In Vitro Activity Assays of Sputtered HAp Coatings with SiC Addition in Various Simulated Biological Fluids
.jpg)
 ,
, 
 ,
,  ,
, 
Abstract
1. Introduction
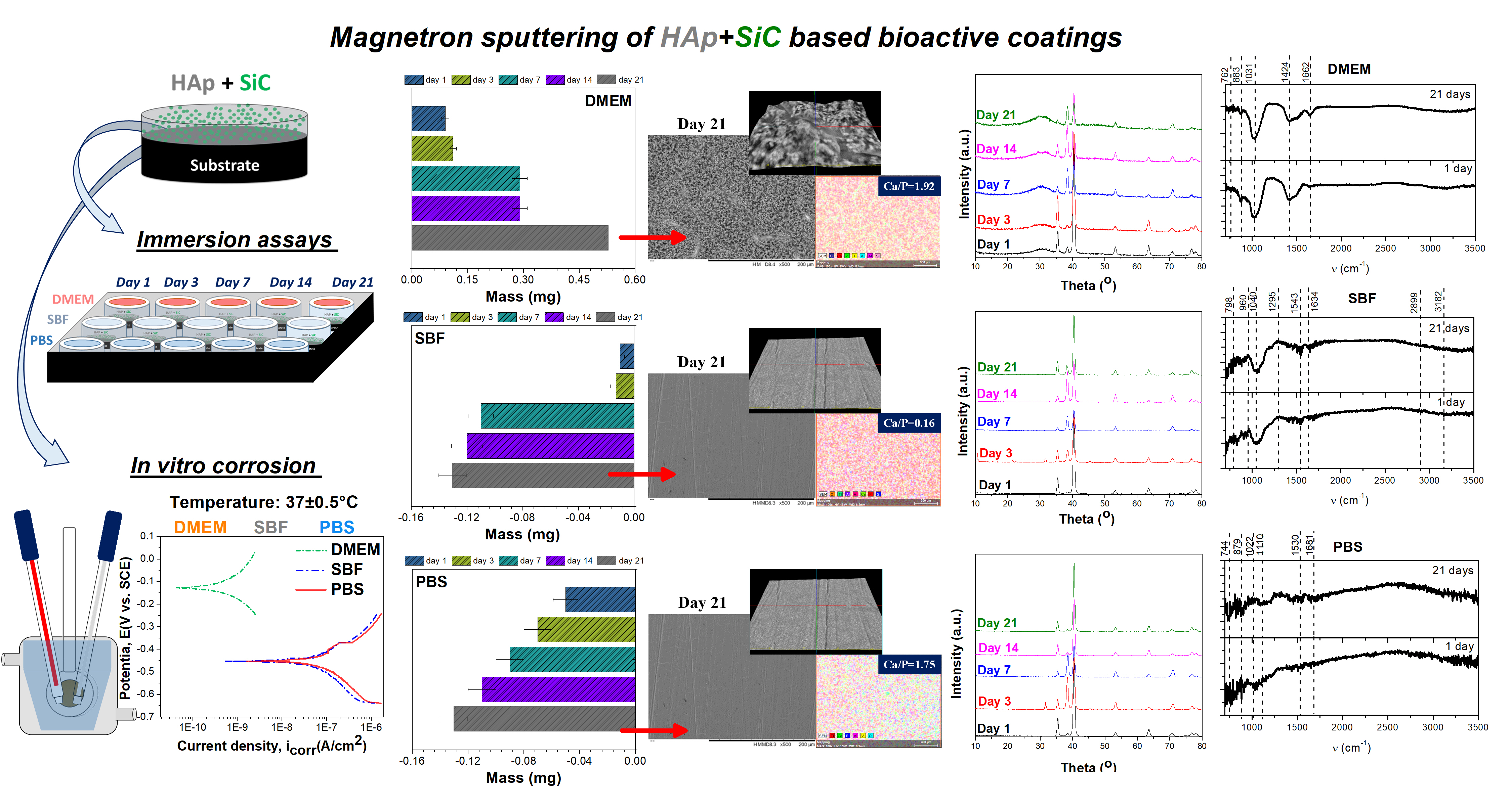
2. Materials and Methods
- Elemental composition and morphology using a scanning electron microscope (SEM, Hitachi TM3030Plus, Tokyo, Japan) equipped with energy dispersive spectrometry (EDS, Bruker);
- Mass evolution, which was monitored gravimetrically using an analytical balance (ALT 100-5AM, Kern, Balingen, Germany) with an accuracy of 0.01 mg;
- Phase composition by grazing-incidence XRD (X-ray diffraction) using Cu Kα radiation (SmartLab, Rigaku, Tokyo, Japan) from 10° to 80° with a step size of 0.02°/min and an incident angle of 2°;
- Chemical binding by Fourier transform infrared spectroscopy at a resolution of 4 cm−1, over the frequency range of 500–4000 cm−1 using an FTIR-6300 spectrophotometer (Jasco, Tokyo, Japan) with a universal Pike MIRacle attenuated total reflectance (ATR) sampling accessory (Pike Technologies, Madison, WI, USA).
- An area of 1 cm2 was exposed to the acellular media, placing the sample in the working electrode (WE—a Teflon sample holder);
- A platinum electrode was used as the counter electrode (CE);
- A saturated calomel (SCE) as the reference electrode (RE).
