2. Materials and Methods
2.1. Materials
ε-Caprolactone (CL, 97% MW = 114.14 g/mol), Stannous Octoate (Sn(Oct)2, 92.5–100%, MW = 405.12 g/mol), 2-hydroxyethyl methacrylate (HEMA, 97%, MW = 130.14 g/mol), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPA, ≥97%, MW = 279.38 g/mol), 4,4′-azobis(cyanovaleric acid) (ACVA, ≥98%, MW = 280.2 g/mol), Rhodamine B (≥95%, MW = 479.01 g/mol), Methacryloyl Chloride (MAC, ≥97%, MW = 104.53 g/mol), Phenol (MW = 94.11 g/mol), Potassium Tert Butoxide (≥98%, MW = 112.21 g/mol), 4 Chloro 3 Nitropyridine (90%, MW = 158.54 g/mol), Diethyl Ether (≥99.7%, MW = 74.12 g/mol), Sodium Sulphate (≥99.0%, MW = 142.04 g/mol), Hydrochloric acid (≥99.8%, MW = 36.46 g/mol), Stannous Chloride (98%, MW = 189.62 g/mol), Sodium hydroxide solution (50% in H2O, MW = 40.00 g/mol), Salicylaldehyde (≥98%, MW = 122.12 g/mol), Potassium carbonate (≥99%, MW = 138.21 g/mol), Propargyl bromide solution (80% in toluene, MW = 118.96 g/mol), Sodium borohydride (MW = 37.83 g/mol), Sodium bicarbonate (≥99.7%, MW = 84.01 g/mol), Hexane (95%, MW = 86.18 g/mol), Acetyl chloride (98%, MW = 78.50 g/mol), Triethylamine (TEA, ≥99.5%, MW = 101.19 g/mol), 4-(Dimethylamino)pyridine (DMAP, ≥99.9%, MW = 122.17 g/mol), 2-methacryloyloxyethylphosphorylcholine (MPC, 97%, MW = 295.27 g/mol), Ethanol (EtOH, ≥99.8% MW = 46.07 g/mol), Dimethyl Sulfoxide (DMSO, MW = 78.13 g/mol), Methanol (MeOH, MW = 32.04 g/mol), Acetonitrile (ACN, MW = 41.05), Dichloromethane (DCM, ≥99.9%, MW = 84.93 g/mol) and DCM anhydrous (dried over molecular sieves), Tetra Deuterated Methanol (MeOD-d4, ≥99.8%, MW = 36.07 g/mol), Deuterated Chloroform (CDCl3, 99.8%, MW = 120.38 g/mol), Sodium Sulphate (Na2SO4, ≥99.5%, MW = 82.03 g/mol), Ethyl Acetate (MW = 88.11 g/mol), Toluene (99.8% MW = 92.14 g/mol), Dimethylformamide (DMF, MW = 73,09 g/mol), Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA, MW = 530.63 g/mol) were purchased from Sigma Aldrich and used as received unless specifically noted.
All reagents and solvents were purchased from commercial sources and used without any further purification. The reactions were carried out under atmospheric air unless otherwise indicated, such as moisture-sensitive ones, for which a static nitrogen atmosphere was used. The reactions were monitored mostly by thin-layer chromatography (TLC), performed on Merck Kieselgel 60 F254 plates. Visualization was accomplished by UV irradiation at 254 nm and subsequently by treatment with the alkaline KMnO4 reactant or with a phosphomolybdic reagent. When necessary, crude compounds were purified through silica gel column chromatography (230–400 mesh). The NMR spectra were recorded on a Bruker 400 spectrometer (1H NMR, 400 MHz; 13C NMR, 100 MHz). The spectra were registered at room temperature in CDCl3 or MeOD-d4, unless otherwise indicated, with tetramethylsilane (TMS, δ = 0.0 ppm) used as the internal standard. The chemical shifts are reported as δ values in parts per million (ppm) in comparison to the internal standards; the coupling constants J are reported in Hz. The following abbreviations were used for spin multiplicity: s = singlet, d = doublet, t = triplet, dd = double doublet, m = multiplet, br s = broad singlet. Electrospray ionization mass spectrometry (ESI-MS) analyses were conducted on an Esquire 3000 Plus spectrometer, using methanol as a solvent. FT-IR spectrometers analysis were conducted on a Varian 640-IR FT-IR spectrometer.
