
Evaluation and Optimization of Poly-d-Lysine as a Non-Natural Cationic Polypeptide for Gene Transfer in Neuroblastoma Cells


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Plasmid/PDL Interaction Assay
2.3. DNase I Protection Assay
2.4. Elaboration of DNA–Poly-d-Lysine Complex
2.5. MTT Assay
2.6. Assays In Vitro of SH-SY5Y Cells
2.7. Cell Count Assay
2.8. Flow Cytometer Assay
2.9. Statistical Analysis
3. Results
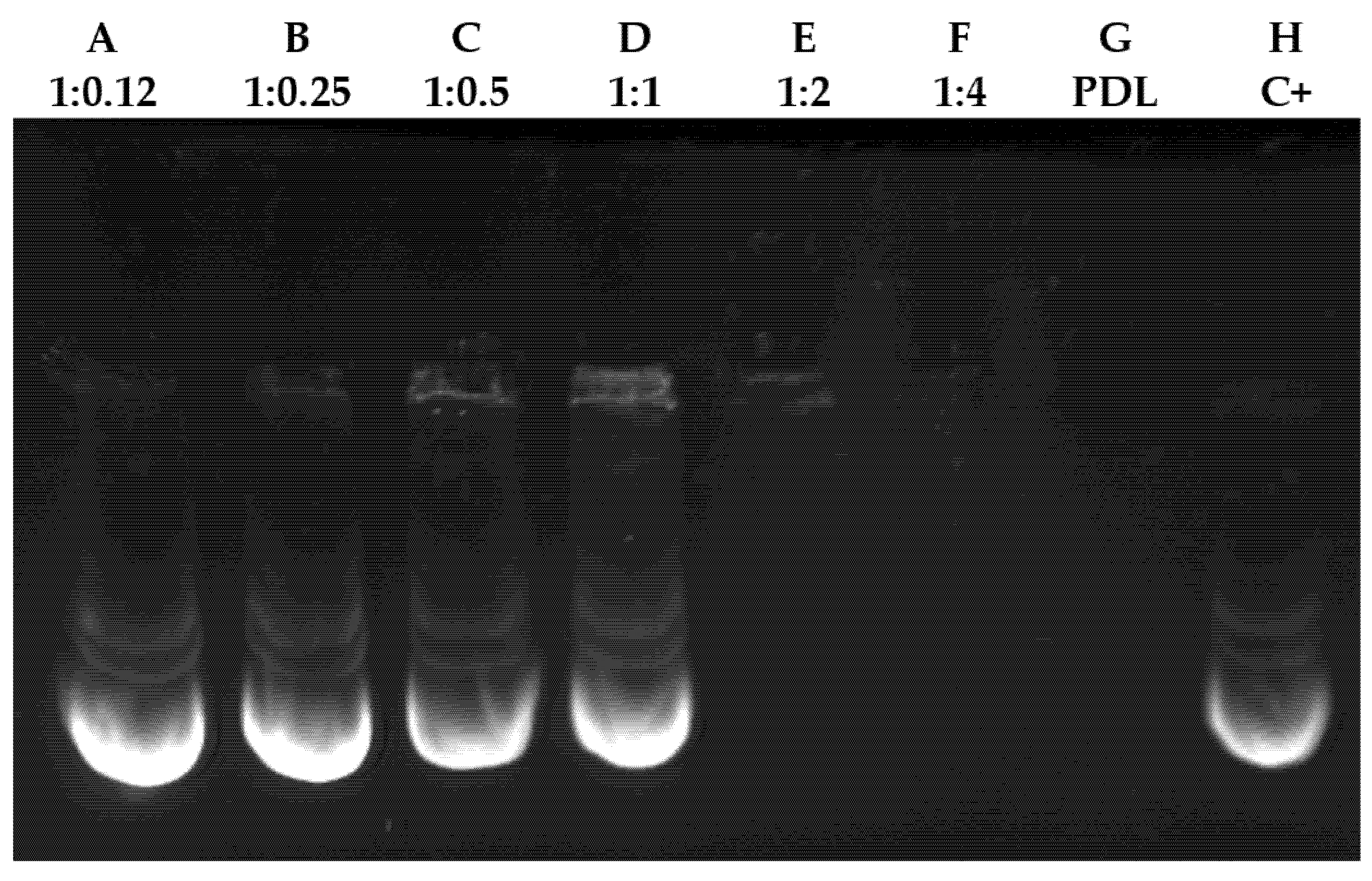
3.1. Gel Retardation Assay
3.2. DNase I Protection and Release Assay
3.3. Cell Viability Evaluated according to PDL Concentration and Duration of Administration
3.4. Cell-Type-Specific Differences in EGFP Expression Using PDL
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wirth, T.; Parker, N.; Ylä-Herttuala, S. History of gene therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef] [PubMed]
- High, K.A.; Roncarolo, M.G. Gene Therapy. N. Engl. J. Med. 2019, 381, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Thorne, B.; Takeya, R.; Vitelli, F.; Swanson, X. Gene Therapy. Adv. Biochem. Eng. Biotechnol. 2018, 165, 351–399. [Google Scholar] [CrossRef] [PubMed]
- Anguela, X.M.; High, K.A. Entering the Modern Era of Gene Therapy. Annu. Rev. Med. 2019, 70, 273–288. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Al Qtaish, N.; Gallego, I.; Villate-Beitia, I.; Sainz-Ramos, M.; Lopez-Mendez, T.B.; Grijalvo, S.; Eritja, R.; Soto-Sánchez, C.; Martínez-Navarrete, G.; Fernandez, E.; et al. Niosome-Based Approach for In Situ Gene Delivery to Retina and Brain Cortex as Immune-Privileged Tissues. Pharmaceutics 2020, 12, 198. [Google Scholar] [CrossRef] [Green Version]
- Mashal, M.; Attia, N.; Martínez-Navarrete, G.; Soto-Sánchez, C.; Fernández, E.; Grijalvo, S.; Eritja, R.; Puras, G.; Pedraz, J.L. Gene delivery to the rat retina by non-viral vectors based on chloroquine-containing cationic niosomes. J. Control. Release Off. J. Control. Release Soc. 2019, 304, 181–190. [Google Scholar] [CrossRef]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [Green Version]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef]
- Delcroix, M.; Riley, L.W. Cell-Penetrating Peptides for Antiviral Drug Development. Pharmaceuticals 2010, 3, 448–470. [Google Scholar] [CrossRef] [Green Version]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Farkhani, S.M.; Valizadeh, A.; Karami, H.; Mohammadi, S.; Sohrabi, N.; Badrzadeh, F. Cell penetrating peptides: Efficient vectors for delivery of nanoparticles, nanocarriers, therapeutic and diagnostic molecules. Peptides 2014, 57, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Akita, H.; Kogure, K.; Gräslund, A.; Langel, U.; Harashima, H.; Futaki, S. Efficient Intracellular Delivery of Nucleic Acid Pharmaceuticals Using Cell-Penetrating Peptides. Acc. Chem. Res. 2012, 45, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xie, G.; Xu, Y.; Ma, L.; Tong, C.; Fan, D.; Du, F.; Yu, H. PEP-1-MsrA ameliorates inflammation and reduces atherosclerosis in apolipoprotein E deficient mice. J. Transl. Med. 2015, 13, 316. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.R.; Li, J.-F.; Lu, S.-W.; Lee, H.-J.; Huang, Y.-W.; Shannon, K.B.; Aronstam, R.S. Cellular Internalization of Quantum Dots Noncovalently Conjugated with Arginine-Rich Cell-Penetrating Peptides. J. Nanosci. Nanotechnol. 2010, 10, 6534–6543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Venkatraman, G.; Batra, S.K. Cell-penetrating peptides and antibodies: A new direction for optimizing radioimmunotherapy. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 973–977. [Google Scholar] [CrossRef]
- Lehto, T.; Kurrikoff, K.; Langel, Ü. Cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv. 2012, 9, 823–836. [Google Scholar] [CrossRef]
- Eguchi, A.; Akuta, T.; Okuyama, H.; Senda, T.; Yokoi, H.; Inokuchi, H.; Fujita, S.; Hayakawa, T.; Takeda, K.; Hasegawa, M.; et al. Protein Transduction Domain of HIV-1 Tat Protein Promotes Efficient Delivery of DNA into Mammalian Cells. J. Biol. Chem. 2001, 276, 26204–26210. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, P.; Bhalla, S.; Usmani, S.S.; Singh, S.; Chaudhary, K.; Raghava, G.P.S.; Gautam, A. CPPsite 2.0: A repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res. 2016, 44, D1098–D1103. [Google Scholar] [CrossRef] [PubMed]
- Osman, G.; Rodriguez, J.; Chan, S.Y.; Chisholm, J.; Duncan, G.; Kim, N.; Tatler, A.L.; Shakesheff, K.M.; Hanes, J.; Suk, J.S.; et al. PEGylated enhanced cell penetrating peptide nanoparticles for lung gene therapy. J. Control. Release Off. J. Control. Release Soc. 2018, 285, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.N.; Cashman, S.M.; Kumar-Singh, R. Cell-penetrating Peptide for Enhanced Delivery of Nucleic Acids and Drugs to Ocular Tissues Including Retina and Cornea. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Characterization of DNA condensates induced by poly(ethylene oxide) and polylysine. Proc. Natl. Acad. Sci. USA 1975, 72, 4288–4292. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.Y.; Wu, C.H. Receptor-mediated in vitro gene transformation by a soluble DNA carrier system. J. Biol. Chem. 1987, 262, 4429–4432. [Google Scholar] [CrossRef]
- Hu, Y.; Xu, B.; Ji, Q.; Shou, D.; Sun, X.; Xu, J.; Gao, J.; Liang, W. A mannosylated cell-penetrating peptide-graft-polyethylenimine as a gene delivery vector. Biomaterials 2014, 35, 4236–4246. [Google Scholar] [CrossRef]
- Isaksson, K.; Akerberg, D.; Andersson, R.; Tingstedt, B. Toxicity and Dose Response of Intra-Abdominally Administered Poly-l-Alpha-Lysine and Poly-l-Glutamate for Postoperative Adhesion Protection. Eur. Surg. Res. Eur. Chir. Forsch. Rech. Chir. Eur. 2010, 44, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.H.; Klibanov, A.L.; Huang, L. Lipophilic polylysines mediate efficient DNA transfection in mammalian cells. Biochim. Biophys. Acta 1991, 1065, 8–14. [Google Scholar] [CrossRef]