3. Results and Discussions
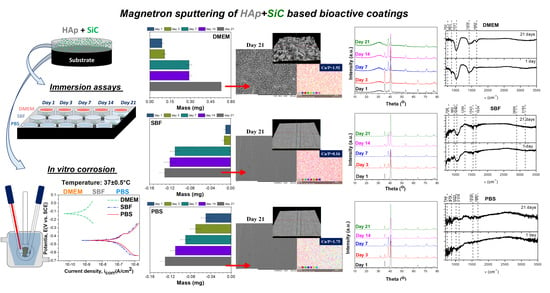
3.1. SEM and EDS after Immersion in DMEM, SBF, and PBS
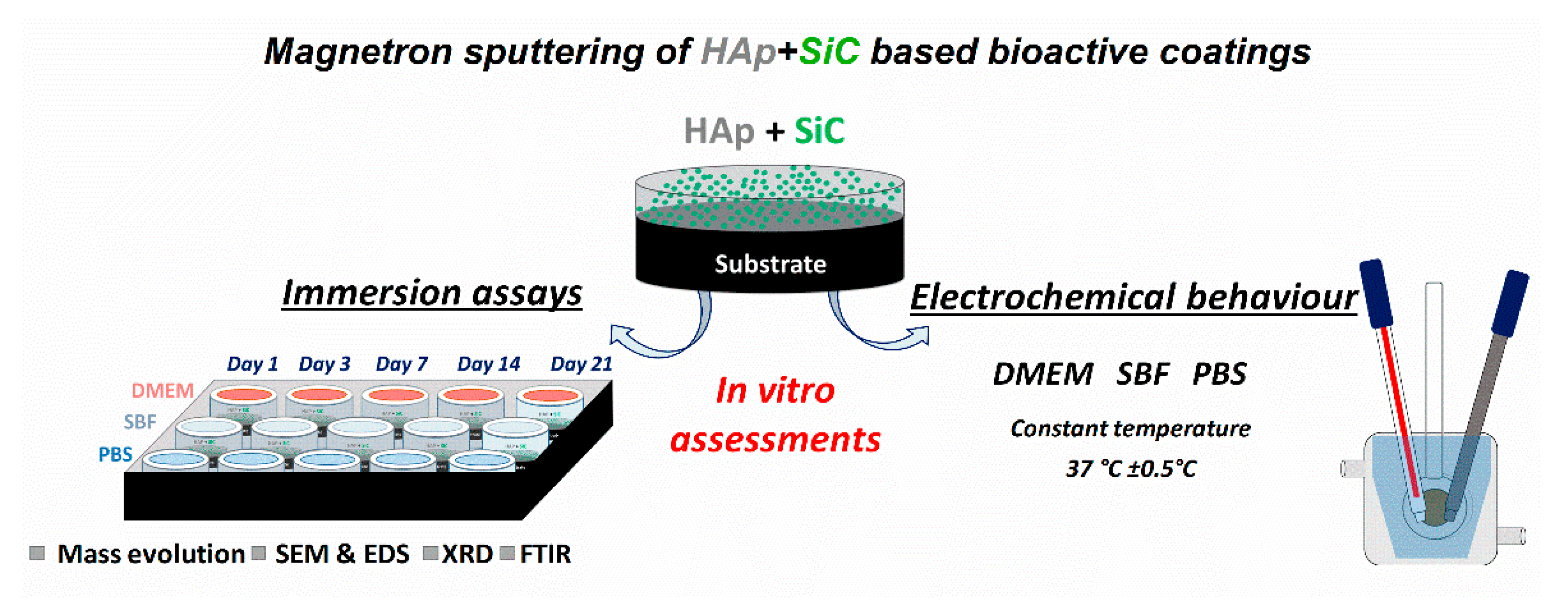
3.2. Mass Evolution in DMEM, SBF, and PBS
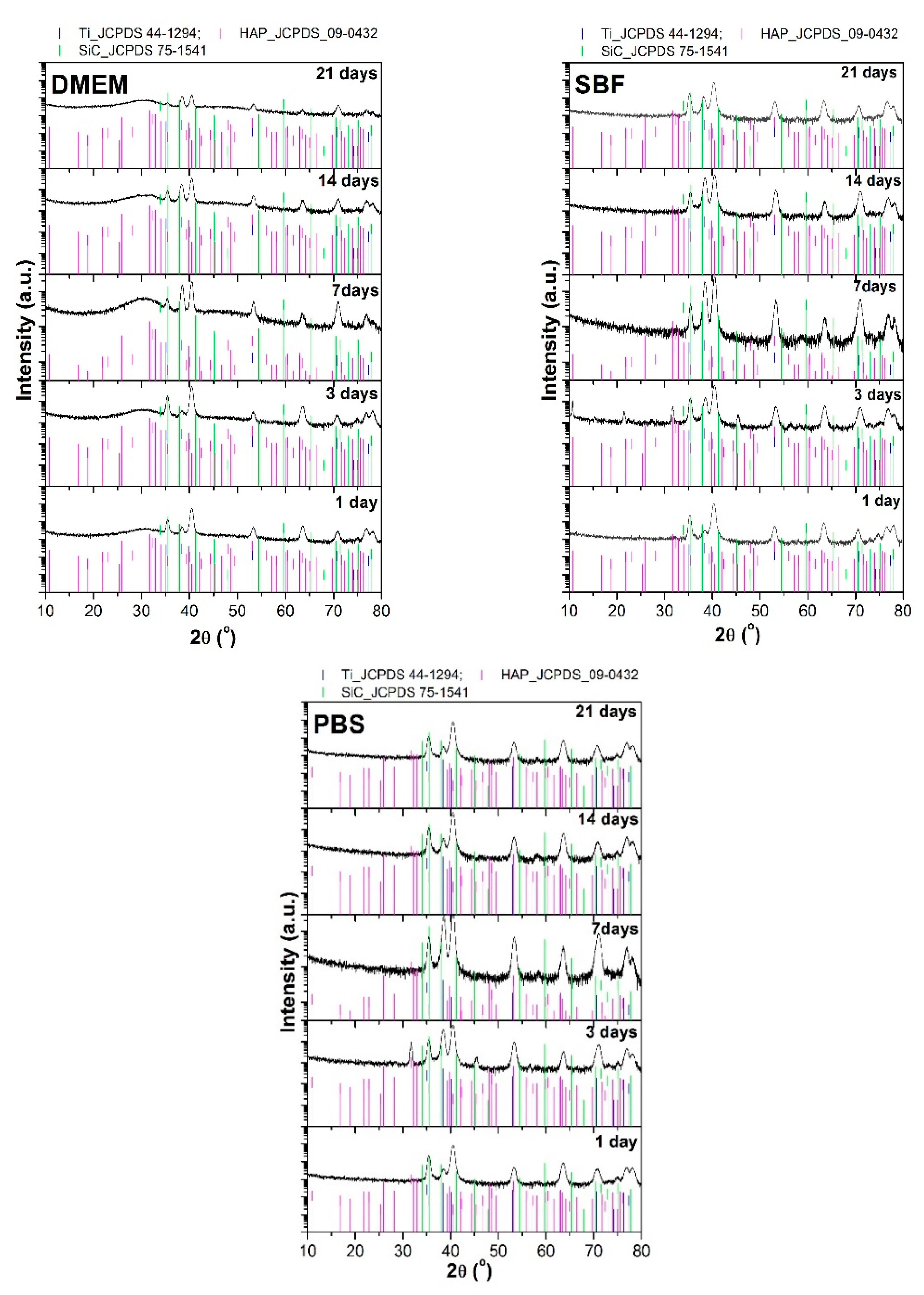
3.3. XRD after Immersion in DMEM, SBF, and PBS
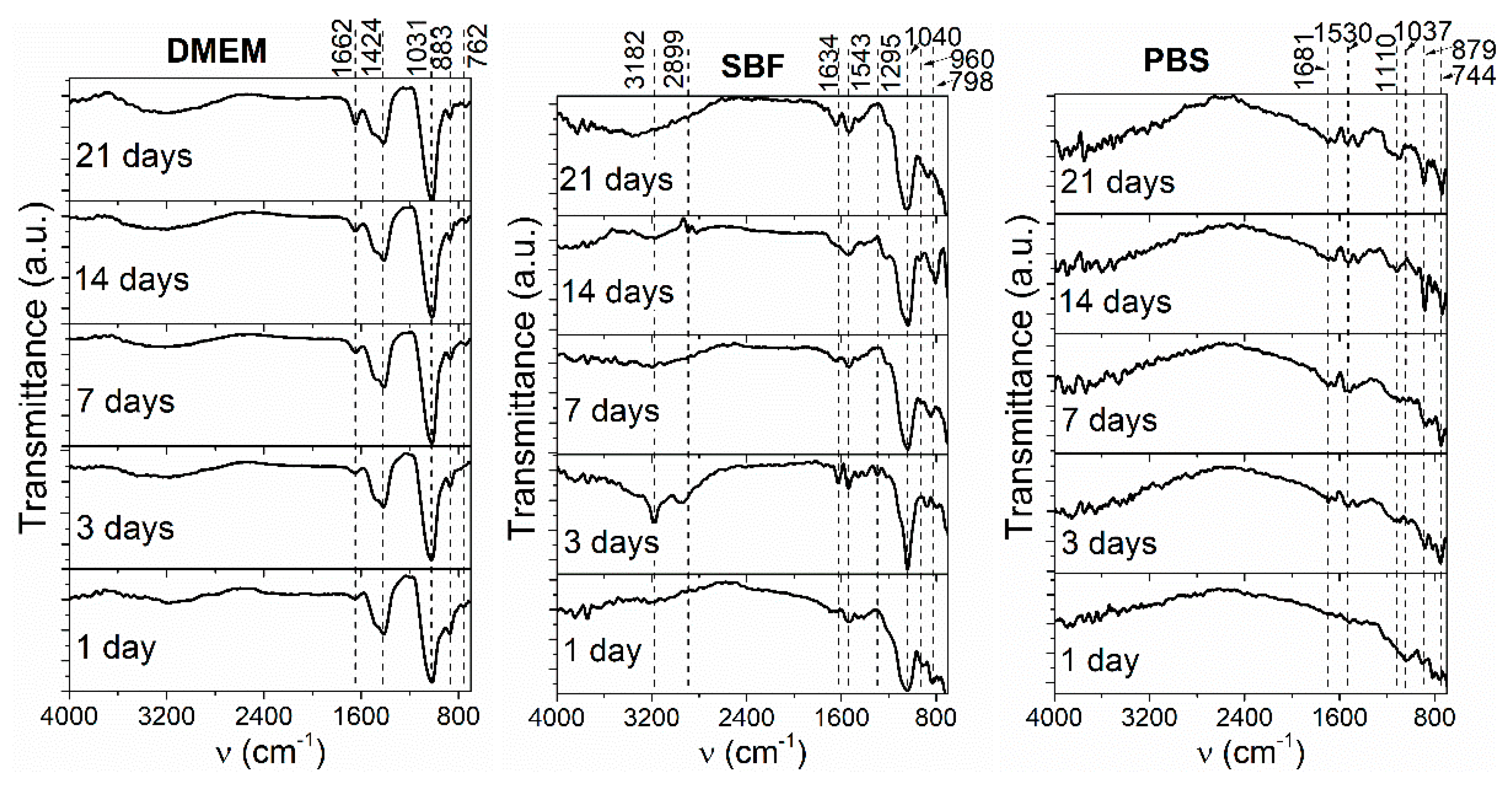
3.4. FTIR after Immersion in DMEM, SBF, and PBS
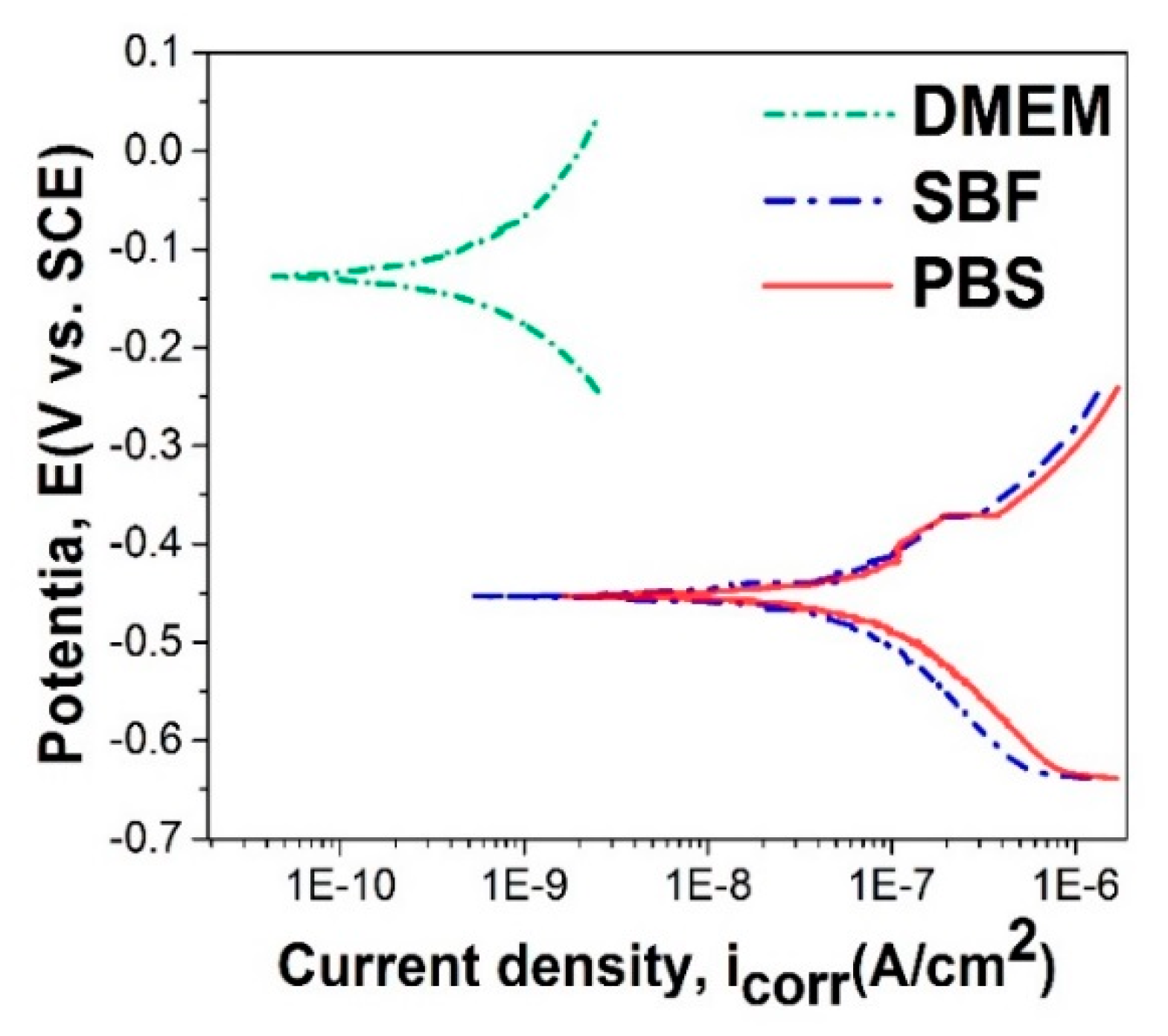
3.5. In Vitro Electrochemical Performance of SiC–HAp Coatings in DMEM, SBF, and PBS
- Coated samples tested in DMEM have a more electropositive value of corrosion potential (Ecorr), indicating a better electrochemical behavior, while in PBS and SBF the values are similar;
- The lowest value of the corrosion current density (icorr) was recorded for coated samples tested in DMEM, followed by those in SBF and then those in PBS;
- The polarization resistance (Rp) is higher for coated samples tested in DMEM, followed by those in PBS and those in SBF.
4. Conclusions
- The in vitro electrochemical measurements highlighted that the Ti6Al4V coated with HAp + SiC films had a better electrochemical behavior in DMEM as compared with PBS and SBF, which present a higher Cl− content as compared with DMEM;
- The immersion assays performed for 21 days showed that in SBF and PBS media, the HAp + SiC films presented signs of degradation and the reported data were comparable (mass loss of ~0.13 mg on day 21), while in DMEM the coatings demonstrated good mineralization abilities (a gained mass of ~0.53 mg on day 21);
- The XRD patterns achieved on the HAp + SiC coatings after their immersion pointed out the appearance of peaks characteristic to a newly formed apatite-based phase, but which either disappear or overlap the substrate over the immersion period;
- FTIR analysis performed on the HAp + SiC coatings showed differences in the spectra shape with respect to the immersion testing media, and characteristic phosphate bands were registered in all three media.
Author Contributions
Funding
Conflicts of Interest
References
- Poitout, D.G. Biomechanics and Biomaterials in Orthopedics; Springer: London, UK, 2016; ISBN 978-1-84882-663-2. [Google Scholar]
- Sugawara, E.; Nikaido, H. Properties of AdeABC and AdeIJK efflux systems of Acinetobacter baumannii compared with those of the AcrAB-TolC system of Escherichia coli. Antimicrob. Agents Chemother. 2014, 58, 7250–7257. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, E.P.; Bazaka, K.; Crawford, R.J. New Functional Biomaterials for Medicine and Healthcare; Woodhead Publishing: Cambridge, UK, 2014; ISBN 9781782422655. [Google Scholar]
- Bhat, S.; Kumar, A. Biomaterials and bioengineering tomorrow’s healthcare. Biomatter 2013, 3, e24717. [Google Scholar] [CrossRef] [PubMed]
- Rau, J.-V.; Cacciotti, I.; Laureti, S.; Fosca, M.; Varvaro, G.; Latini, A. Bioactive, nanostructured Si-substituted hydroxyapatite coatings on titanium prepared by pulsed laser deposition. J. Biomed. Mater. Res. Part B Appl. Biomater. 2015, 103, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V. Calcium orthophosphates: Occurrence, properties, biomineralization, pathological calcification and biomimetic applications. Biomatter 2011, 1, 121–164. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, A.-C.; Stan, G.E.; Maidaniuc, A.; Miculescu, M.; Antoniac, I.V.; Ciocoiu, R.-C.; Voicu, Ș.I.; Mitran, V.; Cîmpean, A.; Miculescu, F. Naturally-derived biphasic calcium phosphates through increased phosphorus-based reagent amounts for biomedical applications. Materials 2019, 12, 381. [Google Scholar] [CrossRef]