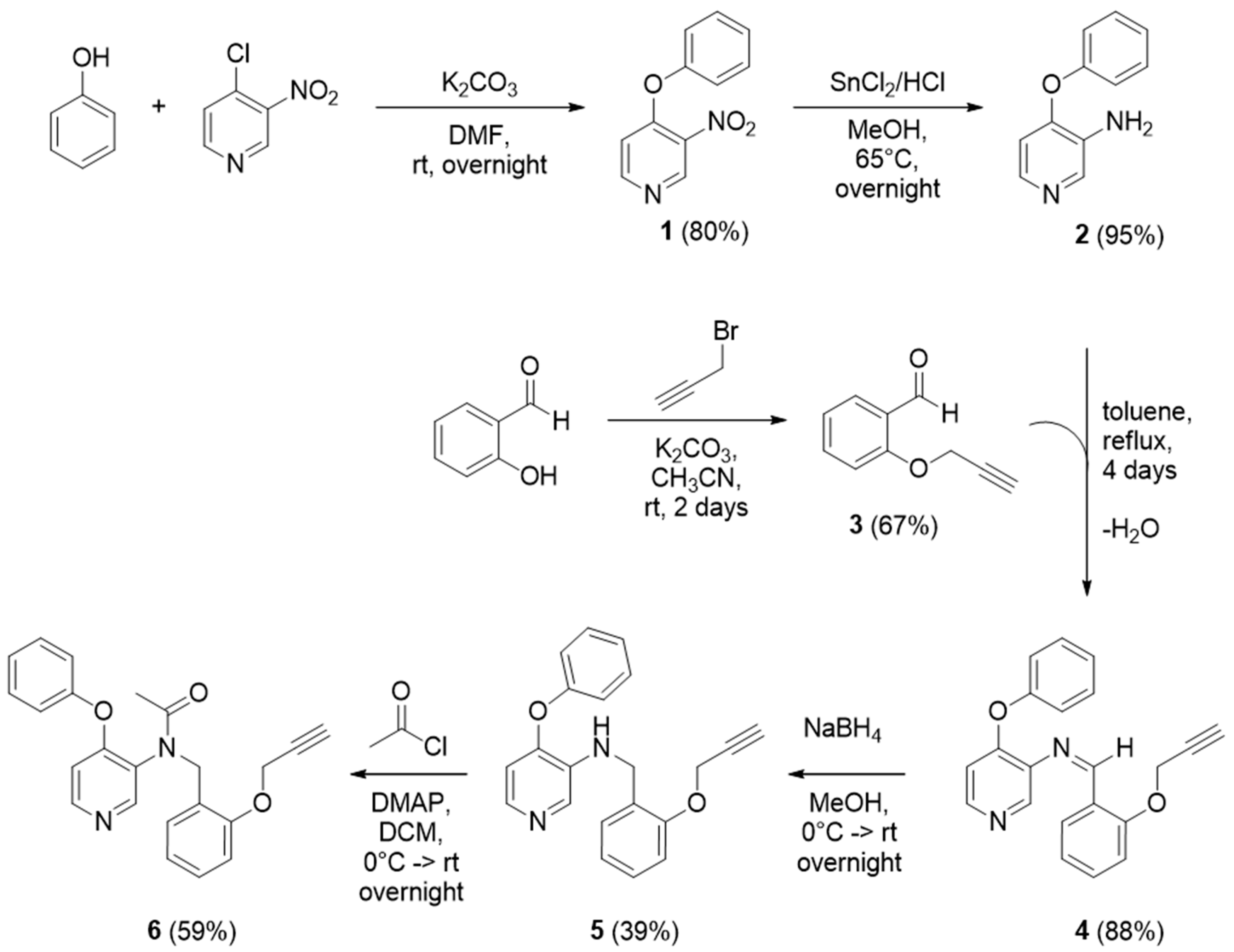
2.2. Synthesis of 3-Nitro-4-phenoxypyridine (1)
For the synthesis of nitro-4-phenoxypyridine,
1 in
Scheme 1, 4.0 g of phenol (1.3 eq., 42.5 mmol) dissolved in 20 mL of DMF were added to a solution of 7.7 g (1.8 eq., 55.8 mmol) of potassium carbonate in 25 mL of DMF at room temperature. After 10 min, 5.0 g (1 eq., 31.5 mmol) of 4-chloro-3-nitropyridine in 25 mL of DMF were slowly added to the solution under magnetic stirring. The mixture was continuously stirred overnight at room temperature. The product was then extracted three times with diethyl ether (3 × 50 mL), washed with brine (3 × 50 mL), and dried over anhydrous sodium sulphate. After filtration, product
2 was recovered by evaporating the solvent at reduced pressure. The orange solid obtained (5.5 g, 80% yield) was analyzed through
1H-NMR (400 MHz, Chloroform-
d) δ 9.11 (s, 1H), 8.53 (d,
J = 5.9 Hz, 1H), 7.53–7.45 (m, 2H), 7.37–7.31 (m, 1H), 7.17–7.12 (m, 2H), 6.78 (d,
J = 5.8 Hz, 1H).
2.3. Synthesis of 4-Phenoxy-3-pyridinamine (2)
For the synthesis of 4-phenoxy-3-pyridinamine 2, 2.8 g of 1 (1 eq., 13 mmol) were dissolved in 20 mL of methanol and added to a solution of 15 g (6.8 eq., 79 mmol) of SnCl2 in 16 mL of 6 N HCl. The mixture was then refluxed overnight at 65 °C. After that, the product was concentrated at reduced pressure in order to remove the methanol. The pH of the residue was increased to 13 with 6 N NaOH and then the precipitated Sn(OH)2 was filtered off over celite, washing with ethyl acetate (100 mL). After reducing the solvent volume with a rotavapor, the product was extracted three times with ethyl acetate/NaHCO3 (aq) (3 × 50 mL). The organic layer was dried over anhydrous sodium sulphate, filtrated and evaporated at reduced pressure to give the resulting product 2 as a pure yellow solid (2.3 g, 95% yield), which was analyzed through 1H-NMR (400 MHz, Chloroform-d) δ 8.15 (s, 1H), 7.89 (d, J = 5.4 Hz, 1H), 7.45–7.36 (m, 2H), 7.25–7.19 (m, 1H), 7.11–7.05 (m, 2H), 6.58 (d, J = 5.4 Hz, 1H), 4.3 (bs, 2H).
2.4. Synthesis of 2-(Prop-2-yn-1-yloxy)benzaldehyde (3)
To a stirring solution of 3.0 mL of salicylaldehyde (1 eq., 30 mmol) in acetonitrile (80 mL), 10.37 g of potassium carbonate (2.5 eq., 75 mmol) were added. The mixture was stirred at room temperature and then 2.76 mL of propargyl bromide (1.1 eq., 31 mmol) were added, allowing it to react for 2 days. After that, the mixture was filtered, the liquid extracted three times with DCM/water (3 × 50 mL) and the organic residue was dried over anhydrous sodium sulfate. The solvent was removed under reduced pressure to obtain the brown pure solid 3 (3.21 g, 67% yield). The final product was analyzed through 1H-NMR (400 MHz, Chloroform-d) δ 10.47 (s, 1H), 7.85 (dd, J = 7.7, 1.8 Hz, 1H), 7.56 (ddd, J = 8.3, 7.3, 1.8 Hz, 1H), 7.11 (dd, J = 8.5, 0.9 Hz, 1H), 7.08 (tt, J = 7.5, 0.9 Hz, 1H), 4.82 (d, J = 2.4 Hz, 2H), 2.58 (t, J = 2.4 Hz, 1H).
