
Quantitative Assessment of Chirality of Protein Secondary Structures and Phenylalanine Peptide Nanotubes

 , and
, and
Abstract
1. Introduction
2. Models and Computational Methods
2.1. Objects of Study
2.1.1. Protein Secondary Structures
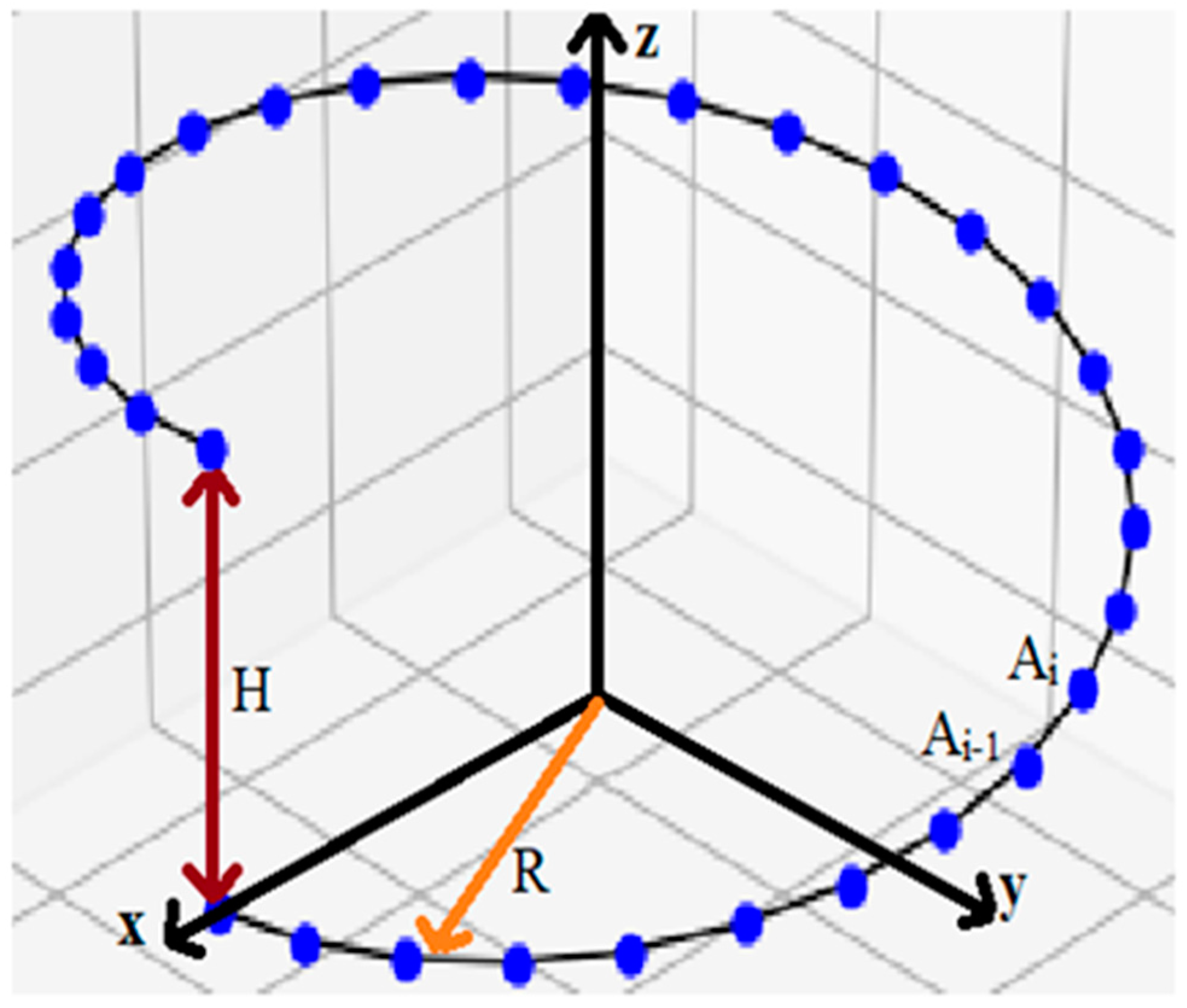
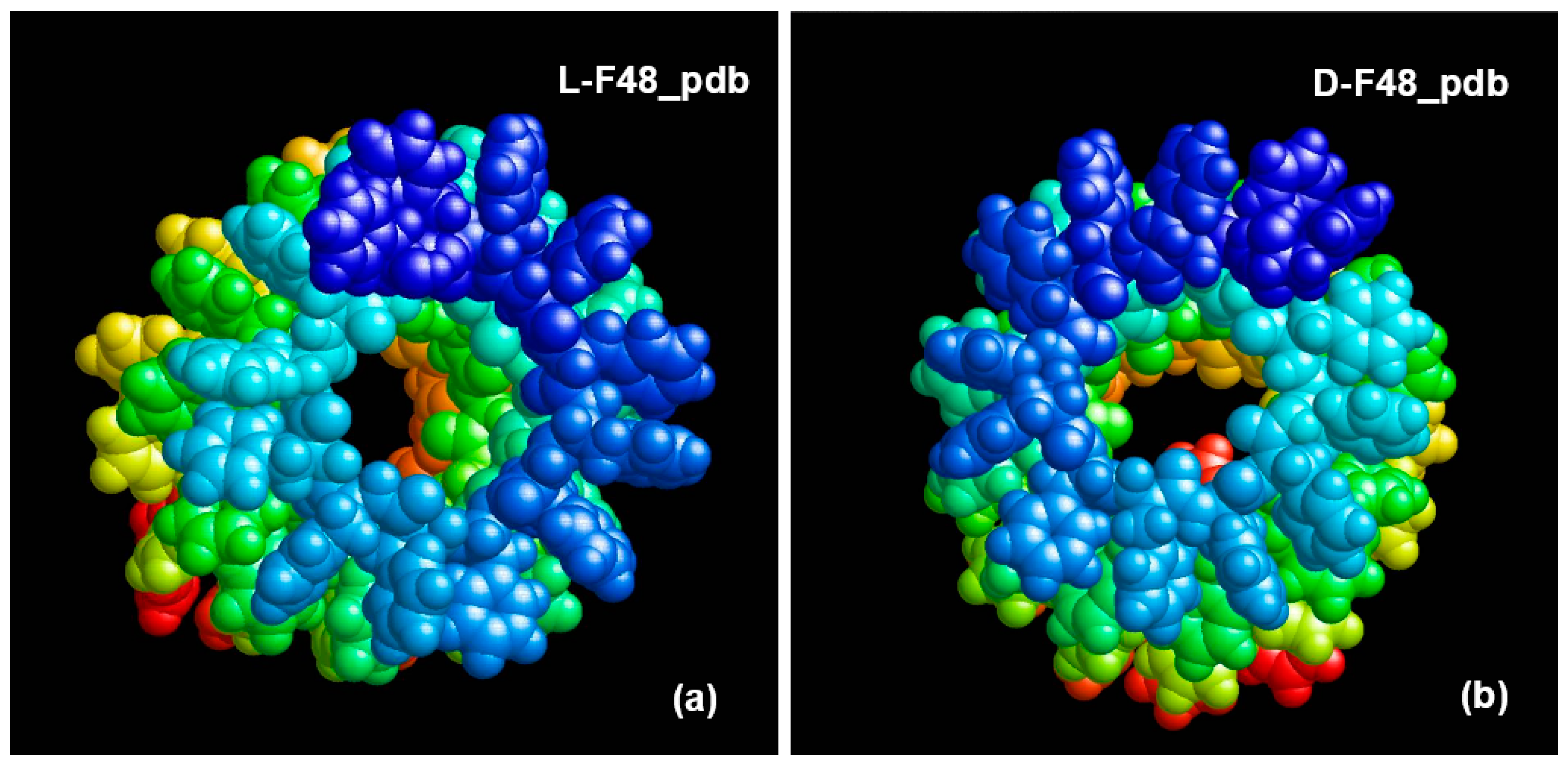
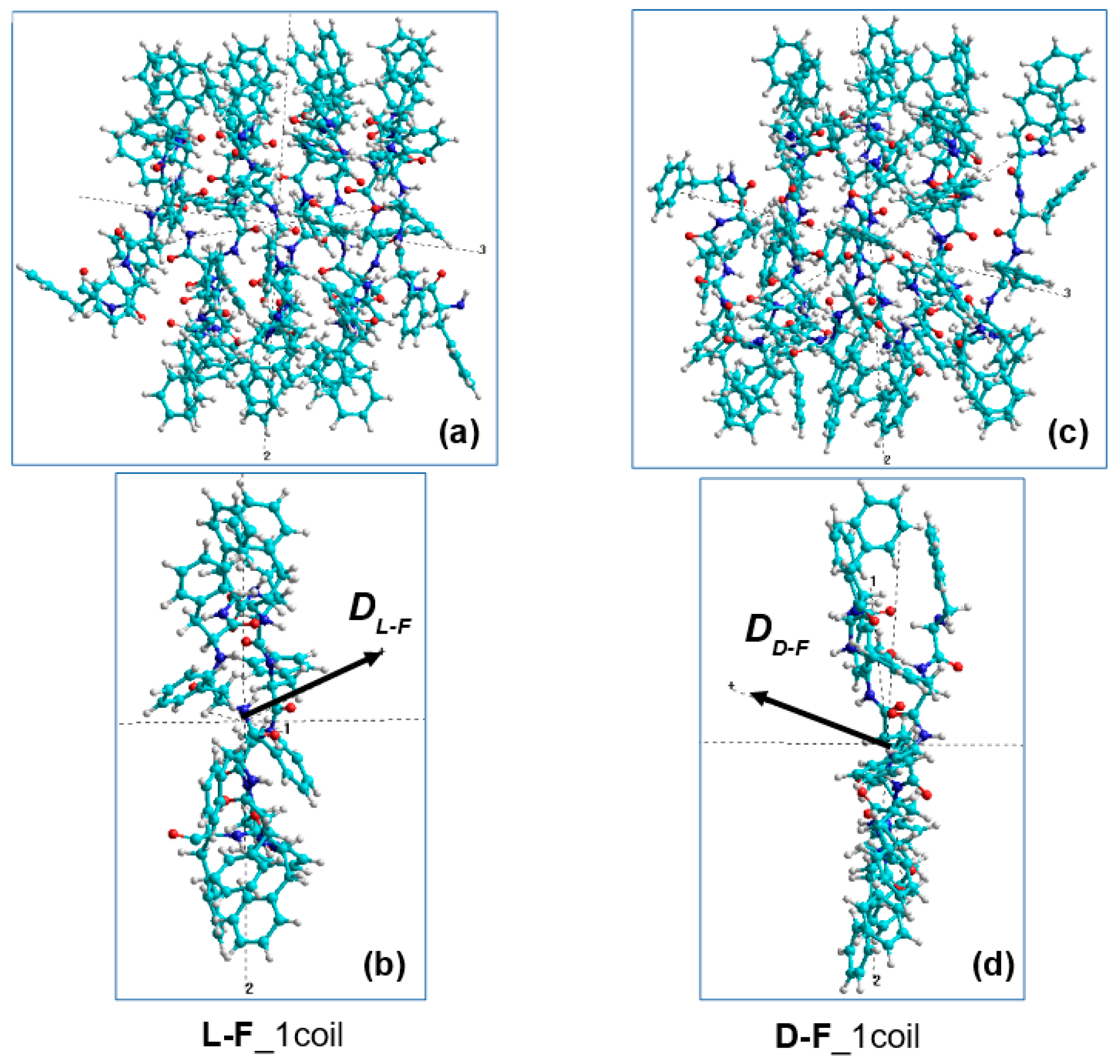
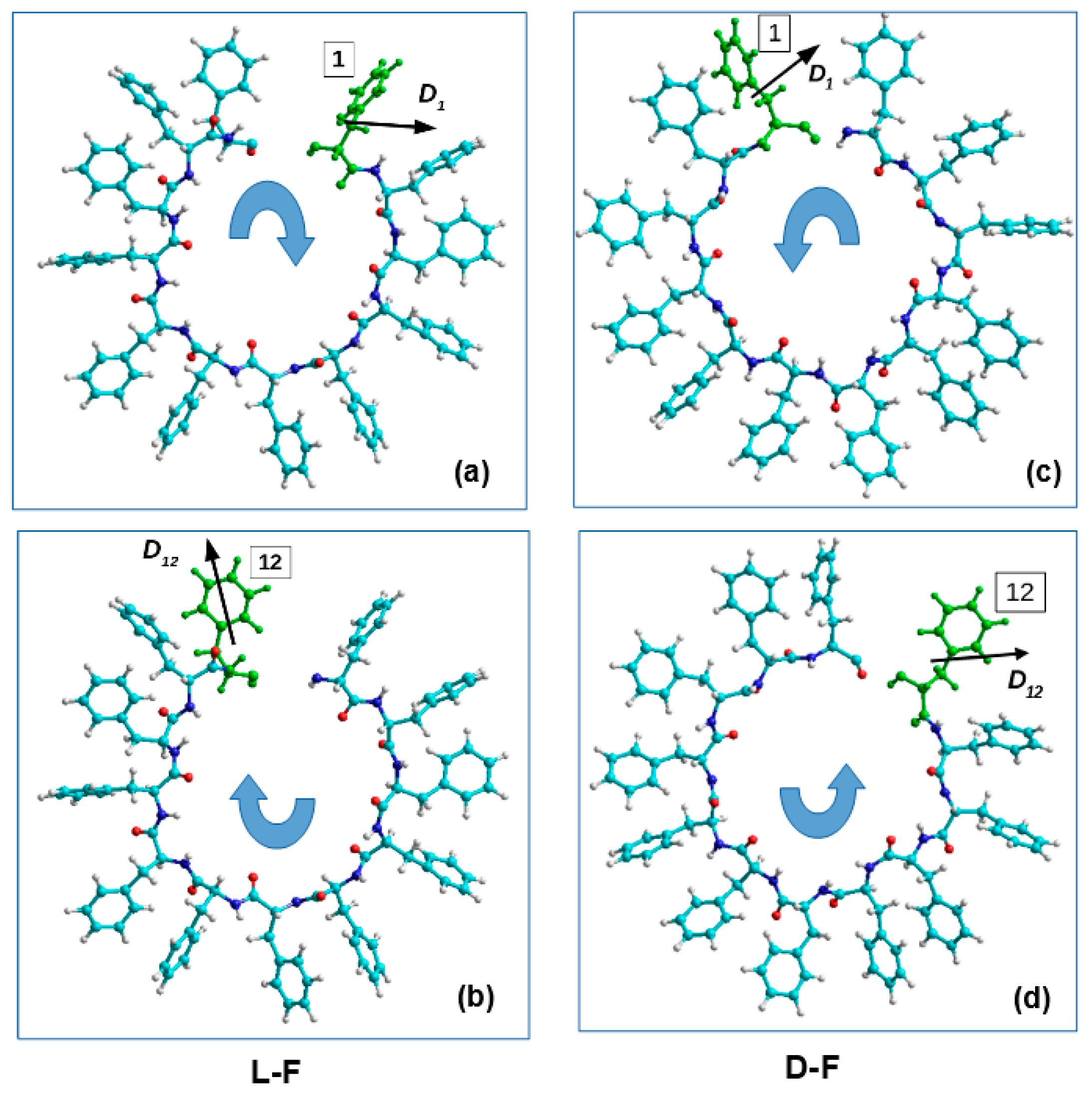
2.1.2. Peptide Nanotubes
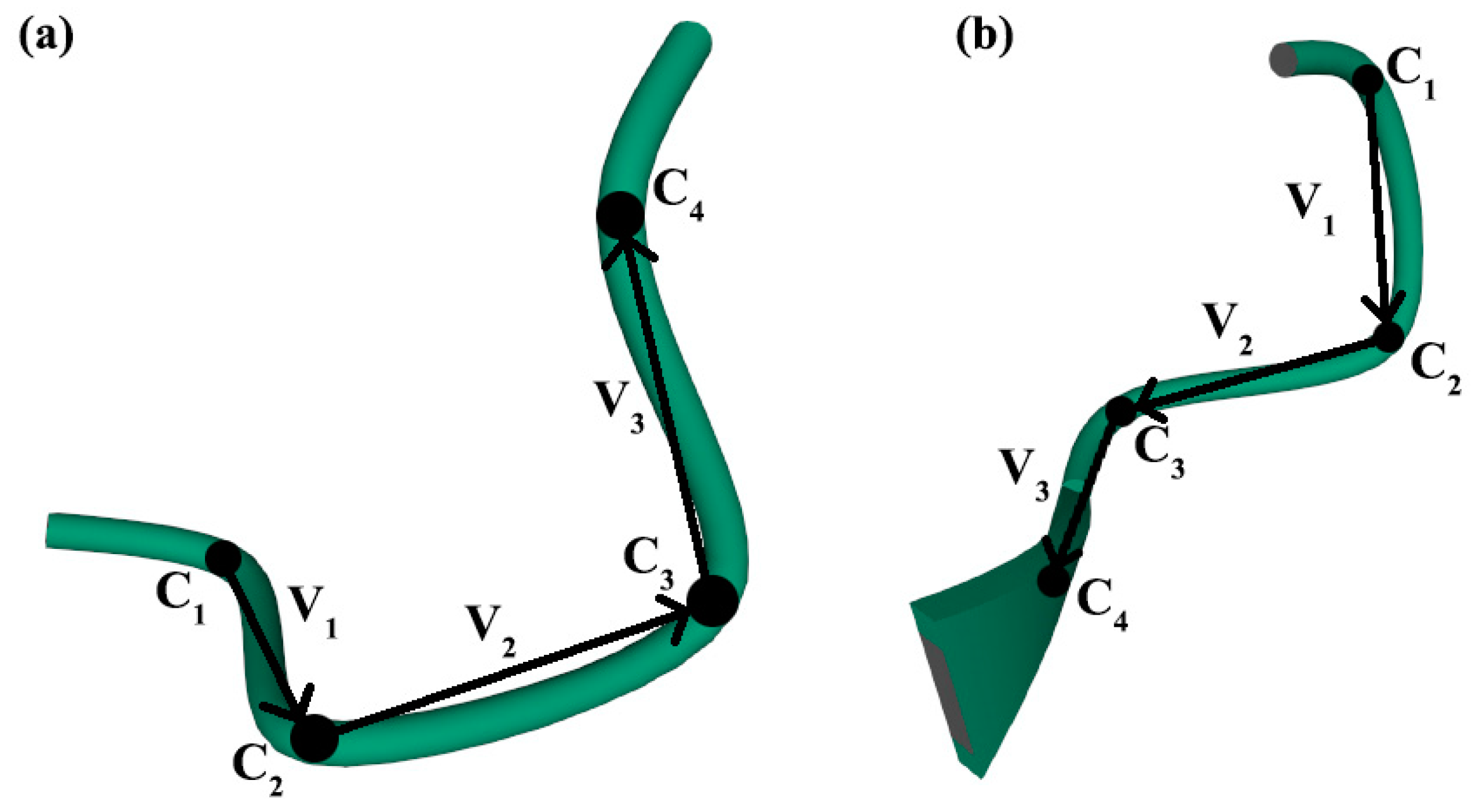
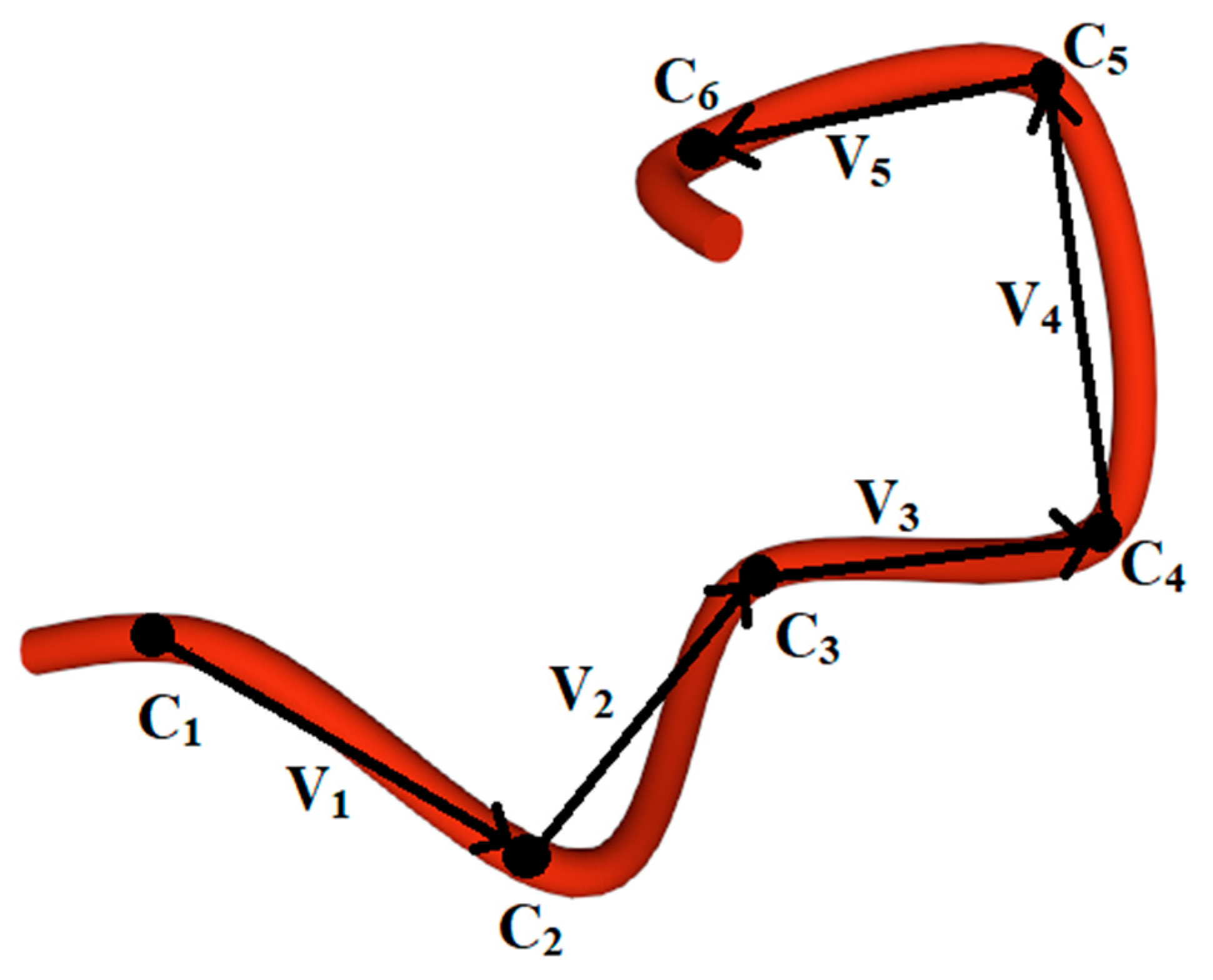
2.2. Methods for Evaluating the Chirality of Regular Helical and Irregular Protein Secondary Structures
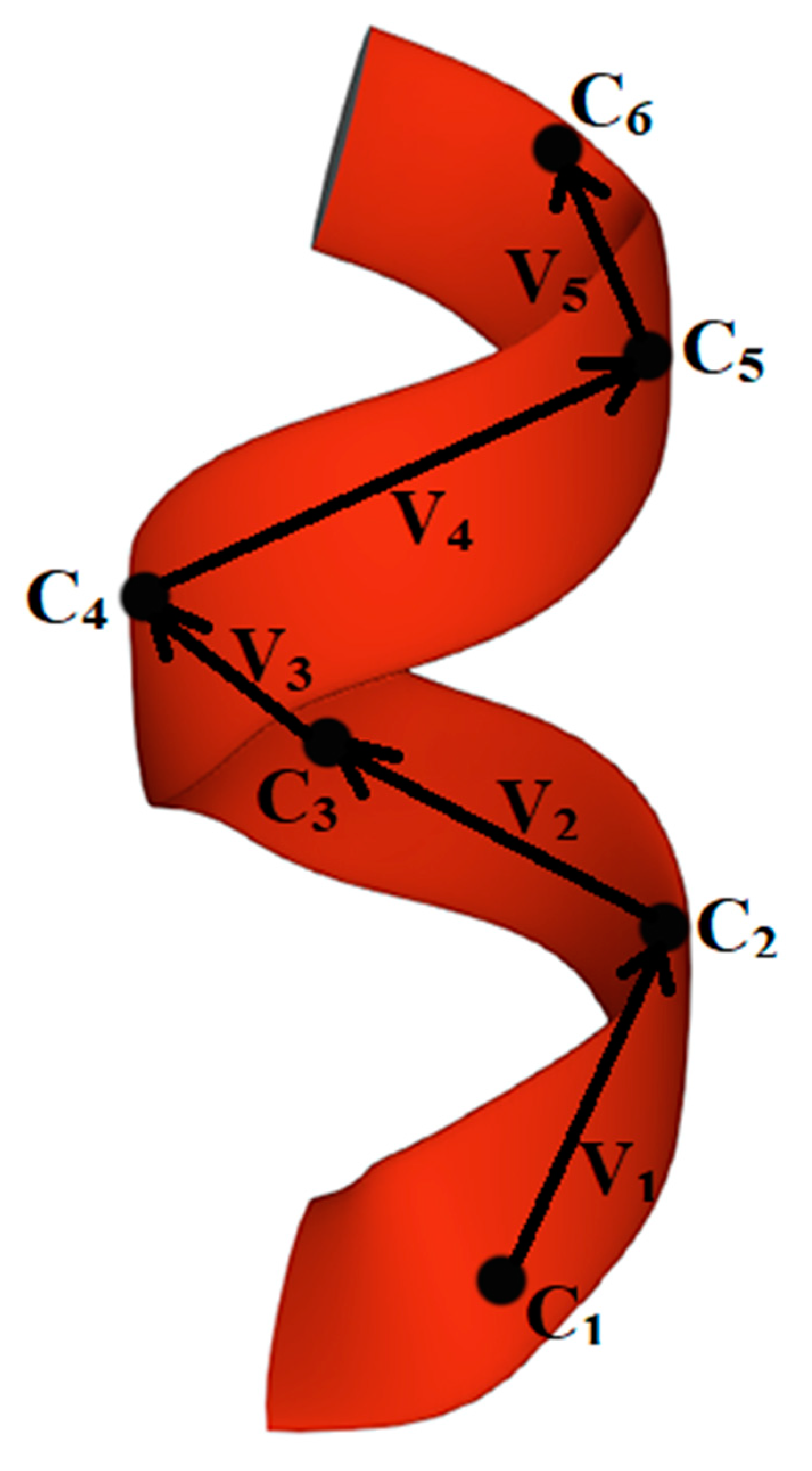
2.3. Method for Calculating the Chirality of Phenylalanine (F) Helical Peptide Nanotubes (PNT) from Successive Dipole Moments of Their Constituent Phenylalanine Molecules
3. Results
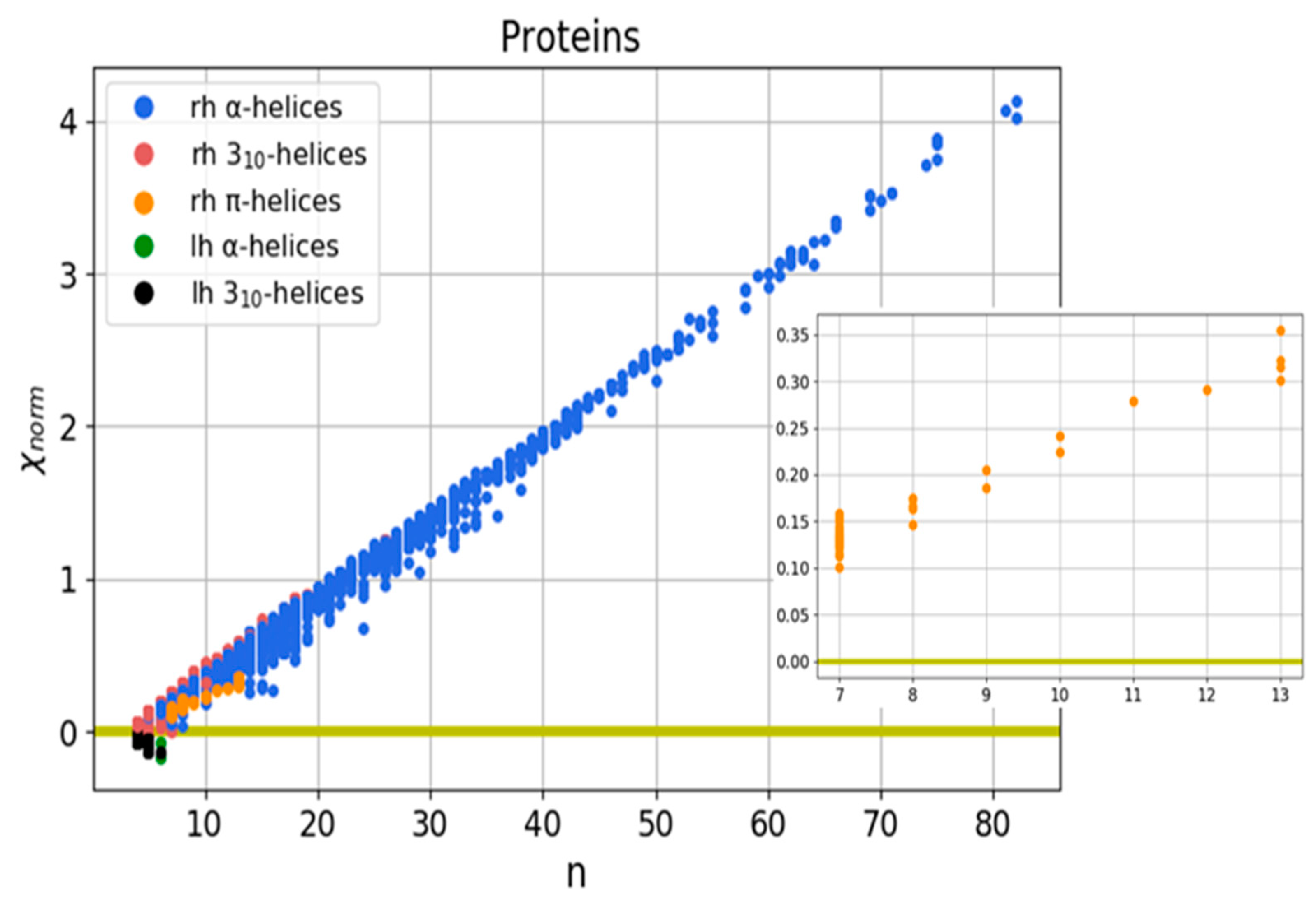
3.1. Helical Protein Secondary Structures
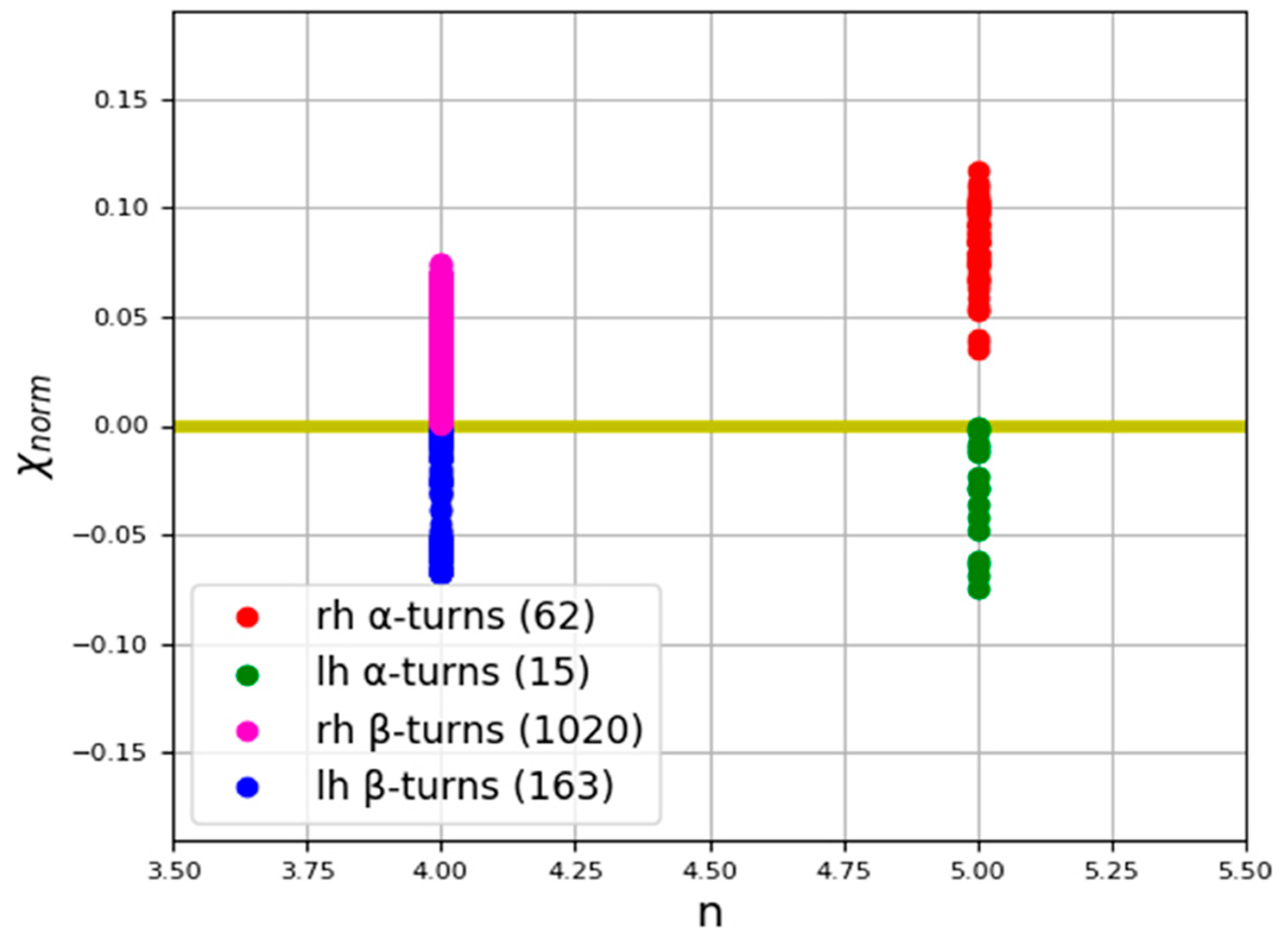
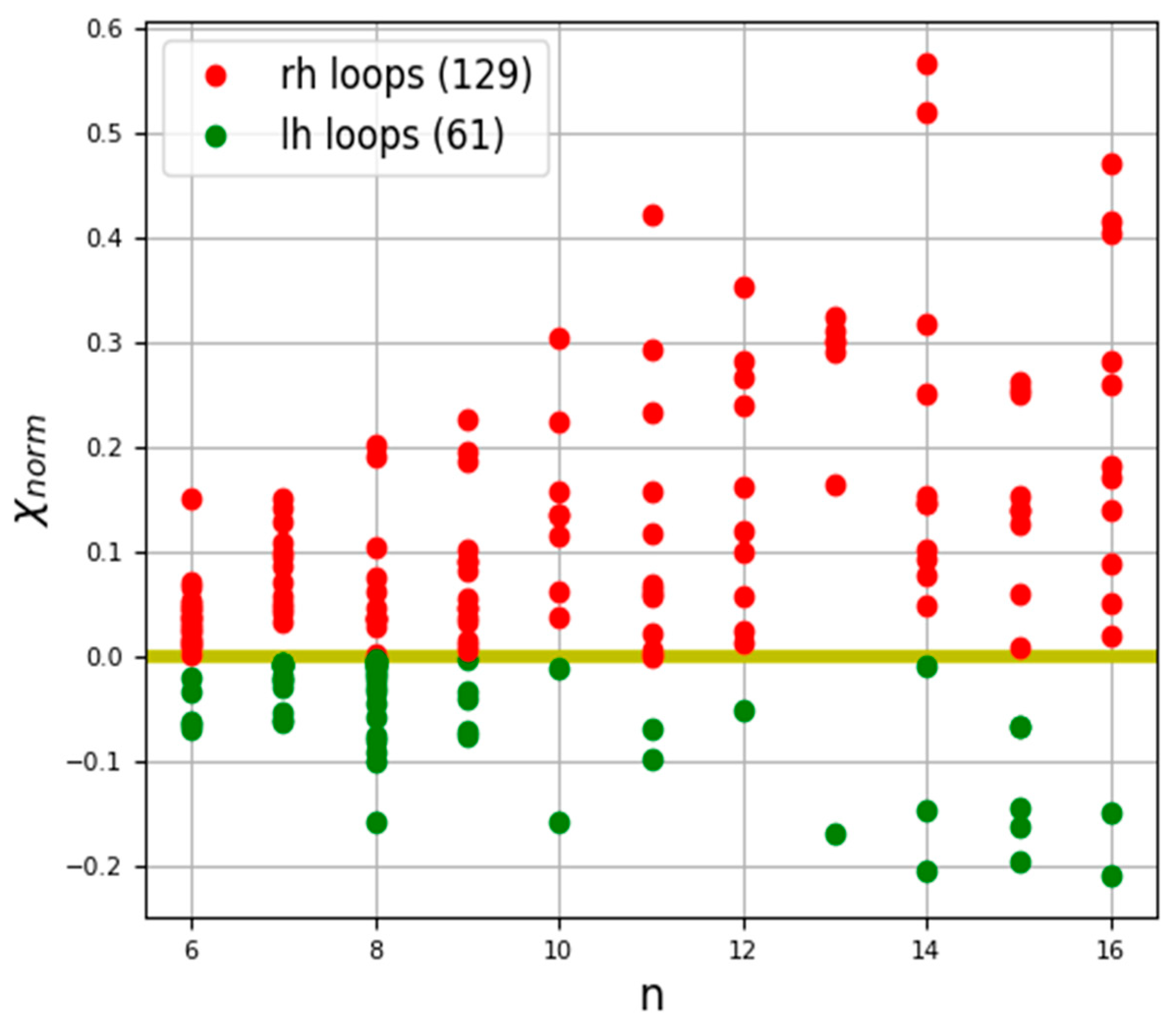
3.2. Irregular Protein Secondary Structures
3.3. Phenylalanine (F) Helix-like Peptide Nanotubes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein PDB ID | Chain/Residues | Length of Helix | Chirality Value | Chirality Sign | Protein Class |
|---|---|---|---|---|---|
| 1A8E | A/124-130 | 7 | 0.1150 | Right-handed | Iron transport |
| 1A8I | A/488-495 | 8 | 0.1632 | Right-handed | Transferase |
| 1BDM | A/217-223 | 7 | 0.1350 | Right-handed | Oxidoreductase |
| 1BG6 | A/297-303 | 7 | 0.1373 | Right-handed | Oxidoreductase |
| 1D3G | A/37-43 | 7 | 0.1283 | Right-handed | Oxidoreductase |
| 1DK8 | A/242-249 | 8 | 0.1711 | Right-handed | Signaling protein |
| 1DYS | A/112-118 | 7 | 0.1250 | Right-handed | Hydrolase |
| 1DZ4 | A/150-156 | 7 | 0.1273 | Right-handed | Oxidoreductase |
| 1E3A | A/138-146 | 9 | 0.2050 | Right-handed | Hydrolase |
| 1EGU | A/292-298 | 7 | 0.1323 | Right-handed | Lyase |
| 1EGU | A/441-447 | 7 | 0.1229 | Right-handed | Lyase |
| 1EI5 | A/177-183 | 7 | 0.1299 | Right-handed | Hydrolase |
| 1EK6 | A/105-111 | 7 | 0.1502 | Right-handed | Isomerase |
| 1EL4 | A/44-51 | 8 | 0.1665 | Right-handed | Luminescent protein |
| 1ELK | A/95-101 | 7 | 0.1421 | Right-handed | Endocytosis/Exocytosis |
| 1EOK | A/111-118 | 8 | 0.1670 | Right-handed | Hydrolase |
| 1EVY | A/257-263 | 7 | 0.1543 | Right-handed | Oxidoreductase |
| 1F3A | A/126-132 | 7 | 0.1235 | Right-handed | Transferase |
| 1F24 | A/214-220 | 7 | 0.1336 | Right-handed | Oxidoreductase |
| 1FQA | A/279-285 | 7 | 0.1304 | Right-handed | Sugar binding protein |
| 1KVE | B/177-183 | 7 | 0.1271 | Right-handed | Toxin |
| 1MRO | A/313-324 | 12 | 0.2911 | Right-handed | Transferase |
| 1MUC | A/70-76 | 7 | 0.1270 | Right-handed | Isomerase |
| 1NCI | A/40-46 | 7 | 0.1125 | Right-handed | Cell adhesin protein |
| 1QGW | C/105-111 | 7 | 0.1287 | Right-handed | Photosynthesis |
| 1QH3 | A/154-160 | 7 | 0.1316 | Right-handed | Hydrolase |
| 1QH8 | A/63-72 | 10 | 0.2242 | Right-handed | Oxidoreductase |
| 1QLM | A/88-94 | 7 | 0.1216 | Right-handed | Hydrolase |
| 1QMG | A/349-355 | 7 | 0.1503 | Right-handed | Oxidoreductase |
| 1QMG | A/490-502 | 13 | 0.3543 | Right-handed | Oxidoreductase |
| 1SMD | A/27-33 | 7 | 0.1276 | Right-handed | Hydrolase |
| 1SUR | A/131-137 | 7 | 0.1303 | Right-handed | Oxidoreductase |
| 1THG | A/424-430 | 7 | 0.1360 | Right-handed | Hydrolase |
| 2SCP | A/56-62 | 7 | 0.1344 | Right-handed | Binding protein |
| 1B16 | A/104-110 | 7 | 0.1277 | Right-handed | Oxidoreductase |
| 1B25 | A/479-485 | 7 | 0.1247 | Right-handed | Oxidoreductase |
| 1BDB | A/112-118 | 7 | 0.1379 | Right-handed | Oxidoreductase |
| 1BXK | A/98-104 | 7 | 0.1439 | Right-handed | Lyase |
| 1C3P | A/97-103 | 7 | 0.1447 | Right-handed | Lyase |
| 1C3W | A/213-219 | 7 | 0.1543 | Right-handed | Ion transport |
| 1C7S | A/641-647 | 7 | 0.0941 | Right-handed | Hydrolase |
| 1C7S | A/801-807 | 7 | 0.1273 | Right-handed | Hydrolase |
| 1CB8 | A/267-273 | 7 | 0.1317 | Right-handed | Lyase |
| 1COJ | A/26-33 | 8 | 0.1745 | Right-handed | Oxidoreductase |
| 1CXP | C/291-297 | 7 | 0.1234 | Right-handed | Oxidoreductase |
| 1CYD | A/104-110 | 7 | 0.1336 | Right-handed | Oxidoreductase |
| 1D3Y | A/253-259 | 7 | 0.1317 | Right-handed | Isomerase |
| 1D8D | A/343-349 | 7 | 0.1146 | Right-handed | Transferase |
| 1DC1 | A/98-104 | 7 | 0.1341 | Right-handed | Hydrolase |
| 1DEK | A/137-145 | 9 | 0.1858 | Right-handed | Transferase |
| 1DJ0 | A/81-87 | 7 | 0.1546 | Right-handed | Lyase |
| 1DOZ | A/265-274 | 10 | 0.2409 | Right-handed | Lyase |
| 1DQA | A/733-740 | 8 | 0.1738 | Right-handed | Oxidoreductase |
| 1DQS | A/142-148 | 7 | 0.1262 | Right-handed | Lyase |
| 1DXR | C/277-283 | 7 | 0.1231 | Right-handed | Photosynthesis |
| 1DXR | H/27-33 | 7 | 0.1234 | Right-handed | Photosynthesis |
| 1DXR | L/129-135 | 7 | 0.1276 | Right-handed | Photosynthesis |
| 1DXR | M/156-162 | 7 | 0.1229 | Right-handed | Photosynthesis |
| 1EA5 | A/396-402 | 7 | 0.1241 | Right-handed | Hydrolase |
| 1EA5 | A/522-528 | 7 | 0.1313 | Right-handed | Hydrolase |
| 1EWF | A/181-187 | 7 | 0.1379 | Right-handed | Antibiotic |
| 1EYZ | A/119-125 | 7 | 0.1426 | Right-handed | Transferase |
| 1F24 | A/140-146 | 7 | 0.1259 | Right-handed | Oxidoreductase |
| 1FDS | A/111-117 | 7 | 0.1373 | Right-handed | Oxidoreductase |
| 1FP3 | A/273-279 | 7 | 0.1241 | Right-handed | Isomerase |
| 1FRP | A/276-282 | 7 | 0.1304 | Right-handed | Hydrolase |
| 1FSW | A/174-180 | 7 | 0.1318 | Right-handed | Hydrolase |
| 1FUR | A/155-161 | 7 | 0.1348 | Right-handed | Hydrolase |
| 1FUR | A/383-389 | 7 | 0.1302 | Right-handed | Hydrolase |
| 1G8K | A/181-187 | 7 | 0.1299 | Right-handed | Oxidoreductase |
| 1G8K | A/242-248 | 7 | 0.1418 | Right-handed | Oxidoreductase |
| 1GAI | A/150-156 | 7 | 0.1346 | Right-handed | Hydrolase |
| 1HFE | S/71-77 | 7 | 0.1228 | Right-handed | Oxidoreductase |
| 1HVB | A/183-189 | 7 | 0.1282 | Right-handed | Hydrolase |
| 1I0H | A/26-32 | 7 | 0.1402 | Right-handed | Oxidoreductase |
| 1LML | A/155-161 | 7 | 0.1243 | Right-handed | Hydrolase |
| 1LST | A/126-132 | 7 | 0.1449 | Right-handed | Amino-acid binding protein |
| 1LST | A/165-171 | 7 | 0.1207 | Right-handed | Amino-acid binding protein |