- Gómez-Morales, J.; Iafisco, M.; Delgado-López, J.M.; Sarda, S.; Drouet, C. Progress on the preparation of nanocrystalline apatites and surface characterization: Overview of fundamental and applied aspects. Prog. Cryst. Growth Charact. Mater. 2013, 59, 1–46. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphates (CaPO4): Occurrence and properties. Morphologie 2017, 101, 125–142. [Google Scholar] [CrossRef]
- Kasemo, B.; Lausmaa, J. Material-tissue interfaces: The role of surface properties and processes. Environ. Health Perspect. 1994, 102, 41–45. [Google Scholar] [CrossRef]
- Karamian, E.; Abdellahi, M.; Khandan, A.; Abdellah, S. Introducing the fluorine doped natural hydroxyapatite-titania nanobiocomposite ceramic. J. Alloy. Compd. 2016, 679, 375–383. [Google Scholar] [CrossRef]
- Dasgupta, S.; Banerjee, S.S.; Bandyopadhyay, A.; Bose, S. Zn- and Mg-doped hydroxyapatite nanoparticles for controlled release of protein. Langmuir 2010, 26, 4958–4964. [Google Scholar] [CrossRef]
- Roy, M.; Bandyopadhyay, A.; Bose, S. Induction plasma sprayed Sr and Mg doped nano hydroxyapatite coatings on Ti for bone implant. J. Biomed. Mater. Res. Part B Appl. Biomater. 2011, 99, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.K.; Bhattacharjee, B.N.; Parkash, O.; Kumar, D.; Rai, S.B. Mg-doped hydroxyapatite nanoplates for biomedical applications: A surfactant assisted microwave synthesis and spectroscopic investigations. J. Alloy. Compd. 2014, 614, 283–288. [Google Scholar] [CrossRef]
- Mansour, S.F.; El-dek, S.I.; Dorozhkin, S.V.; Ahmed, M.K. Physico-mechanical properties of Mg and Ag doped hydroxyapatite/chitosan biocomposites. New J. Chem. 2017, 41, 13773–13783. [Google Scholar] [CrossRef]
- Gozalian, A.; Behnamghader, A.; Daliri, M.; Moshkforoush, A. Synthesis and thermal behavior of Mg-doped calcium phosphate nanopowders via the sol gel method. Sci. Iran. 2011, 18, 1614–1622. [Google Scholar] [CrossRef]
- Kaygili, O.; Keser, S. Sol–gel synthesis and characterization of Sr/Mg, Mg/Zn and Sr/Zn co-doped hydroxyapatites. Mater. Lett. 2015, 141, 161–164. [Google Scholar] [CrossRef]
- Robles-Águila, M.J.; Reyes-Avendaño, J.A.; Mendoza, M.E. Structural analysis of metal-doped (Mn, Fe, Co, Ni, Cu, Zn) calcium hydroxyapatite synthetized by a sol-gel microwave-assisted method. Ceram. Int. 2017, 43, 12705–12709. [Google Scholar] [CrossRef]
- Samani, S.; Hossainalipour, S.M.; Tamizifar, M.; Rezaie, H.R. In vitro antibacterial evaluation of sol-gel-derived Zn-, Ag-, and (Zn + Ag)-doped hydroxyapatite coatings against methicillin-resistant Staphylococcus aureus. J. Biomed. Mater. Res. Part A 2013, 101, 222–230. [Google Scholar] [CrossRef]
- Yuan, Q.; Wu, J.; Qin, C.; Xu, A.; Zhang, Z.; Lin, Y.; Chen, Z.; Lin, S.; Yuan, Z.; Ren, X.; et al. One-pot synthesis and characterization of Zn-doped hydroxyapatite nanocomposites. Mater. Chem. Phys. 2017, 199, 122–130. [Google Scholar] [CrossRef]
- Prosolov, K.A.; Belyavskaya, O.A.; Rau, J.V.; Prymak, O.; Epple, M.; Sharkeev, Y.P. Deposition of polycrystalline zinc substituted hydroxyapatite coatings with a columnar structure by RF magnetron sputtering: Role of in-situ substrate heating. J. Phys. Conf. Ser. 2018, 1115, 032077. [Google Scholar] [CrossRef]
- Vranceanu, D.M.; Parau, A.C.; Cotrut, C.M.; Kiss, A.E.; Constantin, L.R.; Braic, V.; Vladescu, A. In vitro evaluation of Ag doped hydroxyapatite coatings in acellular media. Ceram. Int. 2019, 45, 11050–11061. [Google Scholar] [CrossRef]