2.5. Synthesis of 4-Phenoxy-N-(2-(prop-2-yn-1-yloxy)benzylidene)pyridin-3-amine (4)
To synthesize 4-phenoxy-N-(2-(prop-2-yn-1-yloxy)benzylidene)pyridin-3-amine 4, a mixture of 2.8 g of 3 (1 eq., 17 mmol) and 3.17 g of 2 (1 eq., 17 mmol) in 80 mL of toluene was refluxed for 4 days; a Dean-Stark trap was used to remove the water formed during the reaction. After 4 days, the solution was dried under reduced pressure and the crude, brownish solid 4 (4.9 g, 88% yield) is used directly in the next reduction step.
2.6. Synthesis of 4-Phenoxy-N-(2-(prop-2-yn-1-yloxy)benzyl)pyridin-3-amine (5)
To synthesize 4-phenoxy-N-(2-(prop-2-yn-1-yloxy)benzyl)pyridin-3-amine 5, 4.9 g of 4 (1 eq., 14.9 mmol) were dissolved in methanol (50 mL) and cooled to 0 °C in an ice bath. Then, 2.26 g of sodium borohydride (4 eq., 60 mmol) were slowly added to the solution, the ice bath was removed, and the mixture stirred overnight at room temperature. The methanol was removed under reduced pressure, then the crude was extracted three times with NaHCO3(aq) saturated solution/DCM (3 × 50 mL). The organic layers were combined and dried over anhydrous sodium sulfate, and then the product was recovered by removing the solvent with a rotavapor. The resulting crude (4.79 g) was a yellow oil, purified with a Flash Chromatography silica column, starting with 100% hexane, then increasing the polarity until 100% ethyl acetate and in the end switching to a mixture EA:MeOH (7:3). The fractions were collected and analyzed by TLC. The recovered pure product 5 (2.33 g, 39% yield) was a yellow oil. 1H-NMR (400 MHz, Chloroform-d) δ 8.10 (s, 1H), 7.86 (d, J = 5.3 Hz, 1H), 7.42–7.32 (m, 3H), 7.27 (ddd, J = 9.2, 6.7, 1.7 Hz, 1H), 7.23–7.16 (m, 1H), 7.10–7.03 (m, 2H), 7.04–6.94 (m, 2H), 6.55 (d, J = 5.3 Hz, 1H), 4.73 (d, J = 2.4 Hz, 3H), 4.49 (d, J = 5.8 Hz, 2H), 2.50 (t, J = 2.4 Hz, 1H).
2.7. N-(4-Phenoxypyridin-3-yl)-N-(2-(prop-2-yn-1-yloxy)benzyl)acetamide (6)
To a solution of pure 5 (2.33 g, 1 eq., 7.0 mmol) in DCM (50 mL), 2.6 g of DMAP (3 eq., 21.0 mmol) were added; this mixture was stirred for 5 min, then was cooled down to 0 °C in an ice bath. A solution of 1.5 mL of acetyl chloride (3 eq., 21.0 mmol) in DCM (5 mL) was added dropwise. The mixture was stirred under the ice for 90 min, then it was left stirring at room temperature overnight. The day after, the crude was extracted three times with NaHCO3(aq) saturated solution/DCM (3 × 50 mL). The organic layers were combined and dried over anhydrous sodium sulfate, then the product was recovered by removing the solvent with a rotavapor. The crude (2.1 g) was a brown oil, purified with a Flash Chromatography silica column, starting with hexane: ethyl acetate (7:3), then increasing the polarity until 100% ethyl acetate. The fractions were collected and analyzed by TLC. The recovered pure product 6 (1.54 g, 59% yield) was a yellow oil. 1H-NMR (400 MHz, Chloroform-d) δ 8.28 (d, J = 5.6 Hz, 1H), 8.19 (s, 1H), 7.41 (td, J = 7.2, 1.8 Hz, 3H), 7.30–7.18 (m, 3H), 6.95–6.89 (m, 3H), 6.87 (dd, J = 8.3, 1.0 Hz, 1H), 6.57 (d, J = 5.6 Hz, 1H), 5.20 (d, J = 14.2 Hz, 1H), 4.87 (d, J = 14.2 Hz, 1H), 4.46 (dd, J = 8.3, 2.4 Hz, 2H), 2.41 (t, J = 2.4 Hz, 1H), 1.98 (s, 3H).
2.8. Synthesis of the Methacrylamide-PEG-N3 Monomer
Amine-PEG-N
3 (0.5 g, 0.775 mmol) and TEA (0.153 g, 1.51 mmol) were dissolved in 10 mL of anhydrous DCM. A solution containing 120 mg (1.14 mmol) of methacryloyl chloride in 0.5 mL DCM was added to the previous solution over 10 min under nitrogen atmosphere. The mixture was stirred at room temperature under a nitrogen atmosphere overnight. The solvent was then evaporated, the residue was dissolved in 50 mL of chloroform, washed with 50 mL of HCl 1 M, and then with 50 mL of brine. The organic layer was dried with anhydrous Na
2SO
4, filtered and evaporated to give the PEG-N
3 monomer (MePEG-N
3) as a transparent viscous liquid (0.47 g, yield 85%). The NMR spectrum of the molecule is reported in the
Supporting Material (Figure S5).
1H-NMR (400 MHz, Chloroform-d) δ 6.5 (s, 1H), 5.88 (s, 1H), 5.28 (s, 1H), 3.38–3.65 (m, 44H), 1.96 (s, 3H).
2.9. Synthesis of the 25MPC-mMePEG-N3 Macromolecular Chain Transfer Agent
The RAFT copolymerization of MPC and MePEG-N3 was performed in a 40/60 v/v mixture of ethanol/acetic buffer with CPA as RAFT agent and ACVA as initiator. The MPC and ACVA to CPA mole ratios were set to 25 and 1/3, respectively. On the other hand, two MePEG-N3/CPA mole ratios, i.e., 4 and 6, were adopted to vary the number of azide groups. To synthesize the 25MPC-6MePEG-N3, where 25 and 6 represent the target degree of polymerization for the two repeating units MPC and MePEG-N3, respectively, 0.5 g of MPC, 0.322 g of MePEG-N3, 0.02 g of CPA and 0.0065 g of ACVA were dissolved in ethanol and acetic buffer and the solution was poured in a round bottom flask. The solution was purged by bubbling nitrogen for 20 min and then heated to 65 °C in a thermostatic oil bath for 24 h. Aliquots of the product were withdrawn before the purification and analyzed via 1H-NMR and aqueous gel permeation chromatography (A-GPC). Monomer conversion (XMPC) and the degree of polymerization (n) were evaluated via 1H-NMR spectroscopy dissolving 10 mg of the polymer in 0.6 mL of CD3OD-d4.