| 1MTY | B/140-150 | 11 | 0.2786 | Right-handed | Oxidoreductase |
| 1MTY | B/297-304 | 8 | 0.1463 | Right-handed | Oxidoreductase |
| 1MTY | D/185-191 | 7 | 0.1139 | Right-handed | Oxidoreductase |
| 1MTY | D/202-214 | 13 | 0.3008 | Right-handed | Oxidoreductase |
| 1MTY | D/306-318 | 13 | 0.3226 | Right-handed | Oxidoreductase |
| 1MTY | D/379-385 | 7 | 0.1590 | Right-handed | Oxidoreductase |
| 1ONE | A/67-73 | 7 | 0.1322 | Right-handed | Lyase |
| 1PHN | A/107-113 | 7 | 0.1335 | Right-handed | Electron transport |
| 1QOY | A/26-32 | 7 | 0.1315 | Right-handed | Toxin |
| 1SVF | A/171-177 | 7 | 0.1381 | Right-handed | Viral protein |
| 1UOK | A/393-399 | 7 | 0.1330 | Right-handed | Hydrolase |
| 1YAC | A/114-120 | 7 | 0.1410 | Right-handed | Hydrolase |
| 1YGE | A/261-267 | 7 | 0.1353 | Right-handed | Oxidoreductase |
| 1YGE | A/494-506 | 13 | 0.3152 | Right-handed | Oxidoreductase |
| 1YGE | A/684-690 | 7 | 0.1398 | Right-handed | Oxidoreductase |
| 2EBN | A/257-263 | 7 | 0.1327 | Right-handed | Hydrolase |
| 2HMQ | A/101-107 | 7 | 0.1429 | Right-handed | Oxygen transport |
| 2OLB | A/301-308 | 8 | 0.1504 | Right-handed | Binding protein |
| 4PAN | A/325-331 | 7 | 0.1580 | Right-handed | Signaling protein |
| 5CSM | A/233-239 | 7 | 0.1380 | Right-handed | Isomerase |
| 7A3H | A/146-152 | 7 | 0.1293 | Right-handed | Hydrolase |
| 9GAF | A/186-192 | 7 | 0.1005 | Right-handed | Hydrolase |
| Family or Group | Protein PDB ID | Chain/Residues | Chirality Value | Chirality Sign | Protein Class |
|---|---|---|---|---|---|
| F1 | 1AAP | A/24-28 | 0.0917 | Right-handed | Proteinase Inhibitor (Trypsin) |
| 1ACX | A/82-86 | 0.0735 | Right-handed | Antibacterial Protein | |
| 2AK3 | A/149-153 | 0.0694 | Right-handed | Transferase (Phosphotransferase) | |
| 3COX | A/391-395 | 0.0852 | Right-handed | Oxidoreductase (Oxygen Receptor) | |
| 1DRF | A/152-156 | 0.0837 | Right-handed | Oxidoreductase (Ch-Nh(D)-Nad Or Nadp (A)) | |
| 1ECA | A/38-42 | 0.0884 | Right-handed | Oxygen Transport | |
| 1GD1 | O/47-51 | 0.0668 | Right-handed | Oxidoreductase (Aldehyde(D)-Nad (A)) | |
| 1GOX | A/345-349 | 0.1173 | Right-handed | Oxidoreductase (Oxygen(A)) | |
| 1MBA | A/43-47 | 0.0861 | Right-handed | Oxygen Storage | |
| 1OMD | A/2-6 | 0.0911 | Right-handed | Calcium Binding Protein | |
| 1OVA | A/277-281 | 0.0764 | Right-handed | Serpin | |
| 1OVA | A/318-322 | 0.1015 | Right-handed | Serpin | |
| 1RDG | A/6-10 | 0.0552 | Right-handed | Electron Transfer (Iron-Sulfur Protein) | |
| 1RDG | A/14-18 | 0.0976 | Right-handed | Electron Transfer (Iron-Sulfur Protein) | |
| 1RDG | A/39-43 | 0.0585 | Right-handed | Electron Transfer (Iron-Sulfur Protein) | |
| 2SN3 | A/7-11 | 0.0626 | Right-handed | Toxin | |
| 2SN4 | A/31-35 | 0.0847 | Right-handed | Toxin | |
| 1THB | A/113-117 | 0.0937 | Right-handed | Oxygen Transport | |
| 2ACT | A/85-89 | 0.1009 | Right-handed | Hydrolase (Proteinase) | |
| 2AZA | A/40-44 | 0.0952 | Right-handed | Electron Transport Protein (Cuproprotein) | |
| 2CA2 | A/34-38 | 0.1028 | Right-handed | Lyase (Oxo-Acid) | |
| 2CPP | A/77-81 | 0.0883 | Right-handed | Oxidoreductase (Oxygenase) | |
| 2CPP | A/328-332 | 0.1000 | Right-handed | Oxidoreductase (Oxygenase) | |
| 2CSC | A/59-63 | 0.0674 | Right-handed | Lyase | |
| 2CYP | A/58-62 | 0.0673 | Right-handed | Oxidoreductase (H2O2(A)) | |
| 2ER7 | E/240-244 | 0.0926 | Right-handed | Hydrolase/Hydrolase Inhibitor | |
| 2FCR | A/94-98 | 0.0923 | Right-handed | Electron Transport | |
| 2FCR | A/148-152 | 0.0841 | Right-handed | Electron Transport | |
| 2LTN | A/54-58 | 0.0776 | Right-handed | Lectin | |
| 2LTN | A/125-129 | 0.0523 | Right-handed | Lectin | |
| 2LTN | A/167-171 | 0.0852 | Right-handed | Lectin | |
| 2RHE | A/93-97 | 0.0986 | Right-handed | Immunoglobulin | |
| 2RSP | A/46-50 | 0.1031 | Right-handed | Hydrolase (Aspartyl Proteinase) | |
| 2RSP | A/219-223 | 0.0798 | Right-handed | Hydrolase (Serine Proteinase) | |
| 2TRX | A/59-63 | 0.0993 | Right-handed | Electron Transport | |
| 3BLM | A/50-54 | 0.0742 | Right-handed | Hydrolase | |
| 3CLA | A/97-101 | 0.0833 | Right-handed | Transferase (Acyltransferase) | |
| 3CLA | A/194-198 | 0.0880 | Right-handed | Transferase (Acyltransferase) | |
| 4FGF | A/67-71 | 0.0920 | Right-handed | Growth Factor | |
| 3GRS | A/164-168 | 0.0968 | Right-handed | Oxidoreductase (Flavoenzyme) | |
| 4ENL | A/102-106 | 0.0738 | Right-handed | Carbon-Oxygen Lyase | |
| 5FD1 | A/35-39 | 0.0986 | Right-handed | Electron Transport(Iron-Sulfur) | |
| 5CPA | A/3-7 | 0.1063 | Right-handed | Hydrolase (C-Terminal Peptidase) | |
| 5CPA | A/29-33 | 0.0905 | Right-handed | Hydrolase (C-Terminal Peptidase) | |
| 5P21 | A/145-149 | 0.0840 | Right-handed | Oncogene Protein | |
| 6LDH | A/181-185 | 0.1114 | Right-handed | Oxidoreductase(Choh(D)-Nad(A)) | |
| F2 | 2AK3 | A/137-141 | 0.0723 | Right-handed | Transferase (Phosphotransferase) |
| 3COX | A/453-457 | 0.0649 | Right-handed | Oxidoreductase (Oxygen Receptor) | |
| 1FKF | A/87-91 | 0.0400 | Right-handed | Isomerase | |
| 1GD1 | O/129-133 | 0.0791 | Right-handed | Oxidoreductase (Aldehyde(D)-Nad(A)) | |
| 1GD2 | O/267-271 | 0.0761 | Right-handed | Oxidoreductase (Aldehyde(D)-Nad(A)) | |
| 2TEC | E/261-265 | 0.0750 | Right-handed | Complex(Serine Proteinase-Inhibitor) | |
| 2TRX | A/49-53 | 0.0799 | Right-handed | Electron Transport | |
| 5CPA | A/89-93 | 0.0664 | Right-handed | Hydrolase (C-Terminal Peptidase) | |
| g1 | 4GCR | A/47-51 | −0.0291 | Left-handed | Eye Lens Protein |
| 4GCR | A/136-140 | −0.0283 | Left-handed | Eye Lens Protein | |
| 2CYP | A/35-39 | −0.0422 | Left-handed | Oxidoreductase (H2O2(A)) | |
| 2FBJ | A/48-52 | −0.0411 | Left-handed | Immunoglobulin | |
| 2PRK | A/212-216 | −0.0230 | Left-handed | Serine Proteinase | |
| g2 | 2FBJ | H/100-104 | −0.0355 | Left-handed | Immunoglobulin |
| 2RHE | A/50-54 | −0.0478 | Left-handed | Immunoglobulin | |
| g3 | 1FKF | A/82-86 | 0.0384 | Right-handed | Isomerase |
| 1OVA | A/69-73 | 0.0523 | Right-handed | Serpin | |
| 3GRS | A/55-59 | 0.0351 | Right-handed | Oxidoreductase (Flavoenzyme) | |
| 4BP2 | A/25-29 | −0.0123 | Left-handed | Carboxylic Ester Hydrolase Zymogen | |