- Huang, Y.; Ding, Q.; Pang, X.; Han, S.; Yan, Y. Corrosion behavior and biocompatibility of strontium and fluorine co-doped electrodeposited hydroxyapatite coatings. Appl. Surf. Sci. 2013, 282, 456–462. [Google Scholar] [CrossRef]
- Manzano, M.; Lozano, D.; Arcos, D.; Portal-Núñez, S.; la Orden, C.L.; Esbrit, P.; Vallet-Regí, M. Comparison of the osteoblastic activity conferred on Si-doped hydroxyapatite scaffolds by different osteostatin coatings. Acta Biomater. 2011, 7, 3555–3562. [Google Scholar] [CrossRef] [PubMed]
- Vallet-Regí, M.; Arcos, D. Silicon substituted hydroxyapatites. A method to upgrade calcium phosphate based implants. J. Mater. Chem. 2005, 15, 1509–1516. [Google Scholar] [CrossRef]
- Qiu, Z.-Y.; Noh, I.-S.; Zhang, S.-M. Silicate-doped hydroxyapatite and its promotive effect on bone mineralization. Front. Mater. Sci. 2013, 7, 40–50. [Google Scholar] [CrossRef]
- Martínez-Vázquez, F.J.; Cabañas, M.V.; Paris, J.L.; Lozano, D.; Vallet-Regí, M. Fabrication of novel Si-doped hydroxyapatite/gelatine scaffolds by rapid prototyping for drug delivery and bone regeneration. Acta Biomater. 2015, 15, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Astala, R.; Calderín, L.; Yin, X.; Stott, M.J. Ab initio simulation of Si-doped hydroxyapatite. Chem. Mater. 2006, 18, 413–422. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, J.; Xin, H.; Wang, X.; Zhang, L.; He, F.; Liu, Q.; Zhang, W. Improved dispersion of SiC whisker in nano hydroxyapatite and effect of atmospheres on sintering of the SiC whisker reinforced nano hydroxyapatite composites. Mater. Sci. Eng. C 2018, 91, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, N.C.; Betts, F.; Posner, A.S. Effect of carbonate and biological macromolecules on formation and properties of hydroxyapatite. Calcif. Tissue Res. 1975, 18, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Best, S.M.; Bonfield, W.; Gibson, I.R.; Hing, K.A.; Damien, E.; Revell, P.A. A comparative study on the in vivo behavior of hydroxyapatite and silicon substituted hydroxyapatite granules. J. Mater. Sci. Mater. Med. 2002, 13, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Yamaguchi, S. Novel bioactive materials developed by simulated body fluid evaluation: Surface-modified Ti metal and its alloys. Acta Biomater. 2016, 44, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Vranceanu, D.M.; Cotrut, C.M.; Bramowicz, M.; Titorencu, I.; Kulesza, S.; Kiss, A.; Berbecaru, A.; Pruna, V.; Branzei, M.; Vladescu, A. Osseointegration of sputtered SiC-added hydroxyapatite for orthopaedic applications. Ceram. Int. 2016, 42, 10085–10093. [Google Scholar] [CrossRef]
- Vladescu, A.; Birlik, I.; Braic, V.; Toparli, M.; Celik, E.; Ak Azem, F. Enhancement of the mechanical properties of hydroxyapatite by SiC addition. J. Mech. Behav. Biomed. Mater. 2014, 40, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Azem, F.A.; Kiss, A.; Birlik, I.; Braic, V.; Luculescu, C.; Vladescu, A. The corrosion and bioactivity behavior of SiC doped hydroxyapatite for dental applications. Ceram. Int. 2014, 40, 15881–15887. [Google Scholar] [CrossRef]
- Mertz, W. Human requirements: Basic and optimal. Ann. N. Y. Acad. Sci. 1972, 199, 191–201. [Google Scholar] [CrossRef]
- Carlisle, E.M. Silicon as a trace nutrient. Sci. Total Environ. 1988, 73, 95–106. [Google Scholar] [CrossRef]
- Carlisle, E.M. Silicon as an essential trace element in animal nutrition. In Silicon Biochemistry Ciba Foundation Symposium 121; Evered, D., O’Connor, M., Eds.; John Wiley & Sons: Chichester, UK, 2007; pp. 123–139. [Google Scholar]