The number-average molecular weight (Mn) and dispersity (Đ) were evaluated via A-GPC with a Jasco 2000 chromatograph. A total of 10 mg of sample were dissolved in 2 mL of a 0.05 M Na2SO4/acetonitrile (80/20 v/v) mixture and filtered through a 0.45 μm pore-size nylon membrane. The separation was performed at a flow rate of 0.5 mL min−1 at 35 °C with three Suprema columns (particle size 10 μm and pore sizes of 100, 1000, and 3000 Å, Polymer Standards Service). All the values reported were determined from differential refractive index data and were relative to polyethylene glycol standards. The final product was precipitated several times in acetone and dried under air.
2.10. Synthesis of the Oligo(Caprolactone) Macromonomer
The oligo(caprolactone) macromonomer with 5 ε-caprolactone repeating units, hereinafter HEMA-CL5, was synthesized via ROP of CL in bulk with HEMA as chain initiator and Sn(Oct)2 as catalyst. The HEMA to Sn(Oct)2 molar ratio was set equal to 200 while the monomer to initiator molar ratio was set to 5. In particular, 13.15 g of CL and 13.2 mg of Na2SO4 were weighted in a round-bottom flask and heated to 125 °C in a constant temperature oil bath under stirring. A total of 3 g of HEMA and 56.7 mg of Sn(Oct)2 were stirred until homogenization and injected into the pre-heated round bottom flask. The reaction was allowed to proceed for 2.5 h. The final product was analyzed through 1H-NMR (CDCl3) and GPC (10 mg/mL in THF). Caprolactone conversion (XCL) and the average number of caprolactone units attached to HEMA (q) were evaluated via 1H-NMR while macromonomer number-averaged molecular weight (MnGPC) and dispersity (ĐGPC) were evaluated via GPC. More in detail, the sample was dissolved in THF at 4 mg mL−1 and filtered through a PTFE 0.45 μm pore-size membrane before injection. The separation was performed at a flow rate of 0.5 mL min−1 at 35 °C and with three styrene/divinylbenzene (SDV) columns in series (Polymer Standard Service, Germany; pore size 103, 105, and 106 Å; 300 mm length and 8 mm internal diameter) and a pre-column (50 mm length and 8 mm internal diameter). All the values reported were determined from differential refractive index data and were relative to poly(styrene) standards (from 580 to 3,250,000 g/mol, Polymer Laboratories).
2.11. Synthesis of the Amphiphilic and Biodegradable Block Copolymers
A set of amphiphilic block copolymers were obtained via RAFT polymerization of HEMA-CL5 in the presence of the zwitterionic-PEGylated macromolecular chain transfer agent synthesized previously and using ACVA as initiator. The ACVA to 25MPC-mMePEG-N3 molar ratio was set equal to 1/3, and the HEMA-CL5 to 25MPC-mMePEG-N3 molar ratio (p) was set to 30 or 60. As an example, for the 25MPC-6MePEGN3-30CL5, with 30 units of HEMA-CL5 added to the copolymer, 0.27 g of 25MPC-6MePEGN3, 0.52 g of HEMA-CL5 and 2.3 mg of ACVA were dissolved in 4 mL of methanol and poured in a 5 mL vial. The mixture was purged with nitrogen for 10 min and left to react at 65 °C under stirring for 24 h. The final block copolymer was precipitated in diethyl ether several times, dried under vacuum, and stored at −20 °C. An aliquot of the mixture was analyzed before and after the purification via 1H NMR (10 mg were dissolved in a mixture of 0.7 mL of MeOD). Macromonomer conversion (X) and the degree of polymerization (pNMR) were evaluated from the 1H NMR spectra.
2.12. Block Copolymer Functionalization with the TSPO-Ligand
The N-(4-phenoxypyridin-3-yl)-N-(2-(prop-2-yn-1-yloxy)benzyl)acetamide was conjugated to the block copolymer through click chemistry. Briefly, 0.05 g of 25MPC-6MePEGN3-30CL5 were added to 0.011 g of N-(4-phenoxypyridin-3-yl)-N-(2-(prop-2-yn-1- yloxy)benzyl)acetamide and 1.5 mg of TBTA. The mixture was solubilized in 0.3 mL of a solution of DMSO:MeOH (1/3 v/v). Finally, 0.5 mg of CuI were added to the flask and the final mixture was left to react at 37 °C for 3 days. The final product was analyzed through Fourier Transformed Infrared spectroscopy (FT-IR) to evaluate the intensity of the azide peak before and after the conjugation.
