CrossRef]
- Alifano, P.; Fani, R.; Liò, P.; Lazcano, A.; Bazzicalupo, M.; Carlomagno, M.S.; Bruni, C.B. Histidine biosynthetic pathway and genes: Structure, regulation, and evolution. Microbiol. Rev. 1996, 60, 44–69. [Google Scholar] [CrossRef]
- Kornberg, A.; Lieberman, I.; Simms, E.S. Enzymatic synthesis and properties of 5-phosphoribosylpyrophosphate. J. Biol. Chem. 1955, 215, 389–402. [Google Scholar]
- Nentwich, S.S.; Brinkrolf, K.; Gaigalat, L.; Hüser, A.T.; Rey, D.A.; Mohrbach, T.; Marin, K.; Pühler, A.; Tauch, A.; Kalinowski, J. Characterization of the LacI-type transcriptional repressor RbsR controlling ribose transport in Corynebacterium glutamicum ATCC 13032. Microbiology 2009, 155, 150–164. [Google Scholar] [CrossRef]
- Mashego, M.R.; Rumbold, K.; De Mey, M.; Vandamme, E.; Soetaert, W.; Heijnen, J.J. Microbial metabolomics: Past, present and future methodologies. Biotechnol. Lett. 2007, 29, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schwentner, A.; Feith, A.; Münch, E.; Stiefelmaier, J.; Lauer, I.; Favilli, L.; Massner, C.; Öhrlein, J.; Grund, B.; Hüser, A.; et al. Modular systems metabolic engineering enables balancing of relevant pathways for l -histidine production with Corynebacterium glutamicum. Biotechnol. Biofuels 2019, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, M.T.; Hesek, E.D.; Wilson, C.M.; Gibson, D.G. Vibrio natriegens as a fast-growing host for molecular biology. Nat. Methods 2016, 13, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Hirsch, E.; Bongaerts, J.; Takors, R. Pulse Experiments as a Prerequisite for the Quantification of in Vivo Enzyme Kinetics in Aromatic Amino Acid Pathway of Escherichia coli. Biotechnol. Prog. 2002, 18, 935–941. [Google Scholar] [CrossRef]
- Nöh, K.; Droste, P.; Wiechert, W. Visual workflows for 13 C-metabolic flux analysis. Bioinformatics 2015, 31, 346–354. [Google Scholar] [CrossRef]
- Niedenführ, S.; ten Pierick, A.; van Dam, P.T.N.; Suarez-Mendez, C.A.; Nöh, K.; Wahl, S.A. Natural isotope correction of MS/MS measurements for metabolomics and 13 C fluxomics. Biotechnol. Bioeng. 2015, 113, 1137–1147. [Google Scholar] [CrossRef]
- Hofmann, U.; Maier, K.; Reuss, M.; Mauch, K. Identification of metabolic fluxes in hepatic cells from transient 13 C-labeling experiments: Part II. Flux estimation. Biotechnol. Bioeng. 2007, 100, 355–370. [Google Scholar] [CrossRef]
- Nöh, K.; Wiechert, W. The benefits of being transient: Isotope-based metabolic flux analysis at the short time scale. Appl. Microbiol. Biotechnol. 2011, 91, 1247–1265. [Google Scholar] [CrossRef]
- Bolten, C.J.; Kiefer, P.; Letisse, F.; Portais, J.C.; Wittmann, C. Sampling for metabolome analysis of microorganisms. Anal. Chem. 2007, 79, 3843–3849. [Google Scholar] [CrossRef]
- Dauner, M.; Sauer, U. GC-MS Analysis of Amino Acids Rapidly Provides Rich Information for Isotopomer Balancing. Biotechnol. Prog. 2000, 16, 642–649. [Google Scholar] [CrossRef]
- Rühl, M.; Rupp, B.; Nöh, K.; Wiechert, W.; Sauer, U.; Zamboni, N. Collisional fragmentation of central carbon metabolites in LC-MS/MS increases precision of 13C metabolic flux analysis. Biotechnol. Bioeng. 2012, 109, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Shamir, M.; Bar-On, Y.; Phillips, R.; Milo, R. SnapShot: Timescales in Cell Biology. Cell 2016, 164, 1302–1302.e1. [Google Scholar] [CrossRef] [PubMed]