| g4 | 2ACT | A/188-192 | −0.0636 | Left-handed | Hydrolase (Proteinase) |
| 2LZM | A/27-31 | −0.0613 | Left-handed | Hydrolase (O-Glycosyl) | |
| 3APR | E/11-15 | −0.0684 | Left-handed | Hydrolase/Hydrolase Inhibitor | |
| 2FOX | A/56-60 | −0.0741 | Left-handed | Electron Transport | |
| g5 | 1FX1 | A/71-75 | 0.1042 | Right-handed | Electron Transfer (Flavoprotein) |
| 1YPI | A/25-29 | 0.1020 | Right-handed | Isomerase (Intramolecular Oxidoreductase) | |
| 2FOX | A/40-44 | 0.1004 | Right-handed | Electron Transport | |
| g6 | 1GD1 | O/300-304 | −0.0084 | Left-handed | Oxidoreductase (Aldehyde(D)-Nad(A)) |
| 2LTN | A/100-104 | −0.0026 | Left-handed | Lectin | |
| 4PEP | A/9-13 | −0.0003 | Left-handed | Hydrolase (Acid Proteinase) | |
| g7 | 2AK3 | A/129-133 | 0.0784 | Right-handed | Transferase (Phosphotransferase) |
| 1FX1 | A/72-76 | 0.1092 | Right-handed | Electron Transfer (Flavoprotein) | |
| Other | 1RBP | A/63-67 | −0.0099 | Left-handed | Retinol Transport |
| 2FBJ | L/166-170 | 0.0745 | Right-handed | Immunoglobulin |
| Protein PDB ID | Chain/Residues | Length of Loop | Chirality Value | Chirality Sign | Protein Class |
|---|---|---|---|---|---|
| 1ABE | A/93-99 | 7 | −0.0280 | Left-handed | Binding Protein |
| 1ABE | A/142-148 | 7 | 0.1496 | Right-handed | Binding Protein |
| 1ABE | A/203-208 | 6 | 0.0231 | Right-handed | Binding Protein |
| 1ABE | A/236-248 | 13 | 0.1632 | Right-handed | Binding Protein |
| 1ABE | A/289-294 | 6 | 0.0232 | Right-handed | Binding Protein |
| 1ABE | A/299-304 | 6 | 0.0464 | Right-handed | Binding Protein |
| 2ACT | A/8-13 | 6 | 0.0508 | Right-handed | Hydrolase (Proteinase) |
| 2ACT | A/58-64 | 7 | 0.0462 | Right-handed | Hydrolase (Proteinase) |
| 2ACT | A/89-103 | 15 | −0.1453 | Left-handed | Hydrolase (Proteinase) |
| 2ACT | A/139-144 | 6 | 0.0709 | Right-handed | Hydrolase (Proteinase) |
| 2ACT | A/141-156 | 16 | 0.1703 | Right-handed | Hydrolase (Proteinase) |
| 2ACT | A/182-192 | 11 | 0.0027 | Right-handed | Hydrolase (Proteinase) |
| 2ACT | A/198-205 | 8 | −0.0151 | Left-handed | Hydrolase (Proteinase) |
| 2ACT | A/203-209 | 7 | −0.0605 | Left-handed | Hydrolase (Proteinase) |
| 8ADH | A/14-21 | 8 | −0.0071 | Left-handed | Oxidoreductase (Nad(A)-Choh(D)) |
| 8ADH | A/100-112 | 13 | 0.3005 | Right-handed | Oxidoreductase (Nad(A)-Choh(D)) |
| 8ADH | A/115-122 | 8 | −0.0038 | Left-handed | Oxidoreductase (Nad(A)-Choh(D)) |
| 8ADH | A/122-128 | 7 | −0.0191 | Left-handed | Oxidoreductase (Nad(A)-Choh(D)) |
| 8ADH | A/282-287 | 6 | 0.0022 | Right-handed | Oxidoreductase (Nad(A)-Choh(D)) |
| 3ADK | A/133-142 | 10 | 0.0617 | Right-handed | Transferase(Phosphotransferase) |
| 2ALP | A/217-224 | 8 | −0.0460 | Left-handed | Hydrolase (Serine Proteinase) |
| 3APP | A/41-55 | 15 | 0.2606 | Right-handed | Hydrolase (Acid Proteinase) |
| 3APP | A/129-136 | 8 | 0.0022 | Right-handed | Hydrolase (Acid Proteinase) |
| 3APP | A/139-149 | 11 | 0.4213 | Right-handed | Hydrolase (Acid Proteinase) |
| 3APP | A/184-192 | 9 | 0.0463 | Right-handed | Hydrolase (Acid Proteinase) |
| 2APR | A/8-17 | 10 | −0.1572 | Left-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/18-31 | 14 | −0.2057 | Left-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/43-58 | 16 | 0.0879 | Right-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/61-69 | 9 | 0.1019 | Right-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/76-83 | 8 | −0.0071 | Left-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/90-103 | 14 | −0.1480 | Left-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/129-138 | 10 | 0.0383 | Right-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/189-197 | 9 | 0.0337 | Right-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/203-211 | 9 | −0.0328 | Left-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/216-226 | 11 | 0.0677 | Right-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/227-232 | 6 | 0.1507 | Right-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/233-248 | 16 | 0.0514 | Right-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/243-250 | 8 | −0.0071 | Left-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/261-273 | 13 | −0.1703 | Left-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/280-287 | 8 | −0.0761 | Left-handed | Hydrolase (Aspartic Proteinase) |
| 2APR | A/291-299 | 9 | −0.0724 | Left-handed | Hydrolase (Aspartic Proteinase) |
| 1AZU | A/9-15 | 7 | −0.0069 | Left-handed | Electron Transport (Copper Binding) |
| 1AZU | A/35-46 | 12 | 0.0569 | Right-handed | Electron Transport (Copper Binding) |
| 1AZU | A/67-72 | 6 | 0.0376 | Right-handed | Electron Transport (Copper Binding) |
| 1AZU | A/73-83 | 11 | 0.0646 | Right-handed | Electron Transport (Copper Binding) |
| 1AZU | A/84-92 | 9 | −0.0023 | Left-handed | Electron Transport (Copper Binding) |
| 1AZU | A/112-118 | 7 | 0.0420 | Right-handed | Electron Transport (Copper Binding) |
| 1CYO | A/32-47 | 16 | 0.4146 | Right-handed | Electron Transport |
| 1BP2 | A/23-30 | 8 | 0.0360 | Right-handed | Hydrolase |
| 1BP3 | A/25-39 | 15 | −0.0675 | Left-handed | Hydrolase |
| 1BP4 | A/56-66 | 11 | 0.2318 | Right-handed | Hydrolase |
| 2BP2 | A/23-30 | 8 | 0.0360 | Right-handed | Hydrolase Zymogen |
| 2BP3 | A/25-39 | 15 | −0.0675 | Left-handed | Hydrolase Zymogen |
| 2BP4 | A/61-68 | 8 | −0.0056 | Left-handed | Hydrolase Zymogen |
| 256B | A/16-25 | 10 | 0.1574 | Right-handed | Electron Transport |
| 256B | A/47-58 | 12 | 0.1626 | Right-handed | Electron Transport |
| 351C | A/16-25 | 10 | −0.0117 | Left-handed | Electron Transport |
| 351C | A/51-62 | 12 | 0.0243 | Right-handed | Electron Transport |
| 155C | A/21-28 | 8 | −0.1010 | Left-handed | Electron Transport |
| 155C | A/47-54 | 8 | 0.0381 | Right-handed | Electron Transport |
| 155C | A/83-95 | 13 | 0.3003 | Right-handed | Electron Transport |
| 155C | A/128-133 | 6 | 0.0115 | Right-handed | Electron Transport |
| 2C2C | A/18-33 | 16 | −0.2105 | Left-handed | Electron Transport Protein (Cytochrome) |
| 2C2C | A/30-43 | 14 | 0.1534 | Right-handed | Electron Transport Protein (Cytochrome) |
| 2C2C | A/41-56 | 16 | 0.2603 | Right-handed | Electron Transport Protein (Cytochrome) |
| 2C2C | A/74-89 | 16 | 0.4033 | Right-handed | Electron Transport Protein (Cytochrome) |