2.13. Docking Studies
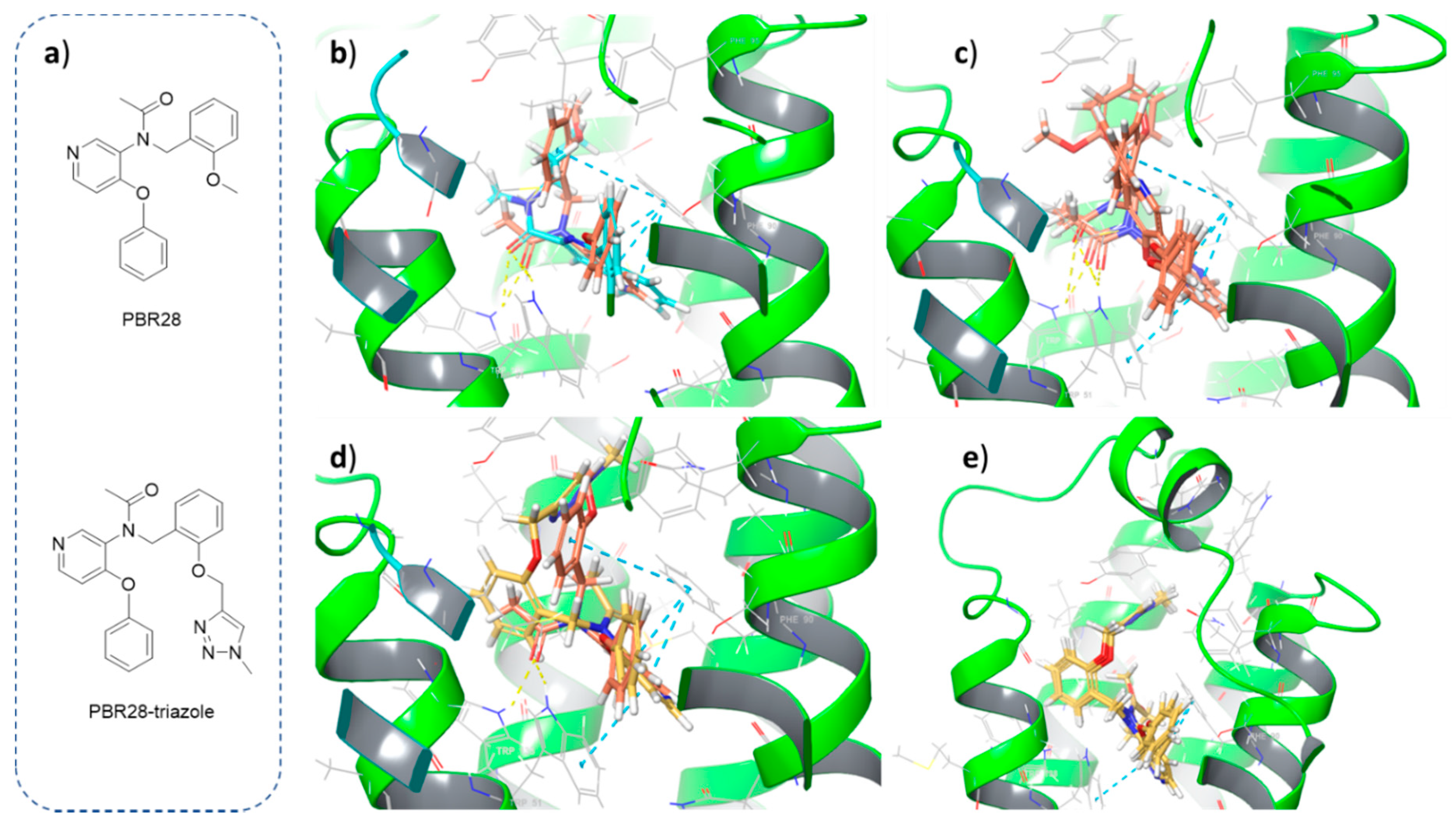
The crystal structure of TSPO co-crystallized with PK11195 was retrieved from Protein Data Bank (PDB ID: 4RYI), prepared and used to dock ligands into the binding site. Both PBR28 and the derivative bearing the triazole ring formed after click chemistry (PBR28-triazole) were docked according to the following procedure. Ligand docking was carried out with Glide, a grid-based algorithm implemented in Maestro Schrödinger’s suite [
18]. The grid was generated around the co-crystallized ligand with a radius of 15 Å. Default Van der Waals scaling was used, with a scaling factor of 1.0 and partial charge cut-off of 0.25. Partial charges were computed with the OPLS-2005 force field. Ligands were then docked into the receptor’s grid applying default scaling of Van Der Waals radii, flexible ligand sampling and standard or extra precision settings.
2.14. Nanoparticle Synthesis
The polymer NPs decorated with the developed TSPO-ligand were synthesized via self-assembly of the zwitterionic PCL-based block copolymer. Briefly, 10 mg of the PBR-functionalized polymer 25MPC-6MePEGPBR-30CL5 were solubilized in 50 μL of DMSO and 150 μL of MeOH and then added dropwise to 3 mL of distilled water under strong stirring. The NP dispersions were dialyzed in regenerated cellulose membranes (Spectra/Por, molecular weight cut-off = 3500 Da) against distilled water for 24 h to remove the organic solvent. The NP volume-average diameter (Dv) and polydispersity (PdI) were evaluated via dynamic light scattering (DLS) on a Zetasizer Nano ZS (Malvern, Instruments). Measurements were taken at a scattering angle of 173° and performed in triplicates on samples previously diluted to 0.1% w/w in distilled water.