- Schwarz, K.; Milne, D.B. Growth-promoting effects of silicon in rats. Nature 1972, 239, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, G.V. Reference values for the concentrations of As, Cd, Co, Cr, Cu, Fe, I, Hg, Mn, Mo, Ni, Pb, Se, and Zn in selected human tissues and body fluids. Biol. Trace Elem. Res. 1987, 12, 263–295. [Google Scholar] [CrossRef] [PubMed]
- Jugdaohsingh, R.; Pedro, L.D.; Watson, A.; Powell, J.J. Silicon and boron differ in their localization and loading in bone. Bone Rep. 2015, 1, 9–15. [Google Scholar] [CrossRef]
- Zhuoer, H.; Environmental, G.; Republic, P. Silicon measurement in bone and other tissues by electrothermal atomic absorption spectrometry. J. Anal. At. Spectrom. 1994, 9, 11–15. [Google Scholar] [CrossRef]
- Carlisle, E.M. Silicon: A requirement in bone formation independent of vitamin D1. Calcif. Tissue Int. 1981, 33, 27–34. [Google Scholar] [CrossRef]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- ISO/FDIS 23317 Implants for Surgery—In Vitro Evaluation for Apatite-Forming Ability of Implant Materials; International Organization for Standardization (ISO): Geneva, Switzerland, 2007.
- Faure, J.; Balamurugan, A.; Benhayoune, H.; Torres, P.; Balossier, G.; Ferreira, J.M.F. Morphological and chemical characterisation of biomimetic bone like apatite formation on alkali treated Ti6Al4V titanium alloy. Mater. Sci. Eng. C 2009, 29, 1252–1257. [Google Scholar] [CrossRef]
- Rohanová, D.; Boccaccini, A.R.; Horkavcová, D.; Bozděchová, P.; Bezdička, P.; Častorálová, M. Is non-buffered DMEM solution a suitable medium for in vitro bioactivity tests? J. Mater. Chem. B 2014, 2, 5068–5076. [Google Scholar] [CrossRef]
- Vladescu, A.; Vranceanu, D.M.; Kulesza, S.; Ivanov, A.N.; Bramowicz, M.; Fedonnikov, A.S.; Braic, M.; Norkin, I.A.; Koptyug, A.; Kurtukova, M.O.; et al. Influence of the electrolyte’s pH on the properties of electrochemically deposited hydroxyapatite coating on additively manufactured Ti64 alloy. Sci. Rep. 2017, 7, 16819. [Google Scholar] [CrossRef]
- Li, P.; Ohtsuki, C.; Kokubo, T.; Nakanishi, K.; Soga, N.; Nakamura, T.; Yamamuro, T. Effects of ions in aqueous media on hydroxyapatite induction by silica gel and its relevance to bioactivity of bioactive glasses and glass-ceramics. J. Appl. Biomater. 1993, 4, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Kushitani, H.; Ohtsuki, C.; Sakka, S.; Yamamuro, T. Effects of ions dissolved from bioactive glass-ceramic on surface apatite formation. J. Mater. Sci. Mater. Med. 1993, 4, 1–4. [Google Scholar] [CrossRef]
- Wu, C.; Xiao, Y. Evaluation of the in vitro bioactivity of bioceramics. Bone Tissue Regen. Insights 2009, 2009, 25–29. [Google Scholar] [CrossRef]
- De Aza, P.N.; Guitian, F.; Merlos, A.; Lora-Tamayo, E.; De Aza, S. Bioceramics—Simulated body fluid interfaces: pH and its influence of hydroxyapatite formation. J. Mater. Sci. Mater. Med. 1996, 7, 399–402. [Google Scholar] [CrossRef]
- Hench, L.L. Bioceramics: From concept to clinic. J. Am. Ceram. Soc. 1991, 74, 1487–1510. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Inorganic chemistry of the dissolution phenomenon: The dissolution mechanism of calcium apatites at the atomic (ionic) level. Comments Inorg. Chem. 1999, 20, 285–299. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. A review on the dissolution models of calcium apatites. Prog. Cryst. Growth Charact. Mater. 2002, 44, 45–61. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphates (CaPO4): Occurrence and properties. Prog. Biomater. 2016, 5, 9–70. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z. Calcium phosphate-based osteoinductive materials. Chem. Rev. 2008, 108, 4742–4753. [Google Scholar] [CrossRef] [PubMed]