| 2CAB | A/6-12 | 7 | 0.0716 | Right-handed | Hydro-Lyase |
| 2CAB | A/17-24 | 8 | 0.1911 | Right-handed | Hydro-Lyase |
| 2CAB | A/78-87 | 10 | 0.1359 | Right-handed | Hydro-Lyase |
| 2CAB | A/98-104 | 7 | 0.0950 | Right-handed | Hydro-Lyase |
| 2CAB | A/108-114 | 7 | −0.0536 | Left-handed | Hydro-Lyase |
| 2CAB | A/128-140 | 13 | 0.2908 | Right-handed | Hydro-Lyase |
| 2CAB | A/197-204 | 8 | −0.0225 | Left-handed | Hydro-Lyase |
| 2CAB | A/230-240 | 11 | −0.0683 | Left-handed | Hydro-Lyase |
| 1CA2 | A/5-16 | 12 | 0.1204 | Right-handed | Lyase (Oxo-Acid) |
| 1CA3 | A/17-23 | 7 | 0.1274 | Right-handed | Lyase (Oxo-Acid) |
| 1CA4 | A/98-103 | 6 | 0.0470 | Right-handed | Lyase (Oxo-Acid) |
| 1CA5 | A/108-114 | 7 | −0.0629 | Left-handed | Lyase (Oxo-Acid) |
| 1CA6 | A/128-140 | 13 | 0.3094 | Right-handed | Lyase (Oxo-Acid) |
| 1CA7 | A/166-172 | 7 | 0.0573 | Right-handed | Lyase (Oxo-Acid) |
| 1CA8 | A/197-204 | 8 | −0.0111 | Left-handed | Lyase (Oxo-Acid) |
| 1CA9 | A/232-239 | 8 | −0.0806 | Left-handed | Lyase (Oxo-Acid) |
| 2CHA | B/70-78 | 9 | 0.0557 | Right-handed | Hydrolase (Serine Proteinase) |
| 2CHA | B/94-102 | 9 | 0.0806 | Right-handed | Hydrolase (Serine Proteinase) |
| 2CHA | B/114-119 | 6 | 0.0369 | Right-handed | Hydrolase (Serine Proteinase) |
| 2CHA | C/217-224 | 8 | −0.0060 | Left-handed | Hydrolase (Serine Proteinase) |
| 3CNA | A/13-21 | 9 | 0.0381 | Right-handed | Lectin (Agglutinin) |
| 3CNA | A/97-104 | 8 | −0.0102 | Left-handed | Lectin (Agglutinin) |
| 3CNA | A/116-123 | 8 | 0.0756 | Right-handed | Lectin (Agglutinin) |
| 3CNA | A/147-155 | 9 | 0.0144 | Right-handed | Lectin (Agglutinin) |
| 3CNA | A/160-165 | 6 | 0.0127 | Right-handed | Lectin (Agglutinin) |
| 3CNA | A/199-209 | 11 | 0.0003 | Right-handed | Lectin (Agglutinin) |
| 3CNA | A/222-235 | 14 | 0.1464 | Right-handed | Lectin (Agglutinin) |
| 3CNA | A/229-237 | 9 | 0.0108 | Right-handed | Lectin (Agglutinin) |
| 3CPA | A/128-141 | 14 | 0.0767 | Right-handed | Hydrolase (C-Terminal Peptidase) |
| 3CPA | A/142-156 | 15 | 0.2551 | Right-handed | Hydrolase (C-Terminal Peptidase) |
| 3CPA | A/156-166 | 11 | 0.1575 | Right-handed | Hydrolase (C-Terminal Peptidase) |
| 3CPA | A/205-213 | 9 | 0.0898 | Right-handed | Hydrolase (C-Terminal Peptidase) |
| 3CPA | A/231-237 | 7 | −0.0073 | Left-handed | Hydrolase (C-Terminal Peptidase) |
| 3CPA | A/244-250 | 7 | −0.0284 | Left-handed | Hydrolase (C-Terminal Peptidase) |
| 3CPA | A/272-285 | 14 | −0.0102 | Left-handed | Hydrolase (C-Terminal Peptidase) |
| 5CPV | A/18-23 | 6 | 0.0092 | Right-handed | Calcium Binding |
| 5CPV | A/64-77 | 14 | 0.2505 | Right-handed | Calcium Binding |
| 1CRN | A/33-44 | 12 | −0.0524 | Left-handed | Plant Protein |
| 1CTX | A/1-15 | 15 | 0.0597 | Right-handed | Toxin |
| 1CTX | A/26-35 | 10 | 0.1348 | Right-handed | Toxin |
| 1CYC | A/18-32 | 15 | −0.1967 | Left-handed | Electron Transport |
| 1CYC | A/30-43 | 14 | 0.0486 | Right-handed | Electron Transport |
| 1CYC | A/40-54 | 15 | 0.1398 | Right-handed | Electron Transport |
| 1CYC | A/70-84 | 15 | 0.1522 | Right-handed | Electron Transport |
| 3CYT | O/18-32 | 15 | −0.1637 | Left-handed | Electron Transport (Heme Protein) |
| 3CYT | O/34-43 | 10 | 0.1153 | Right-handed | Electron Transport (Heme Protein) |
| 3CYT | O/40-54 | 15 | 0.1250 | Right-handed | Electron Transport (Heme Protein) |
| 3CYT | O/70-84 | 15 | 0.2509 | Right-handed | Electron Transport (Heme Protein) |
| 1ECD | A/33-42 | 10 | 0.3042 | Right-handed | Oxygen Transport |
| 1ECD | A/41-49 | 9 | 0.0899 | Right-handed | Oxygen Transport |
| 1EST | A/69-80 | 12 | 0.0984 | Right-handed | Hydrolase |
| 1EST | A/94-104 | 11 | 0.1166 | Right-handed | Hydrolase |
| 1EST | A/112-118 | 7 | −0.0091 | Left-handed | Hydrolase |
| 1EST | A/165-178 | 14 | 0.3179 | Right-handed | Hydrolase |
| 1EST | A/216-226 | 11 | 0.0211 | Right-handed | Hydrolase |
| 7FAB | L/24-29 | 6 | 0.0311 | Right-handed | Immune System |
| 7FAB | L/122-132 | 11 | −0.0979 | Left-handed | Immune System |
| 7FAB | L/168-173 | 6 | −0.0200 | Left-handed | Immune System |
| 7FAB | L/182-187 | 6 | 0.0160 | Right-handed | Immune System |
| 7FAB | H/72-77 | 6 | 0.0406 | Right-handed | Immune System |
| 7FAB | H/99-105 | 7 | −0.0067 | Left-handed | Immune System |
| 1DUR | A/12-23 | 12 | 0.2808 | Right-handed | Electron Transport |
| 1DUR | A/30-41 | 12 | 0.0120 | Right-handed | Electron Transport |
| 1DUR | A/39-50 | 12 | 0.2652 | Right-handed | Electron Transport |
| 5NLL | A/54-61 | 8 | −0.0590 | Left-handed | Electron Transport |
| 2GCH | F/70-78 | 9 | 0.1866 | Right-handed | Hydrolase (Serine Proteinase) |
| 2GCH | F/94-101 | 8 | 0.0378 | Right-handed | Hydrolase (Serine Proteinase) |
| 2GCH | F/112-118 | 7 | −0.0220 | Left-handed | Hydrolase (Serine Proteinase) |
| 2GCH | G/165-176 | 12 | 0.3527 | Right-handed | Hydrolase (Serine Proteinase) |
| 2GCH | G/217-224 | 8 | −0.0086 | Left-handed | Hydrolase (Serine Proteinase) |
| 1GPD | G/47-52 | 6 | 0.0284 | Right-handed | Oxidoreductase |
| 1GPD | G/76-82 | 7 | 0.0872 | Right-handed | Oxidoreductase |
| 1GPD | G/121-129 | 9 | 0.0052 | Right-handed | Oxidoreductase |
| 1GPD | G/183-198 | 16 | −0.1486 | Left-handed | Oxidoreductase |
| 3GRS | A/83-89 | 7 | −0.0102 | Left-handed | Oxidoreductase (Flavoenzyme) |
| 3GRS | A/139-147 | 9 | −0.0760 | Left-handed | Oxidoreductase (Flavoenzyme) |
| 3GRS | A/162-172 | 11 | 0.0576 | Right-handed | Oxidoreductase (Flavoenzyme) |
| 3GRS | A/239-245 | 7 | −0.0094 | Left-handed | Oxidoreductase (Flavoenzyme) |
| 3GRS | A/256-261 | 6 | −0.0633 | Left-handed | Oxidoreductase (Flavoenzyme) |
| 3GRS | A/268-274 | 7 | −0.0219 | Left-handed | Oxidoreductase (Flavoenzyme) |
| 3GRS | A/300-307 | 8 | −0.0017 | Left-handed | Oxidoreductase (Flavoenzyme) |
| 3GRS | A/315-320 | 6 | −0.0687 | Left-handed | Oxidoreductase (Flavoenzyme) |
| 3GRS | A/331-337 | 7 | 0.0505 | Right-handed | Oxidoreductase (Flavoenzyme) |
| 3GRS | A/404-415 | 12 | 0.2396 | Right-handed | Oxidoreductase (Flavoenzyme) |
| 3GRS | A/465-472 | 8 | 0.0467 | Right-handed | Oxidoreductase (Flavoenzyme) |
| 1HIP | A/20-26 | 7 | 0.1417 | Right-handed | Electron Transfer (Iron-Sulfur Protein) |