- Kim, Y.H.; Baek, N.S.; Han, Y.H.; Chung, M.-A.; Jung, S.-D. Enhancement of neuronal cell adhesion by covalent binding of poly-d-lysine. J. Neurosci. Methods 2011, 202, 38–44. [Google Scholar] [CrossRef]
- Barrett, L.B.; Logan, A.; Berry, M.; Ying, W.; Gonzalez, A.M.; Baird, A.; Seymour, L.W. Targeted transfection of neuronal cells using a poly(d-lysine)-cholera-toxin b chain conjugate. Biochem. Soc. Trans. 1999, 27, 851–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, L.B.; Berry, M.; Ying, W.-B.; Hodgkin, M.N.; Seymour, L.W.; Gonzalez, A.-M.; Read, M.L.; Baird, A.; Logan, A. CTb targeted non-viral cDNA delivery enhances transgene expression in neurons. J. Gene Med. 2004, 6, 429–438. [Google Scholar] [CrossRef]
- Kim, T.-G.; Kang, S.-Y.; Kang, J.-H.; Cho, M.-Y.; Kim, J.-I.; Kim, S.-H.; Kim, J.-S. Gene Transfer into Human Hepatoma Cells by Receptor-Associated Protein/Polylysine Conjugates. Bioconjugate Chem. 2004, 15, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Mirmiran, A.; Schmitt, C.; Lefebvre, T.; Manceau, H.; Daher, R.; Oustric, V.; Poli, A.; Lacapère, J.-J.; Moulouel, B.; Puy, H.; et al. Erythroid-Progenitor-Targeted Gene Therapy Using Bifunctional TFR1 Ligand-Peptides in Human Erythropoietic Protoporphyria. Am. J. Hum. Genet. 2019, 104, 341–347. [Google Scholar] [CrossRef] [Green Version]
- Konstan, M.W.; Davis, P.B.; Wagener, J.S.; Hilliard, K.A.; Stern, R.C.; Milgram, L.J.H.; Kowalczyk, T.H.; Hyatt, S.L.; Fink, T.L.; Gedeon, C.R.; et al. Compacted DNA Nanoparticles Administered to the Nasal Mucosa of Cystic Fibrosis Subjects Are Safe and Demonstrate Partial to Complete Cystic Fibrosis Transmembrane Regulator Reconstitution. Hum. Gene Ther. 2004, 15, 1255–1269. [Google Scholar] [CrossRef] [PubMed]
- Sela, M.; Zisman, E. Different roles of D-amino acids in immune phenomena. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1997, 11, 449–456. [Google Scholar] [CrossRef]
- Verdurmen, W.P.R.; Bovee-Geurts, P.H.; Wadhwani, P.; Ulrich, A.S.; Hällbrink, M.; van Kuppevelt, T.H.; Brock, R. Preferential Uptake of L- versus D-Amino Acid Cell-Penetrating Peptides in a Cell Type-Dependent Manner. Chem. Biol. 2011, 18, 1000–1010. [Google Scholar] [CrossRef] [Green Version]
- Louis, C.U.; Shohet, J.M. Neuroblastoma: Molecular pathogenesis and therapy. Annu. Rev. Med. 2015, 66, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ara, T.; Declerck, Y.A. Mechanisms of invasion and metastasis in human neuroblastoma. Cancer Metastasis Rev. 2006, 25, 645–657. [Google Scholar] [CrossRef]