- Ishino, S.; Kuga, T.; Yamaguchi, K.; Shirahata, K.; Araki, K. 13 C NMR Studies of Histidine Fermentation with a Corynebacterium glutamicum Mutant. Agric. Biol. Chem. 1986, 50, 307–310. [Google Scholar] [CrossRef]
- Chim-Anage, P.; Shioya, S.; Suga, K.-I. Optimum conditions for histidine production by fed-batch culture of Brevibacterium flavum. J. Ferment. Bioeng. 1990, 70, 386–391. [Google Scholar] [CrossRef]
- Lu, W.; Kwon, Y.K.; Rabinowitz, J.D. Isotope Ratio-Based Profiling of Microbial Folates. J. Am. Soc. Mass Spectrom. 2007, 18, 898–909. [Google Scholar] [CrossRef]
- Malykh, E.A.; Butov, I.A.; Ravcheeva, A.B.; Krylov, A.A.; Mashko, S.V.; Stoynova, N.V. Specific features of l-histidine production by Escherichia coli concerned with feedback control of AICAR formation and inorganic phosphate/metal transport. Microb. Cell Fact. 2018, 17, 42. [Google Scholar] [CrossRef]
- Kim, P.B.; Nelson, J.W.; Breaker, R.R. An Ancient Riboswitch Class in Bacteria Regulates Purine Biosynthesis and One-Carbon Metabolism. Mol. Cell 2015, 57, 317–328. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Description | YP/Shis [mol mol−1] | Comment |
|---|---|---|---|
| C. glutamicum WT | Wildtype strain ATCC 13032 | - | - |
| C. glutamicum HIS1 | C. glutamicum hisGG233H-T235Q Ptuf (hisD-hisC-hisB-cg2302-cg2301) Ptuf (hisH-hisA-impA-PsodA(hisF-hisI-cg2294)) Ptuf(cg0911-hisN) PdapA-A16 (hisEATG-hisGG233H-T235Q) | 0.065 ± 0.004 | HIS7 in [32] |
| C. glutamicum HIS2 | C. glutamicum HIS1 containing pJC4 purA purB | 0.054 ± 0.002 | HIS8 in [32] |
| C. glutamicum HIS3 | C. glutamicum HIS2 containing pEC-XT99A_gcv_OP1-Cjk | 0.086 ± 0.001 | HIS9 in [32] |
| Time (min) | A (%) | B (%) | C (%) | D (%) |
|---|---|---|---|---|
| 0 | 89.75 | 2.75 | 5 | 2.5 |
| 36 | 62.25 | 30.25 | 5 | 2.5 |
| 40 | 1.75 | 90.75 | 5 | 2.5 |
| 45 | 1.75 | 90.75 | 5 | 2.5 |
| 50 | 89.75 | 2.75 | 5 | 2.5 |
| 70 | 89.75 | 2.75 | 5 | 2.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feith, A.; Schwentner, A.; Teleki, A.; Favilli, L.; Blombach, B.; Takors, R. Streamlining the Analysis of Dynamic 13C-Labeling Patterns for the Metabolic Engineering of Corynebacterium glutamicum as l-Histidine Production Host. Metabolites 2020, 10, 458. https://doi.org/10.3390/metabo10110458
Feith A, Schwentner A, Teleki A, Favilli L, Blombach B, Takors R. Streamlining the Analysis of Dynamic 13C-Labeling Patterns for the Metabolic Engineering of Corynebacterium glutamicum as l-Histidine Production Host. Metabolites. 2020; 10(11):458. https://doi.org/10.3390/metabo10110458
Chicago/Turabian StyleFeith, André, Andreas Schwentner, Attila Teleki, Lorenzo Favilli, Bastian Blombach, and Ralf Takors. 2020. "Streamlining the Analysis of Dynamic 13C-Labeling Patterns for the Metabolic Engineering of Corynebacterium glutamicum as l-Histidine Production Host" Metabolites 10, no. 11: 458. https://doi.org/10.3390/metabo10110458
APA StyleFeith, A., Schwentner, A., Teleki, A., Favilli, L., Blombach, B., & Takors, R. (2020). Streamlining the Analysis of Dynamic 13C-Labeling Patterns for the Metabolic Engineering of Corynebacterium glutamicum as l-Histidine Production Host. Metabolites, 10(11), 458. https://doi.org/10.3390/metabo10110458