| 1HIP | A/28-41 | 14 | 0.0932 | Right-handed | Electron Transfer (Iron-Sulfur Protein) |
| 1HIP | A/43-49 | 7 | 0.0998 | Right-handed | Electron Transfer (Iron-Sulfur Protein) |
| 1HIP | A/44-59 | 16 | 0.1820 | Right-handed | Electron Transfer (Iron-Sulfur Protein) |
| 6LDH | A/173-188 | 16 | 0.2820 | Right-handed | Oxidoreductase (Choh(D)-Nad(A)) |
| 6LDH | A/192-200 | 9 | 0.0461 | Right-handed | Oxidoreductase (Choh(D)-Nad(A)) |
| 6LDH | A/203-218 | 16 | 0.0187 | Right-handed | Oxidoreductase (Choh(D)-Nad(A)) |
| 6LDH | A/212-225 | 14 | 0.1010 | Right-handed | Oxidoreductase (Choh(D)-Nad(A)) |
| 6LDH | A/219-226 | 8 | 0.1033 | Right-handed | Oxidoreductase (Choh(D)-Nad(A)) |
| 6LDH | A/239-246 | 8 | −0.0174 | Left-handed | Oxidoreductase (Choh(D)-Nad(A)) |
| 6LDH | A/275-285 | 11 | 0.0584 | Right-handed | Oxidoreductase (Choh(D)-Nad(A)) |
| 1LH1 | A/41-53 | 13 | 0.3229 | Right-handed | Oxygen Transport |
| 1LH2 | A/47-54 | 8 | 0.0285 | Right-handed | Oxygen Transport |
| 2LHB | A/46-59 | 14 | 0.5669 | Right-handed | Oxygen Transport |
| 2LHB | A/55-64 | 10 | 0.2227 | Right-handed | Oxygen Transport |
| 7LYZ | A/18-25 | 8 | −0.0331 | Left-handed | Hydrolase (O-Glycosyl) |
| 7LYZ | A/36-42 | 7 | 0.0318 | Right-handed | Hydrolase (O-Glycosyl) |
| 7LYZ | A/44-52 | 9 | −0.0407 | Left-handed | Hydrolase (O-Glycosyl) |
| 7LYZ | A/60-75 | 16 | 0.1404 | Right-handed | Hydrolase (O-Glycosyl) |
| 2LZM | A/134-139 | 6 | 0.0657 | Right-handed | Hydrolase (O-Glycosyl) |
| 1MBN | A/40-47 | 8 | 0.2021 | Right-handed | Oxygen Storage |
| 1MBS | A/37-50 | 14 | 0.5199 | Right-handed | Oxygen Transport |
| 1MBS | A/49-54 | 6 | 0.0358 | Right-handed | Oxygen Transport |
| 1MBS | A/78-84 | 7 | 0.1077 | Right-handed | Oxygen Transport |
| 2MHB | A/40-48 | 9 | 0.2250 | Right-handed | Oxygen Transport |
| 2MHB | B/39-54 | 16 | 0.4710 | Right-handed | Oxygen Transport |
| 2MHB | B/47-57 | 11 | 0.2937 | Right-handed | Oxygen Transport |
| 1NXB | A/6-13 | 8 | −0.1576 | Left-handed | Neurotoxin (Post-Synaptic) |
| 2PAB | A/49-54 | 6 | −0.0652 | Left-handed | Transport (Thyroxine,Retinol) In Serum |
| 9PAP | A/8-13 | 6 | 0.0535 | Right-handed | Hydrolase (Sulfhydryl Proteinase) |
| 9PAP | A/60-67 | 8 | −0.0304 | Left-handed | Hydrolase (Sulfhydryl Proteinase) |
| 9PAP | A/86-100 | 15 | 0.0073 | Right-handed | Hydrolase (Sulfhydryl Proteinase) |
| 9PAP | A/138-153 | 14 | 0.1452 | Right-handed | Hydrolase (Sulfhydryl Proteinase) |
| 9PAP | A/175-185 | 11 | 0.0051 | Right-handed | Hydrolase (Sulfhydryl Proteinase) |
| 9PAP | A/191-198 | 8 | 0.0624 | Right-handed | Hydrolase (Sulfhydryl Proteinase) |
| 9PAP | A/198-203 | 6 | −0.0338 | Left-handed | Hydrolase (Sulfhydryl Proteinase) |
| 1PLC | A/6-13 | 8 | −0.0920 | Left-handed | Electron Transport |
| 1PLC | A/41-55 | 15 | 0.1393 | Right-handed | Electron Transport |
| 1PLC | A/63-68 | 6 | 0.0171 | Right-handed | Electron Transport |
| 1PLC | A/84-92 | 9 | 0.1941 | Right-handed | Electron Transport |
References
- Zhang, S.; Holmes, T.; DiPersio, C.; Hynes, R.O.; Su, X.; Rich, A. Self-complementary oligopeptide matrices support mammalian cell attachment. Biomaterials 1995, 16, 1385–1393. [Google Scholar] [CrossRef]
- Boyle, A.L. Applications of de novo designed peptides. In Book Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Elsevier: Amsterdam, The Netherlands, 2018; pp. 51–86. [Google Scholar] [CrossRef]
- Ellis-Behnke, R.G.; Liang, Y.-X.; You, S.-W.; Tay, D.K.C.; Zhang, S.; So, K.-F.; Schneider, G.E. Nano neuro knitting: Peptide nanofiber scaffold for brain repair and axon regeneration with functional return of vision. Proc. Natl. Acad. Sci. USA 2006, 103, 5054–5059. [Google Scholar] [CrossRef] [PubMed]
- Gelain, F.; Bottai, D.; Vescovi, A.; Zhang, S. Designer Self-Assembling Peptide Nanofiber Scaffolds for Adult Mouse Neural Stem Cell 3-Dimensional Cultures. PLoS ONE 2006, 1, e119. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.T.; Kearney, W.R.; Franklin, S.J. Lanthanide-binding helix-turn-helix peptides: Solution structure of a designed metallonuclease. Proc. Natl. Acad. Sci. USA 2003, 100, 3725–3730. [Google Scholar] [CrossRef]
- Kovacic, R.T.; Welch, J.T.; Franklin, S.J. Sequence-Selective DNA Cleavage by a Chimeric Metallopeptide. J. Am. Chem. Soc. 2003, 125, 6656–6662. [Google Scholar] [CrossRef]
- Reches, M.; Porat, Y.; Gazit, E. Amyloid Fibril Formation by Pentapeptide and Tetrapeptide Fragments of Human Calcitonin. J. Biol. Chem. 2002, 277, 35475–35480. [Google Scholar] [CrossRef]
- Tverdislov, V.A.; Malyshko, E.V. On regularities in the spontaneous formation of structural hierarchies in chiral systems of nonliving and living matter. Phys. Uspekhi 2019, 62, 354–363. [Google Scholar] [CrossRef]
- Tverdislov, V.A.; Malyshko, E.V. Chiral Dualism as an Instrument of Hierarchical Structure Formation in Molecular Biology. Symmetry 2020, 12, 587. [Google Scholar] [CrossRef]
- Guichard, G.; Benkirane-Jessel, N.; Zeder-Lutz, G.; van Regenmortel, M.H.; Briand, J.P.; Muller, S. Antigenic mimicry of natural L-peptides with retro-inverso-peptidomimetics. Proc. Natl. Acad. Sci. USA 1994, 91, 9765–9769. [Google Scholar] [CrossRef]
- Nanda, V.; Andrianarijaona, A.; Narayanan, C. The role of protein homochirality in shaping the energy landscape of folding. Protein Sci. 2007, 16, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Nagy-Smith, K.; Beltramo, P.; Moore, E.; Tycko, R.; Furst, E.M.; Schneider, J.P. Molecular, Local, and Network-Level Basis for the Enhanced Stiffness of Hydrogel Networks Formed from Coassembled Racemic Peptides: Predictions from Pauling and Corey. ACS Cent. Sci. 2017, 3, 586–597. [Google Scholar] [CrossRef]
- McAulay, K.; Dietrich, B.; Su, H.; Scott, M.T.; Rogers, S.; Al-Hilaly, Y.K.; Cui, H.; Serpell, L.C.; Seddon, A.; Draper, E.R.; et al. Using chirality to influence supramolecular gelation. Chem. Sci. 2019, 10, 7801–7806. [Google Scholar] [CrossRef]
- Bera, S.; Xue, B.; Rehak, P.; Jacoby, G.; Ji, W.; Shimon, L.J.W.; Beck, R.; Král, P.; Cao, Y.; Gazit, E. Self-Assembly of Aromatic Amino Acid Enantiomers into Supramolecular Materials of High Rigidity. ACS Nano 2020, 14, 1694–1706. [Google Scholar] [CrossRef]