- Kumar, M.D.; Dravid, A.; Kumar, A.; Sen, D. Gene therapy as a potential tool for treating neuroblastoma—a focused review. Cancer Gene Ther. 2016, 23, 115–124. [Google Scholar] [CrossRef]
- Rubio-Zapata, H.A.; Rembao-Bojorquez, J.D.; Arango-Rodriguez, M.L.; Dupouy, S.; Forgez, P.; Martinez-Fong, D. NT-polyplex: A new tool for therapeutic gene delivery to neuroblastoma tumors. Cancer Gene Ther. 2009, 16, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Chabner, B.A.; Roberts, T.G. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef]
- Lukashev, A.N.; Zamyatnin, A.A. Viral vectors for gene therapy: Current state and clinical perspectives. Biochem. Biokhimiia 2016, 81, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Gong, H.; Yin, D.; Lu, S.; Fu, Q. High-Molecular-Weight Polyethyleneimine Conjuncted Pluronic for Gene Transfer Agents. Chem. Pharm. Bull. 2011, 59, 1094–1101. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, S.J.; Sørensen, A.H.; Hansen, L.H.; Oregaard, G.; Veal, D. Direct Detection and Quantification of Horizontal Gene Transfer by Using Flow Cytometry and gfp as a Reporter Gene. Curr. Microbiol. 2003, 47, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Zagorovsky, K.; Chou, L.Y.T.; Chan, W.C.W. Controlling DNA–nanoparticle serum interactions. Proc. Natl. Acad. Sci. USA 2016, 113, 13600–13605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Montañez, E.; López-Téllez, J.F.; Acevedo, M.J.; Pavía, J.; Khan, Z.U. Efficiency of gene transfection reagents in NG108-15, SH-SY5Y and CHO-K1 cell lines. Methods Find. Exp. Clin. Pharmacol. 2010, 32, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Futaki, S.; Niwa, M.; Tanaka, S.; Ueda, K.; Sugiura, Y. Possible Existence of Common Internalization Mechanisms among Arginine-rich Peptides. J. Biol. Chem. 2002, 277, 2437–2443. [Google Scholar] [CrossRef] [Green Version]
- van de Wetering, P.; Schuurmans-Nieuwenbroek, N.M.; Hennink, W.E.; Storm, G. Comparative Transfection Studies of Human Ovarian Carcinoma Cells in Vitro, Ex Vivo and in Vivo with Poly(2-(Dimethylamino)Ethyl Methacrylate)-Based Polyplexes. J. Gene Med. 1999, 1, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, V.; Rojas-Sánchez, L.; Białas, N.; Schulze, N.; Epple, M. Calcium phosphate nanoparticle-mediated transfection in 2D and 3D mono- and co-culture cell models. Acta Biomater. 2019, 84, 391–401. [Google Scholar] [CrossRef]
- Mashal, M.; Attia, N.; Puras, G.; Martínez-Navarrete, G.; Fernández, E.; Pedraz, J.L. Retinal gene delivery enhancement by lycopene incorporation into cationic niosomes based on DOTMA and polysorbate 60. J. Control. Release Off. J. Control. Release Soc. 2017, 254, 55–64. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Giralt, E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef] [Green Version]
- Kardani, K.; Bolhassani, A. Cppsite 2.0: An Available Database of Experimentally Validated Cell-Penetrating Peptides Predicting their Secondary and Tertiary Structures. J. Mol. Biol. 2021, 433, 166703. [Google Scholar] [CrossRef]