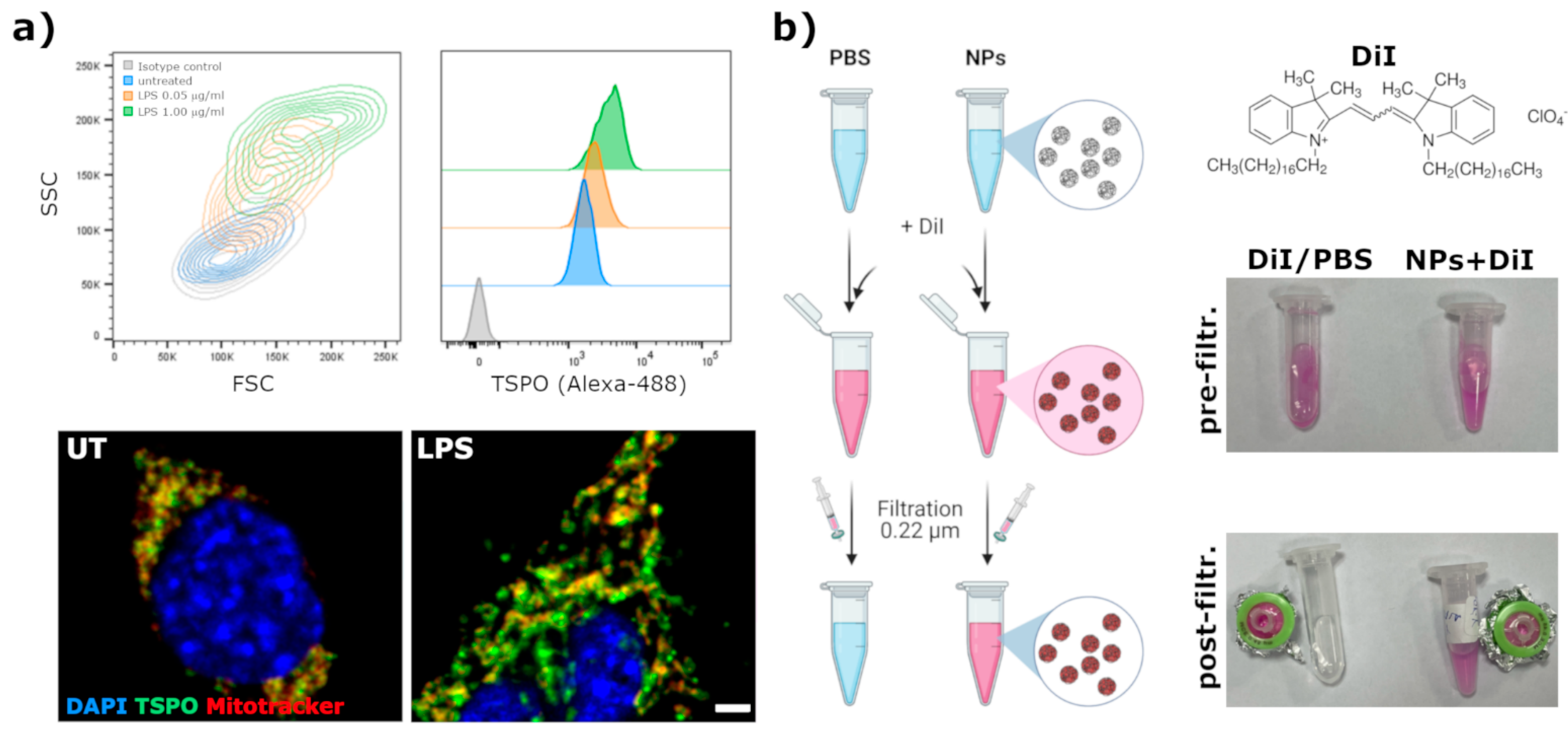
2.15. Nanoparticle Staining
In order to visualize by fluorescent microscopy the internalization of NPs in cells, a fluorescent lipophilic dye was encapsulated. DiI (1,1′-Dioctadecyl-3,3,3′,3-tetramethylindocarbocyanine perchlorate, cat. # 468495, from Sigma-Aldrich, St. Louis, MO, USA) was used for this purpose. NPs (at 1 mg/mL in PBS) were incubated for 30 min at RT with DiI (0.2 mg/mL final concentration). Afterward, by taking advantage of the poorly water solubility of DiI and the high affinity of the NPs core for lipophilic compounds, the not encapsulated dye was removed from the solution by filtration through a 0.22 μm syringe methyl-cellulose filter.
2.16. In Vitro Tests
BV2 immortalized microglia cell line was used to assess the uptake of fluorescently labeled nanoparticles. BV2 cells were cultured at 37 °C, 5% CO2 with complete Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM glutamine and 1% penicillin/streptomycin. To verify the endogenous expression and modulation of TSPO, BV2 cells were plated in 24-well tissue culture plates (15,000 cells/well). One day later, cells were treated for 24 h with LPS (from Sigma-Aldrich) at a final concentration of 0.05 or 1 μg/mL. Afterward, cells were detached with 0.05% Trypsin-EDTA, fixed with 4% phosphate-buffered paraformaldheyde (PFA) for 20 min and then stained with rabbit anti-TSPO (Abcam) antibody diluted 1:500 in BSA3% Triton 0.3% in PBS for 2 h at 4 °C. After one wash in PBS, cells were stained with anti-rabbit-Alexafluor-488 secondary antibody (Molecular Probes) at 1:1000 in BSA3% Triton 0.3% in PBS for 30 min at RT. TSPO signal was evaluated by flow cytometry with a BD Fortessa II cytometer. To visualize the mitochondria network, cells were plated on glass coverslips in 24 well plates and treated as described above. After the LPS treatment, cells were incubated for 15 min with Mitotracker-DR (Molecular Probes) at 150 nM at 37 °C in medium and then washed and fixed with 4% PFA. TSPO was stained following the same procedure applied for flow cytometry; nuclei were stained with DAPI 0.1 μg/mL in PBS for 10 min and then coverslips were mounted with Mowiol. The extent of co-localization between the mitochondria network and TSPO was analyzed at an SP5 laser scanning confocal microscope (Leica).
To highlight TSPO in live cells, lentiviral vectors expressing human TSPO (NM_000714) fused at the C-terminal with GFP reporter gene (from OriGene) were produced and titered as previously described [
19]. These vectors were used to infect BV2 cells at MOI 5, to generate a cell line stably expressing TSPO-GFP fusion protein. Single-cell clones were generated by cell sorting and one clone expressing TSPO-GFP at high levels (hereafter called BV2-TSPO clone#4) was expanded and used for subsequent experiments.