- Chen, K.; Sheng, Y.; Wang, J.; Wang, W. Chirality-Dependent Adsorption between Amphipathic Peptide and POPC Membrane. Int. J. Mol. Sci. 2019, 20, 4760. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Zhang, Y.; Xing, C.; Yang, L.; Zhao, C.; Dou, X.; Feng, C.L. Effect of Stereochemistry on Chirality and Gelation Properties of Supramolecular Self-Assemblies. Chemistry 2021, 27, 3119–3129. [Google Scholar] [CrossRef]
- Qing, G.; Zhao, S.; Xiong, Y.; Lv, Z.; Jiang, F.; Liu, Y.; Chen, H.; Zhang, M.; Sun, T. Chiral Effect at Protein/Graphene Interface: A Bioinspired Perspective to Understand Amyloid Formation. J. Am. Chem. Soc. 2014, 136, 10736–10742. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Zhao, J.; Wang, H.; Li, B.; Li, K.; Shi, X.; Wan, K.; Ai, J.; Lv, J.; Wang, D.; et al. Chiral gold nanoparticles enantioselectively rescue memory deficits in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 4790. [Google Scholar] [CrossRef]
- Zheng, Y.; Mao, K.; Chen, S.; Zhu, H. Chirality Effects in Peptide Assembly Structures. Front. Bioeng. Biotechnol. 2021, 9, 703004. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Hu, Y.; Cao, B.; Peng, R.; Ding, J. Effects of surface molecular chirality on adhesion and differentiation of stem cells. Biomaterials 2013, 34, 9001–9009. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Shi, L.; Yue, H.; Gao, X. Recognition at chiral interfaces: From molecules to cells. Colloids Surf. B Biointerfaces 2020, 195, 111268. [Google Scholar] [CrossRef]
- Krause, E.; Bienert, M.; Schmieder, P.; Wenschuh, H. The Helix-Destabilizing Propensity Scale of d-Amino Acids: The Influence of Side Chain Steric Effects. J. Am. Chem. Soc. 2000, 122, 4865–4870. [Google Scholar] [CrossRef]
- Punitha, V.; Raman, S.S.; Parthasarathi, R.; Subramanian, V.; Rao, J.R.; Nair, B.U.; Ramasami, T. Molecular Dynamics Investigations on the Effect of d Amino Acid Substitution in a Triple-Helix Structure and the Stability of Collagen. J. Phys. Chem. B 2009, 113, 8983–8992. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yu, L.; Zou, Y.; Yang, Y.; Wang, C. Steric Dependence of Chirality Effect in Surface-Mediated Peptide Assemblies Identified with Scanning Tunneling Microscopy. Nano Lett. 2019, 19, 5403–5409. [Google Scholar] [CrossRef]
- Fairman, R.; Anthony-Cahill, S.J.; DeGrado, W.F. The helix-forming propensity of D-alanine in a right-handed. alpha.-helix. J. Am. Chem. Soc. 1992, 114, 5458–5459. [Google Scholar] [CrossRef]
- Hu, K.; Jiang, Y.; Xiong, W.; Li, H.; Zhang, P.-Y.; Yin, F.; Zhang, Q.; Geng, H.; Jiang, F.; Li, Z.; et al. Tuning peptide self-assembly by an in-tether chiral center. Sci. Adv. 2018, 4, eaar5907. [Google Scholar] [CrossRef]
- Hu, K.; Geng, H.; Zhang, Q.; Liu, Q.; Xie, M.; Sun, C.; Li, W.; Lin, H.; Jiang, F.; Wang, T.; et al. An In-tether Chiral Center Modulates the Helicity, Cell Permeability, and Target Binding Affinity of a Peptide. Angew. Chem. 2016, 128, 8145–8149. [Google Scholar] [CrossRef]
- Gil, A.M.; Casanovas, J.; Mayans, E.; Jiménez, A.I.; Puiggalí, J.; Alemán, C. Heterochirality Restricts the Self-Assembly of Phenylalanine Dipeptides Capped with Highly Aromatic Groups. J. Phys. Chem. B 2020, 124, 5913–5918. [Google Scholar] [CrossRef]
- Kralj, S.; Bellotto, O.; Parisi, E.; Garcia, A.M.; Iglesias, D.; Semeraro, S.; Deganutti, C.; D’Andrea, P.; Vargiu, A.V.; Geremia, S.; et al. Heterochirality and Halogenation Control Phe-Phe Hierarchical Assembly. ACS Nano 2020, 14, 16951–16961. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, L.; Rao, H.; Wang, Y.; Li, Q.; Qi, W.; Yang, X.; Su, R.; He, Z. Role of molecular chirality and solvents in directing the self-assembly of peptide into an ultra-pH-sensitive hydrogel. J. Colloid Interface Sci. 2020, 577, 388–396. [Google Scholar] [CrossRef]
- Adzhubei, A.A.; Sternberg, M.; Makarov, A.A. Polyproline-II Helix in Proteins: Structure and Function. J. Mol. Biol. 2013, 425, 2100–2132. [Google Scholar] [CrossRef] [PubMed]
- Zarrinpar, A.; Bhattacharyya, R.P.; Lim, W.A. The structure and function of proline recognition domains. Sci. STKE 2003, 179, re8. [Google Scholar] [CrossRef] [PubMed]
- Platé, N.A.; Shibaev, V.P. Comb-Shaped Polymers and Liquid Crystals; Springer: Boston, MA, USA, 2012. [Google Scholar] [CrossRef]
- Livolant, F.; Leforestier, A. Condensed phases of DNA: Structures and phase transitions. Prog. Polym. Sci. 1996, 21, 1115–1164. [Google Scholar] [CrossRef]
- Chakraborty, D.; Mugnai, M.; Thirumalai, D. On the Emergence of Orientational Order in Folded Proteins with Implications for Allostery. Symmetry 2021, 13, 770. [Google Scholar] [CrossRef]
- Sidorova, A.E.; Levashova, N.T.; Malyshko, E.; Tverdislov, V. Autowave Self-Organization in the Folding of Proteins. Mosc. Univ. Phys. Bull. 2019, 74, 213–226. [Google Scholar] [CrossRef]
- Abrusán, G.; Marsh, J.A. Alpha Helices Are More Robust to Mutations than Beta Strands. PLoS Comput. Biol. 2016, 12, e1005242. [Google Scholar] [CrossRef]
- Pauling, L.; Corey, R.B. The Pleated Sheet, A New Layer Configuration of Polypeptide Chains. Proc. Natl. Acad. Sci. USA 1951, 37, 2451–2456. [Google Scholar] [CrossRef]
- Tonlolo, C.; Benedetti, E. The polypeptide 310-helix. Trends Biochem. Sci. 1991, 16, 350–353. [Google Scholar] [CrossRef]
- Kendrew, J.C.; Bodo, G.; Dintzis, H.M.; Parrish, R.G.; Wyckoff, H.; Phillips, D.C. A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis. Nature 1958, 181, 662–666. [Google Scholar] [CrossRef]
- Armen, R.; Alonso, D.O.; Daggett, V. The role of α-, 310-, and π-helix in helix→coil transitions. Protein Sci. 2003, 12, 1145–1157. [Google Scholar] [CrossRef]
- Cooley, R.B.; Arp, D.J.; Karplus, P.A. Evolutionary Origin of a Secondary Structure: π-Helices as Cryptic but Widespread Insertional Variations of α-Helices That Enhance Protein Functionality. J. Mol. Biol. 2010, 404, 232–246. [Google Scholar] [CrossRef]
- Dasgupta, B.; Chakrabarti, P. pi-Turns: Types, systematics and the context of their occurrence in protein structures. BMC Struct. Biol. 2008, 8, 39. [Google Scholar] [CrossRef]