- Šupová, M. Substituted hydroxyapatites for biomedical applications: A review. Ceram. Int. 2015, 41, 9203–9231. [Google Scholar] [CrossRef]
- Orly, I.; Gregoire, M.; Menanteau, J.; Heughebaert, M.; Kerebel, B. Chemical changes in hydroxyapatite biomaterial under in vivo and in vitro biological conditions. Calcif. Tissue Int. 1989, 45, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Berndt, C.C.; Gross, K.A.; Kucuk, A. Material fundamentals and clinical performance of plasma-sprayed hydroxyapatite coatings: A review. J. Biomed. Mater. Res. 2001, 58, 570–592. [Google Scholar] [CrossRef] [PubMed]
- Pietak, A.M.; Reid, J.W.; Stott, M.J.; Sayer, M. Silicon substitution in the calcium phosphate bioceramics. Biomaterials 2007, 28, 4023–4032. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, J.H.; Kim, Y.T.; Riu, D.H.; Jung, S.J.; Lee, Y.J.; Chung, S.C.; Kim, Y.H. Synthesis of Si, Mg substituted hydroxyapatites and their sintering behaviors. Biomaterials 2003, 24, 1389–1398. [Google Scholar] [CrossRef]
- Barrère, F.; van der Valk, C.M.; Dalmeijer, R.A.J.; van Blitterswijk, C.A.; de Groot, K.; Layrolle, P. In vitro and in vivo degradation of biomimetic octacalcium phosphate and carbonate apatite coatings on titanium implants. J. Biomed. Mater. Res. Part A 2003, 64, 378–387. [Google Scholar] [CrossRef]
- Raynaud, S.; Champion, E.; Lafon, J.P.; Bernache-Assollant, D. Calcium phosphate apatites with variable Ca/P atomic ratio III. Mechanical properties and degradation in solution of hot pressed ceramics. Biomaterials 2002, 23, 1081–1089. [Google Scholar] [CrossRef]
- Wenisch, S.; Stahl, J.-P.; Horas, U.; Heiss, C.; Kilian, O.; Trinkaus, K.; Hild, A.; Schnettler, R. In vivo mechanisms of hydroxyapatite ceramic degradation by osteoclasts: Fine structural microscopy. J. Biomed. Mater. Res. 2003, 67, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V. Dissolution mechanism of calcium apatites in acids: A review of literature. World J. Methodol. 2012, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Descamps, M.; Dejou, J.; Koubi, G.; Hardouin, P.; Lemaitre, J.; Proust, J.-P. The biodegradation mechanism of calcium phosphate biomaterials in bone. J. Biomed. Mater. Res. 2002, 63, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Yao, H.; Li, G.; Li, Y. Biomimetic synthesis of needle-like nano-hydroxyapatite templated by double-hydrophilic block copolymer. J. Mater. Sci. 2010, 45, 1930–1936. [Google Scholar] [CrossRef]
- Wongwitwichot, P.; Kaewsrichan, J.; Chua, K.H.; Ruszymah, B.H.I. Comparison of TCP and TCP/HA hybrid scaffolds for osteoconductive activity. Open Biomed. Eng. J. 2010, 4, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Johnson, I.; Liu, H. A study on factors affecting the degradation of magnesium and a magnesium-yttrium alloy for biomedical applications. PLoS ONE 2013, 8, e65603. [Google Scholar] [CrossRef] [PubMed]
- Díaz, E.; Sandonis, I.; Valle, M.B. In vitro degradation of poly(caprolactone)/nHA composites. J. Nanomater. 2014, 2014, 802435. [Google Scholar] [CrossRef]
- Sumathi, S.; Gopal, B. In vitro degradation of multisubstituted hydroxyapatite and fluorapatite in the physiological condition. J. Cryst. Growth 2015, 422, 36–43. [Google Scholar] [CrossRef]