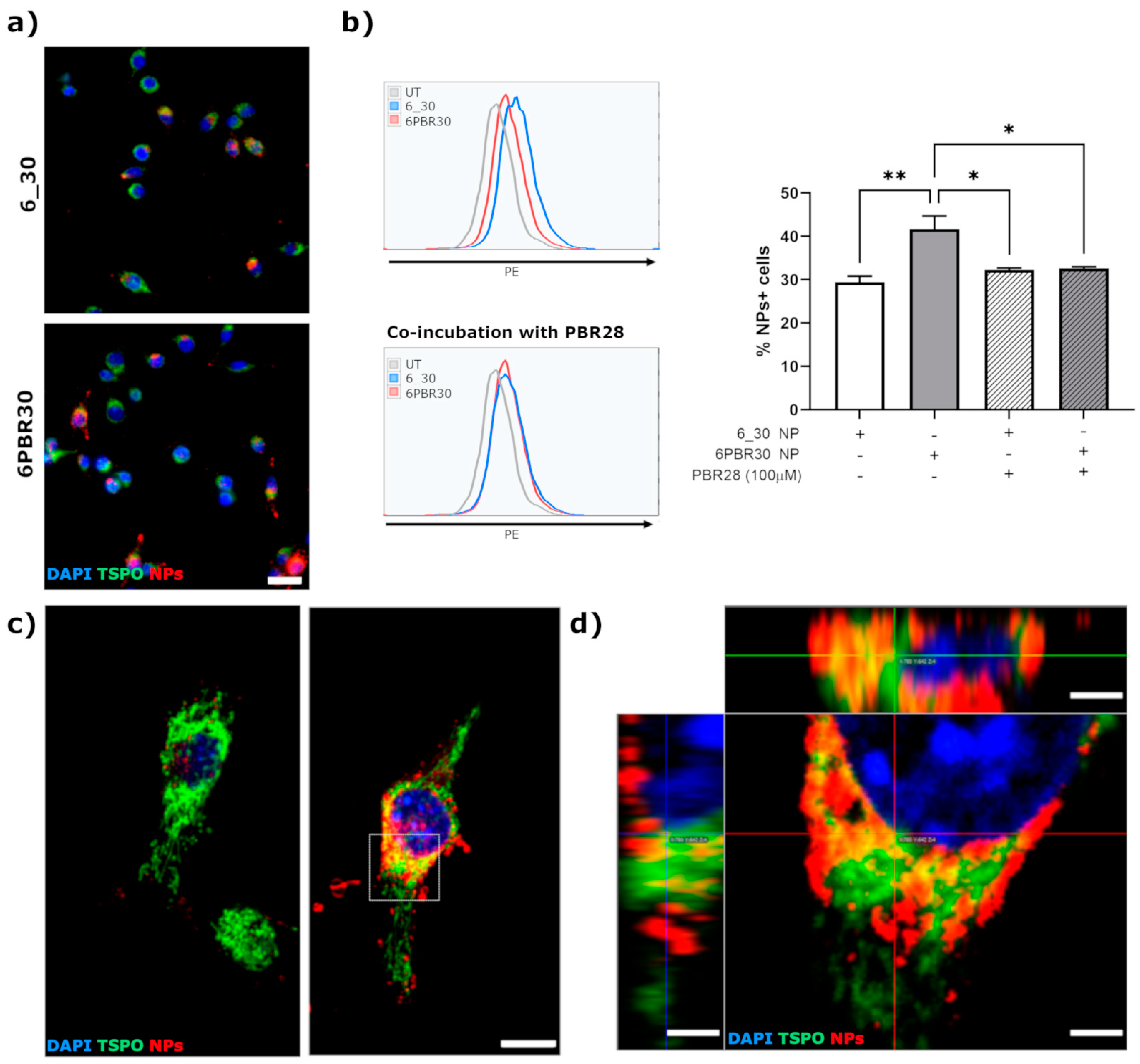
For the assays, BV2-TSPO clone#4 cells were detached from the culture dishes, centrifuged at 150× g for 5 min and plated in 24-well tissue culture plates (15,000 cells/well) on glass coverslips. Twenty-four hours after plating, the medium was replaced with complete DMEM with different NP formulations (either not functionalized -6_30 NP- or modified by conjugation with PBR28-derivative -6PBR30 NP) at 5 μg/mL. At 4 h after NPs administration, the medium was removed and cells were washed with PBS and then fixed with 4% phosphate-buffered paraformaldheyde. Nuclei were stained with DAPI and then glass coverslips were mounted with Mowiol. High-resolution confocal microscope images were acquired at SP5 Leica confocal microscope equipped with UV-diode, Ne-He and Argon laser sources. No DiI signal was detected in cells incubated with the eluate obtained from the DiI diluted in PBS, confirming that no free DiI could be detected in the absence of nanoparticles upon filtration with 0.45 μm filter (data not shown).
Quantification of NPs internalization was performed by flow cytometry, by collecting the cells through trypsinization at 4 h after administration of NPs (at 1 μg/mL). The competition pharmacological assay consisted of co-incubation of NPs (either 6_30 or 6PBR30) tested at 1 μg/mL with 100 μM PBR28. Cells were analyzed by flow cytometry with a BD Facs Lyric cytometer.
4. Discussion and Conclusions
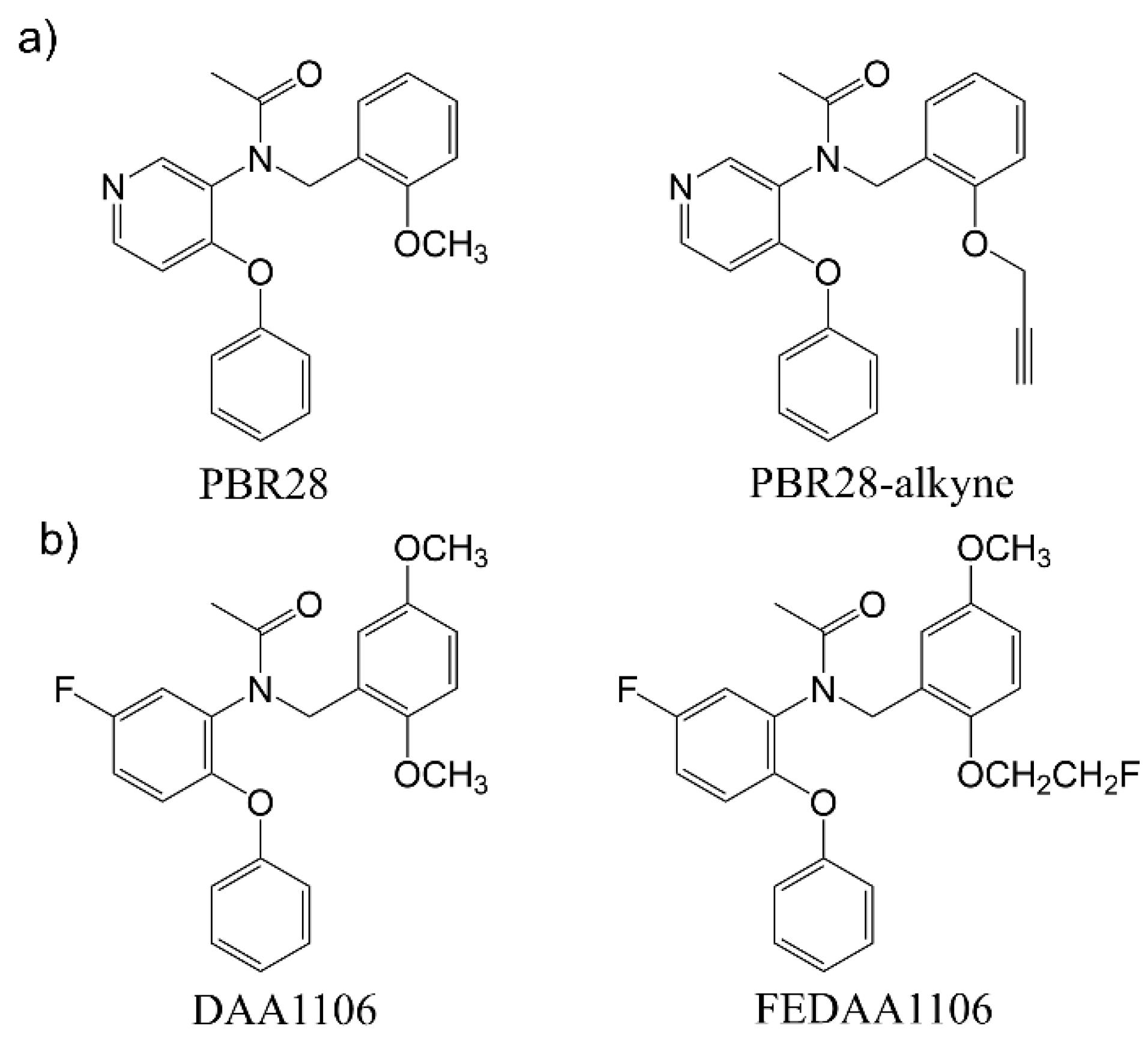
In this work, we successfully added a functional moiety to the high-affinity TSPO ligand PBR28 allowing for the generation of a PBR28-derivative suitable for conjugation with azide-functionalized polymer nanoparticles via click chemistry. First, we exploited in silico docking studies to exclude potential interferences with the binding of PBR28 to TSPO, caused by the triazole ring that is formed after the click reaction. Even if these analyses were not quantitative, we demonstrated that these in silico studies could be exploited as a guide for the rational design of small molecule putative TSPO-ligand candidates with optimal structural features. In fact, the good correlation that we observed between the poses of a reference TSPO-ligand, such as PK11195 (co-crystallized with the TSPO protein used for the docking studies) or PBR28 itself, and the PBR28-triazole derivative were successfully translated into experimental evidence of the suitability of the PBR28-alkyne, designed and synthesized in this study, for functionalizing the surface of polymeric NPs achieving efficient internalization in TSPO-expressing cells, such as BV2 microglia-like cell line.
Since its discovery in 1977 [
26], radiolabeled ligands specific for TSPO were used in PET scans for many years to image the areas of enriched neuroinflammation in the diseased central nervous system of patients as well as rodent models of several neurodegenerative diseases, including Multiple Sclerosis, Alzheimer’s and Parkinson’s disease and ALS. A striking increase in TSPO expression was reported in several tumors, with a correlation with the level of malignancy, suggesting that TSPO ligands may be exploited also to identify cancerous cells and potentially target them with therapeutics. Accordingly, the development of bivalent compounds, derived from the conjugation of a TSPO ligand with a chemotherapic or a drug delivery system, is regarded as a promising strategy to achieve cell-selective targeting of therapeutics, testified by the increasing number of studies in the literature, reporting the exploitation of a diverse range of small molecule TSPO-ligands as a targeting moiety, ranging from TSPO-ligand chemotherapic (doxorubicin or cytarabine) [