- Fodje, M.; Al-Karadaghi, S. Occurrence, conformational features and amino acid propensities for the π-helix. Protein Eng. Des. Sel. 2002, 15, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Religa, T.L.; Johnson, C.M.; Vu, D.; Brewer, S.H.; Dyer, R.B.; Fersht, A.R. The helix turn helix motif as an ultrafast independently folding domain: The pathway of folding of Engrailed homeodomain. Proc. Natl. Acad. Sci. USA 2007, 104, 9272–9277. [Google Scholar] [CrossRef] [PubMed]
- Pavone, V.; Gaeta, G.; Lombardi, A.; Nastri, F.; Maglio, O.; Isernia, C.; Saviano, M. Discovering protein secondary structures: Classification and description of isolated α-turns. Biopolymers 1996, 38, 705–721. [Google Scholar] [CrossRef]
- Hutchinson, E.G.; Thornton, J. A revised set of potentials for beta-turn formation in proteins. Protein Sci. 1994, 3, 2207–2216. [Google Scholar] [CrossRef]
- Donate, L.E.; Rufino, S.D.; Canard, L.H.; Blundell, T.L. Conformational analysis and clustering of short and medium size loops connecting regular secondary structures: A database for modeling and prediction. Protein Sci. 1996, 5, 2600–2616. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.; Chavan, A.G.; Fraga, K.J.; Tsai, J. An amino acid code for irregular and mixed protein packing. Proteins 2015, 83, 2147–2161. [Google Scholar] [CrossRef]
- Rose, G.D.; Glerasch, L.M.; Smith, J.A. Turns in Peptides and Proteins. Adv. Protein Chem. 1985, 37, 1–109. [Google Scholar] [CrossRef]
- Marcelino, A.M.C.; Gierasch, L.M. Roles of β-turns in protein folding: From peptide models to protein engineering. Biopolymers 2008, 89, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Koch, O.; Klebe, G. Turns revisited: A uniform and comprehensive classification of normal, open, and reverse turn families minimizing unassigned random chain portions. Proteins Struct. Funct. Bioinform. 2009, 74, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Munoz, V.; Henry, E.; Hofrichter, J.; Eaton, W.A. A statistical mechanical model for β-hairpin kinetics. Proc. Natl. Acad. Sci. USA 1998, 95, 5872–5879. [Google Scholar] [CrossRef]
- Dasgupta, B.; Pal, L.; Basu, G.; Chakrabarti, P. Expanded turn conformations: Characterization and sequence-structure correspondence in α-turns with implications in helix folding. Proteins Struct. Funct. Bioinform. 2004, 55, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Skipper, L. Proteins. Overview. In Encyclopedia of Analytical Science; Elsevier: Amsterdam, The Netherlands, 2005; pp. 344–352. [Google Scholar] [CrossRef]
- Leszczynski, J.F.; Rose, G.D. Loops in Globular Proteins: A Novel Category of Secondary Structure. Science 1986, 234, 849–855. [Google Scholar] [CrossRef]
- Wang, X.; Wang, M.; Tong, Y.; Shan, L.; Wang, J. Probing the folding capacity and residual structures in 1–79 residues fragment of staphylococcal nuclease by biophysical and NMR methods. Biochimie 2006, 88, 1343–1355. [Google Scholar] [CrossRef]
- Neuhaus, F.C. Role of the omega loop in specificity determination in subsite 2 of the D-alanine:D-alanine (D-lactate) ligase from Leuconostoc mesenteroides: A molecular docking study. J. Mol. Graph. Model. 2011, 30, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Likhachev, I.; Bystrov, V. Assembly of a phenylalanine nanotube with a molecular dynamic manipulator. Math. Biol. Bioinform. 2021, 16, 244–255. [Google Scholar] [CrossRef]
- Zelenovskiy, P.S.; Nuraeva, A.; Kopyl, S.; Arkhipov, S.G.; Vasilev, S.G.; Bystrov, V.S.; Gruzdev, D.A.; Waliczek, M.; Svitlyk, V.; Shur, V.Y.; et al. Chirality-Dependent Growth of Self-Assembled Diphenylalanine Microtubes. Cryst. Growth Des. 2019, 19, 6414–6421. [Google Scholar] [CrossRef]
- Bystrov, V.S.; Zelenovskiy, P.; Nuraeva, A.; Kopyl, S.; Zhulyabina, O.A.; Tverdislov, V. Molecular modeling and computational study of the chiral-dependent structures and properties of self-assembling diphenylalanine peptide nanotubes. J. Mol. Model. 2019, 25, 199. [Google Scholar] [CrossRef] [PubMed]
- Bystrov, V.; Coutinho, J.; Zelenovskiy, P.; Nuraeva, A.; Kopyl, S.; Zhulyabina, O.; Tverdislov, V. Structures and Properties of the Self-Assembling Diphenylalanine Peptide Nanotubes Containing Water Molecules: Modeling and Data Analysis. Nanomaterials 2020, 10, 1999. [Google Scholar] [CrossRef]
- German, H.W.; Uyaver, S.; Hansmann, A.U.H.E. Self-Assembly of Phenylalanine-Based Molecules. J. Phys. Chem. A 2015, 119, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Adler-Abramovich, L.; Vaks, L.; Carny, O.; Trudler, D.; Magno, A.; Caflisch, A.; Frenkel, D.; Gazit, E. Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nat. Chem. Biol. 2012, 8, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Bystrov, V.; Sidorova, A.; Lutsenko, A.; Shpigun, D.; Malyshko, E.; Nuraeva, A.; Zelenovskiy, P.; Kopyl, S.; Kholkin, A. Modeling of Self-Assembled Peptide Nanotubes and Determination of Their Chirality Sign Based on Dipole Moment Calculations. Nanomaterials 2021, 11, 2415. [Google Scholar] [CrossRef]
- Petitjean, M. On the root mean square quantitative chirality and quantitative symmetry measures. J. Math. Phys. 1999, 40, 4587–4595. [Google Scholar] [CrossRef][Green Version]
- Peng, X.-L.; Fang, K.-T.; Hu, Q.-N.; Liang, Y.-Z. Impersonality of the Connectivity Index and Recomposition of Topological Indices According to Different Properties. Molecules 2004, 9, 1089–1099. [Google Scholar] [CrossRef]
- Yaffe, D.; Cohen, Y. Neural Network Based Temperature-Dependent Quantitative Structure Property Relations (QSPRs) for Predicting Vapor Pressure of Hydrocarbons. J. Chem. Inf. Comput. Sci. 2001, 41, 463–477. [Google Scholar] [CrossRef]
- McClelland, H.E.; Jurs, P.C. Quantitative Structure−Property Relationships for the Prediction of Vapor Pressures of Organic Compounds from Molecular Structures. J. Chem. Inf. Comput. Sci. 2000, 40, 967–975. [Google Scholar] [CrossRef]
- Zhao, T.; Zhang, Q.; Long, H.; Xu, L. Graph Theoretical Representation of Atomic Asymmetry and Molecular Chirality of Benzenoids in Two-Dimensional Space. PLoS ONE 2014, 9, e102043. [Google Scholar] [CrossRef] [PubMed]
- Mezey, P.G. The proof of the metric properties of a fuzzy chirality measure of molecular electron density clouds. J. Mol. Struct. Theochem 1998, 455, 183–190. [Google Scholar] [CrossRef]
- Gilat, G.; Schulman, L. Chiral interaction, magnitude of the effects and application to natural selection of L-enantiomer. Chem. Phys. Lett. 1985, 121, 13–16. [Google Scholar] [CrossRef]