- Sheikh, Z.; Zhang, Y.L.; Grover, L.; Merle, G.E.; Tamimi, F.; Barralet, J. In vitro degradation and in vivo resorption of dicalcium phosphate cement based grafts. Acta Biomater. 2015, 26, 338–346. [Google Scholar] [CrossRef]
- Porter, A.E.; Best, S.M.; Bonfield, W. Ultrastructural comparison of hydroxyapatite and silicon-substituted hydroxyapatite for biomedical applications. J. Biomed. Mater. Res. Part A 2004, 68, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K. A bound form of silicon in glycosaminoglycans and polyuronides. Proc. Natl. Acad. Sci. USA 1973, 70, 1608–1612. [Google Scholar] [CrossRef] [PubMed]
- Hench, L.L.; Xynos, I.D.; Polak, J.M. Bioactive glasses for in situ tissue regeneration. J. Biomater. Sci. Polym. Ed. 2004, 15, 543–562. [Google Scholar] [CrossRef] [PubMed]
- Drouet, C. Apatite formation: Why it may not work as planned, and how to conclusively identify apatite compounds. Biomed. Res. Int. 2013, 2013, 490946. [Google Scholar] [CrossRef] [PubMed]
- Balas, F.; Pérez-Pariente, J.; Vallet-Regí, M. In vitro bioactivity of silicon-substituted hydroxyapatites. J. Biomed. Mater. Res. Part A 2003, 66, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, B.; Alshemary, A.Z.; Evis, Z. Co-doped hydroxyapatites as potential materials for biomedical applications. Microchem. J. 2019, 144, 443–453. [Google Scholar] [CrossRef]
- Ouadfel, M.A.; Keffous, A.; Brighet, A.; Gabouze, N.; Hadjersi, T.; Cheriet, A.; Kechouane, M.; Boukezzata, A.; Boukennous, Y.; Belkacem, Y.; et al. Si-rich a-Si1−xCx thin films by d.c. magnetron co-sputtering of silicon and silicon carbide: Structural and optical properties. Appl. Surf. Sci. 2013, 265, 94–100. [Google Scholar] [CrossRef]
- Bianco, A.; Cacciotti, I.; Lombardi, M.; Montanaro, L. Si-substituted hydroxyapatite nanopowders: Synthesis, thermal stability and sinterability. Mater. Res. Bull. 2009, 44, 345–354. [Google Scholar] [CrossRef]
- Czikó, M.; Bogya, E.-S.; Barabás, R.; Bizo, L.; Stefan, R. In vitro biological activity comparison of some hydroxyapatite-based composite materials using simulated body fluid. Open Chem. 2013, 11, 400372. [Google Scholar] [CrossRef]
- Rehman, I.; Bonfield, W. Characterization of hydroxyapatite and carbonated apatite by photo acoustic FTIR spectroscopy. J. Mater. Sci. Mater. Med. 1997, 8, 1–4. [Google Scholar] [CrossRef]
- Nakata, K.; Kubo, T.; Numako, C.; Onoki, T.; Nakahira, A. Synthesis and characterization of silicon-doped hydroxyapatite. Mater. Trans. 2009, 50, 1046–1049. [Google Scholar] [CrossRef]
- Narayan, R.; Bose, S.; Bandyopadhyay, A. Biomaterials Science: Processing, Properties, and Applications V; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; ISBN 9781119190134. [Google Scholar]
- Berzina-Cimdina, L.; Borodajenko, N. Research of calcium phosphates using fourier transform infrared spectroscopy. In Infrared Spectroscopy—Materials Science, Engineering and Technology; IntechOpen: London, UK, 2012; pp. 123–148. [Google Scholar]
- Durdu, S.; Deniz, Ö.F.; Kutbay, I.; Usta, M. Characterization and formation of hydroxyapatite on Ti6Al4V coated by plasma electrolytic oxidation. J. Alloy. Compd. 2013, 551, 422–429. [Google Scholar] [CrossRef]
- Mohan, L.; Durgalakshmi, D.; Geetha, M.; Narayanan, T.S.N.S.; Asokamani, R. Electrophoretic deposition of nanocomposite (HAp + TiO2) on titanium alloy for biomedical applications. Ceram. Int. 2012, 38, 3435–3443. [Google Scholar] [CrossRef]