27,
28] to TSPO-ligand dendrimers [
9,
29] or iron-oxide nanoparticle [
7,
30] conjugates. In line with these studies, in our work we demonstrated that the functionalization with a TSPO-targeted ligand enhances the internalization of polymer nanoparticles in TSPO-expressing cells; moreover, the internalized 6PBR30 NPs displayed a high degree of co-localization with the mitochondrial network. However, differently from other works where the mitochondria were highlighted, in our study we exploited a fluorescently tagged TSPO to obtain a more precise evaluation of the extent of co-localization between the PBR28-conjugated NPs and the target receptor. We observed that even if there are some regions in the cellular cytoplasm displaying good co-localization between the NP signal and TSPO, there are some NP signals that do not co-localize. This is not surprising and is in line with what has been reported by other authors (where only partial co-localization between TSPO-targeted NPs and mitochondria was reported [
9,
30]). In previous studies, we reported that our polymeric NPs can be internalized in microglia cells even without surface functionalization, through the interaction with scavenger receptors that are highly expressed by microglia/macrophages [
22]. Based on the results of the confocal analyses performed with 6PBR30, and since TSPO is mainly localized on mitochondria, it can be hypothesized that the 6PBR30 NPs (as most likely also other TSPO-ligand functionalized NPs), are retained on the mitochondria network through the interaction of TSPO after cellular internalization through other mechanisms. This could represent an interesting mechanism by which TSPO-targeting could be exploited to enhance nanoparticle-mediated drug delivery to the mitochondrial compartment. Recent studies have highlighted increased pharmacological potency of apocynin (an inhibitor of NOX2, an enzyme involved in reactive oxygen species production) when targeted to mitochondria [
31]. Interestingly, TSPO and the ER-resident NOX2 subunit gp91phox co-localize in primary microglia, pointing to a possible interaction between TSPO and NOX2 in these cells.
A polymorphism in the TSPO gene, evidenced in about 10% of the population, affects the binding of PBR28 [
1]. Even if this could be envisaged as a possible limitation that could hinder the widespread applicability of the nanoparticle platform developed in our study, PBR28 is still widely used as a PET tracer (especially for tracking of neuroinflammation in neurodegenerative disorders) thanks to high affinity for the receptor and optimal biodistribution profile. So, exploiting PBR28-derivatives such as the one generated in this study, albeit only after the genetic screening of the patients to exclude poor binders, could represent an additional tool exploitable for the generation of cell- (including mitochondria-) selective targeted therapeutics.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}