- Lentz, T.B.; Gray, S.J.; Samulski, R.J. Viral vectors for gene delivery to the central nervous system. Neurobiol. Dis. 2012, 48, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Marrano, P.; Irwin, M.S.; Thorner, P.S. Heterogeneity of MYCN amplification in neuroblastoma at diagnosis, treatment, relapse, and metastasis. Genes Chromosomes Cancer 2017, 56, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M. Amplification of N-myc as a prognostic marker for patients with neuroblastoma. Semin. Cancer Biol. 1993, 4, 13–18. [Google Scholar] [PubMed]
- Doronin, I.I.; Vishnyakova, P.A.; Kholodenko, I.V.; Ponomarev, E.D.; Ryazantsev, D.Y.; Molotkovskaya, I.M.; Kholodenko, R.V. Ganglioside GD2 in reception and transduction of cell death signal in tumor cells. BMC Cancer 2014, 14, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorokin, M.; Kholodenko, I.; Kalinovsky, D.; Shamanskaya, T.; Doronin, I.; Konovalov, D.; Mironov, A.; Kuzmin, D.; Nikitin, D.; Deyev, S.; et al. RNA Sequencing-Based Identification of Ganglioside GD2-Positive Cancer Phenotype. Biomedicines 2020, 8, 142. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | ||
|---|---|---|---|---|---|
| Time | Ratio 1:2 | Ratio 1:4 | C+ | C− | |
| 3rd day (48 h) | A | X | X | X | - |
| B | X | X | X | - | |
| C | X | X | X | - | |
| 5th day (96 h) | A | - | - | - | - |
| B | X | X | X | - | |
| C | X | X | X | - | |
| 7th day (144 h) | A | - | - | - | - |
| B | - | - | - | - | |
| C | X | X | X | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez-Martos, M.; Martinez-Navarrete, G.; Bernabeu-Zornoza, A.; Humphreys, L.; Fernandez, E. Evaluation and Optimization of Poly-d-Lysine as a Non-Natural Cationic Polypeptide for Gene Transfer in Neuroblastoma Cells. Nanomaterials 2021, 11, 1756. https://doi.org/10.3390/nano11071756
Sanchez-Martos M, Martinez-Navarrete G, Bernabeu-Zornoza A, Humphreys L, Fernandez E. Evaluation and Optimization of Poly-d-Lysine as a Non-Natural Cationic Polypeptide for Gene Transfer in Neuroblastoma Cells. Nanomaterials. 2021; 11(7):1756. https://doi.org/10.3390/nano11071756
Chicago/Turabian StyleSanchez-Martos, Miguel, Gema Martinez-Navarrete, Adela Bernabeu-Zornoza, Lawrence Humphreys, and Eduardo Fernandez. 2021. "Evaluation and Optimization of Poly-d-Lysine as a Non-Natural Cationic Polypeptide for Gene Transfer in Neuroblastoma Cells" Nanomaterials 11, no. 7: 1756. https://doi.org/10.3390/nano11071756
APA StyleSanchez-Martos, M., Martinez-Navarrete, G., Bernabeu-Zornoza, A., Humphreys, L., & Fernandez, E. (2021). Evaluation and Optimization of Poly-d-Lysine as a Non-Natural Cationic Polypeptide for Gene Transfer in Neuroblastoma Cells. Nanomaterials, 11(7), 1756. https://doi.org/10.3390/nano11071756