- Ślósarczyk, A.; Paszkiewicz, Z.; Zima, A. The effect of phosphate source on the sintering of carbonate substituted hydroxyapatite. Ceram. Int. 2010, 36, 577–582. [Google Scholar] [CrossRef]
- Barinov, S.M.; Rau, J.V.; Cesaro, S.N.; Ďurišin, J.; Fadeeva, I.V.; Ferro, D.; Medvecky, L.; Trionfetti, G. Carbonate release from carbonated hydroxyapatite in the wide temperature rage. J. Mater. Sci. Mater. Med. 2006, 17, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V. Calcium orthophosphate-based biocomposites and hybrid biomaterials. J. Mater. Sci. 2009, 44, 2343–2387. [Google Scholar] [CrossRef]
- Krishna, D.S.R.; Siddharthan, A.; Seshadri, S.K.; Kumar, T.S.S. A novel route for synthesis of nanocrystalline hydroxyapatite from eggshell waste. J. Mater. Sci. Mater. Med. 2007, 18, 1735–1743. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Composition | Human Blood Plasma | Acellular Media | |||
|---|---|---|---|---|---|
| SBF | DMEM [46,47] | PBS | |||
| Ion Concentration (mM) | Na+ | 142 | 142 | 154.5 | 137 |
| K+ | 5 | 5 | 5.4 | 2.7 | |
| Mg2+ | 1.5 | 1.5 | 0.8 | − | |
| Ca2+ | 2.5 | 2.5 | 1.8 | − | |
| Cl− | 103 | 148.8 | 120.5 | − | |
| HCO3− | 27 | 4.2 | 44 | − | |
| HPO4− | 1 | 1 | 0.9 | 10 | |
| SO42− | 0.5 | − | 0.8 | − | |
| Buffer | TRIS and 1 M HCl | No | NaHCO3 | ||
| pH | 7.2–7.4 | 7.4 | 7.4 | 7.4 | |
| Amino Acids | N/A | No | 0.084 g/L L-Arginine · HCl | No | |
| 0.042 g/L L-Histidine · HCl · H2O | |||||
| 0.105 g/L L-Isoleucine | |||||
| 0.105 g/L L-Leucine | |||||
| 0.146 g/L L-Lysine · HCl | |||||
| 0.03 g/L L-Methionine | |||||
| 0.066 g/L L-Phenylalanine | |||||
| 0.042 g/L L-Serine | |||||
| 0.095 g/L L-Threonine | |||||
| 0.016 g/L L-Tryptophan | |||||
| 0.10379 g/L L-Tyrosine, 2Na · 2H2O | |||||
| 0.094 g/L L-Valine | |||||
| Vitamins | N/A | No | 0.004 g/L Choline Chloride | No | |
| 0.004 g/L Folic Acid | |||||
| 0.0072 g/L Myo-Inositol | |||||
| 0.004 g/L Niacinamide | |||||
| 0.004 g/L D-Pantothenic Acid · ½Ca | |||||
| 0.004 g/L Pyridoxal · HC | |||||
| 0.0004 g/L Riboflavin | |||||
| 0.004 g/L Thiamine · HCl | |||||
| Other Components | N/A | No | 1.0 g/L D-Glucose | No | |
| 0.0159 g/L Phenol Red · Na | |||||
| 0.11 g/L Pyruvic Acid · Na | |||||
| Electrolyte | Ecorr (mV) | icorr (nA/cm2) | Rp (kΩ∙cm2) |
|---|---|---|---|
| DMEM | −126 | 1.90 | 54,652.6 |
| SBF | −453 | 89.73 | 474.4 |
| PBS | −454 | 108.02 | 364.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlădescu, A.; Pârâu, A.; Pană, I.; Cotruț, C.M.; Constantin, L.R.; Braic, V.; Vrânceanu, D.M. In Vitro Activity Assays of Sputtered HAp Coatings with SiC Addition in Various Simulated Biological Fluids. Coatings 2019, 9, 389. https://doi.org/10.3390/coatings9060389
Vlădescu A, Pârâu A, Pană I, Cotruț CM, Constantin LR, Braic V, Vrânceanu DM. In Vitro Activity Assays of Sputtered HAp Coatings with SiC Addition in Various Simulated Biological Fluids. Coatings. 2019; 9(6):389. https://doi.org/10.3390/coatings9060389
Chicago/Turabian StyleVlădescu, Alina, Anca Pârâu, Iulian Pană, Cosmin M. Cotruț, Lidia R. Constantin, Viorel Braic, and Diana M. Vrânceanu. 2019. "In Vitro Activity Assays of Sputtered HAp Coatings with SiC Addition in Various Simulated Biological Fluids" Coatings 9, no. 6: 389. https://doi.org/10.3390/coatings9060389
APA StyleVlădescu, A., Pârâu, A., Pană, I., Cotruț, C. M., Constantin, L. R., Braic, V., & Vrânceanu, D. M. (2019). In Vitro Activity Assays of Sputtered HAp Coatings with SiC Addition in Various Simulated Biological Fluids. Coatings, 9(6), 389. https://doi.org/10.3390/coatings9060389