- Zabrodsky, H.; Peleg, S.; Avnir, D. Continuous symmetry measures. J. Am. Chem. Soc. 1992, 114, 7843–7851. [Google Scholar] [CrossRef]
- Pinsky, M.; Dryzun, C.; Casanova, D.; Alemany, P.; Avnir, D. Analytical methods for calculating Continuous Symmetry Measures and the Chirality Measure. J. Comput. Chem. 2008, 29, 2712–2721. [Google Scholar] [CrossRef] [PubMed]
- Luzanov, A.V.; Nerukh, D. Simple One-electron Invariants of Molecular Chirality. J. Math. Chem. 2006, 41, 417–435. [Google Scholar] [CrossRef]
- Raos, G. Degrees of Chirality in Helical Structures. Macromol. Theory Simul. 2002, 11, 739–750. [Google Scholar] [CrossRef]
- Ramachandran, G.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Baruch-Shpigler, Y.; Wang, H.; Tuvi-Arad, I.; Avnir, D. Chiral Ramachandran Plots I: Glycine. Biochemistry 2017, 56, 5635–5643. [Google Scholar] [CrossRef]
- Wang, H.; Avnir, D.; Tuvi-Arad, I. Chiral Ramachandran Plots II: General Trends and Protein Chirality Spectra. Biochemistry 2018, 57, 6395–6403. [Google Scholar] [CrossRef] [PubMed]
- Mannige, R.V. An exhaustive survey of regular peptide conformations using a new metric for backbone handedness (h). PeerJ 2017, 5, 3327. [Google Scholar] [CrossRef]
- Robinson, S.W.; Afzal, A.M.; Leader, D.P. Bioinformatics: Concepts, Methods, and Data. In Handbook of Pharmacogenomics and Stratified Medicine, 1st ed.; Padmanabhan, S., Ed.; Academic Press: New York, NY, USA, 2014; pp. 259–287. [Google Scholar]
- Johansson, M.U.; Zoete, V.; Michielin, O.; Guex, N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinform. 2012, 13, 173. [Google Scholar] [CrossRef]
- Richardson, J.S. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 1981, 34, 167–339. [Google Scholar] [CrossRef]
- Lewis, P.N.; Momany, F.A.; Scheraga, H.A. Energy Parameters in Polypeptides. VI. Conformational Energy Analysis of the N-Acetyl N′-Methyl Amides of the Twenty Naturally Occurring Amino Acids. Isr. J. Chem. 1973, 11, 121–152. [Google Scholar] [CrossRef]
- Wilmot, C.; Thornton, J. Analysis and prediction of the different types of β-turn in proteins. J. Mol. Biol. 1988, 203, 221–232. [Google Scholar] [CrossRef]
- De Brevern, A.G. Extension of the classical classification of β-turns. Sci. Rep. 2016, 6, 33191. [Google Scholar] [CrossRef] [PubMed]
- Ting, D.; Wang, G.; Shapovalov, M.; Mitra, R.; Jordan, M.; Dunbrack, R.L. Neighbor-Dependent Ramachandran Probability Distributions of Amino Acids Developed from a Hierarchical Dirichlet Process Model. PLoS Comput. Biol. 2010, 6, e1000763. [Google Scholar] [CrossRef]
- Shapovalov, M.; Slobodan, V.; Dunbrack, R.L. A new clustering and nomenclature for beta turns derived from high-resolution protein structures. PLoS Comput. Biol. 2019, 15, e1006844. [Google Scholar] [CrossRef]
- Fang, C.; Shang, Y.; Xu, N. Improving Protein Gamma-Turn Prediction Using Inception Capsule Networks. Sci. Rep. 2018, 8, 15741. [Google Scholar] [CrossRef]
- Fiser, A.; Do, R.K.G.; Šali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed]
- Duddy, W.J.; Nissink, J.W.M.; Allen, F.H.; Milner-White, E.J. Mimicry by asx- and ST-turns of the four main types of β-turn in proteins. Protein Sci. 2008, 13, 3051–3055. [Google Scholar] [CrossRef]
- Eswar, N.; Ramakrishnan, C. Secondary structures without backbone: An analysis of backbone mimicry by polar side chains in protein structures. Protein Eng. Des. Sel. 1999, 12, 447–455. [Google Scholar] [CrossRef]
- Sidorova, A.E.; Lutsenko, A.O.; Shpigun, D.K.; Malyshko, E.V.; Tverdislov, V.A. Methods to Determine the Chirality Sign for Helical and Superhelical Protein Structures. Biophysics 2021, 66, 357–363. [Google Scholar] [CrossRef]
- Sidorova, A.; Malyshko, E.; Lutsenko, A.; Shpigun, D.; Bagrova, O. Protein Helical Structures: Defining Handedness and Localization Features. Symmetry 2021, 13, 879. [Google Scholar] [CrossRef]
- Nicholson, H.; Anderson, D.E.; Pin, S.D.; Matthews, B.W. Analysis of the interaction between charged side chains and the.alpha.-helix dipole using designed thermostable mutants of phage T4 lysozyme. Biochemistry 1991, 30, 9816–9828. [Google Scholar] [CrossRef]
- Ludwig, M.L.; Pattridge, K.A.; Metzger, A.L.; Dixon, M.M.; Eren, M.; Feng, Y.; Swenson, R.P. Control of Oxidation−Reduction Potentials in Flavodoxin from Clostridium beijerinckii: The Role of Conformation Changes. Biochemistry 1997, 36, 1259–1280. [Google Scholar] [CrossRef] [PubMed]
- Nataraj, D.V.; Srinivasan, N.; Sowdhamini, R.; Ramakrishnan, C. α-Turns in protein structure. Curr. Sci. 1995, 69, 434–447. [Google Scholar]
- Taylor, N.R.; Cleasby, A.; Singh, O.; Skarzynski, T.; Wonacott, A.J.; Smith, P.W.; Sollis, S.L.; Howes, P.D.; Cherry, P.C.; Bethell, R.; et al. Dihydropyrancarboxamides Related to Zanamivir: A New Series of Inhibitors of Influenza Virus Sialidases. 2. Crystallographic and Molecular Modeling Study of Complexes of 4-Amino-4H-pyran-6-carboxamides and Sialidase from Influenza Virus Types A and B. J. Med. Chem. 1998, 41, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.; Dodson, E.J. Crystallographic refinement of the structure of actinidin at 1.7 Å resolution by fast Fourier least-squares methods. Acta Crystallogr. 1980, A36, 559–572. [Google Scholar] [CrossRef]
- Likhachev, I.V.; Balabaev, N.K.; Galzitskaya, O.V. Elastic and Non-elastic Properties of Cadherin Ectodomain: Comparison with Mechanical System. Adv. Intell. Syst. Comput. 2019, 938, 555–566. [Google Scholar] [CrossRef]
- Glyakina, A.V.; Likhachev, I.V.; Balabaev, N.K.; Galzitskaya, O.V. Comparative mechanical unfolding studies of spectrin domains R15, R16 and R17. J. Struct. Biol. 2018, 201, 162–170. [Google Scholar] [CrossRef]
- Lemak, A.S.; Balabaev, N.K. A Comparison Between Collisional Dynamics and Brownian Dynamics. Mol. Simul. 1995, 15, 223–231. [Google Scholar] [CrossRef]
- Lemak, A.S.; Balabaev, N.K. Molecular dynamics simulation of a polymer chain in solution by collisional dynamics method. J. Comput. Chem. 1996, 17, 1685–1695. [Google Scholar] [CrossRef]
- HyperChem Download—Sophisticated Molecular Modeling Environment. Available online: https://hyperchem.software.informer.com/ (accessed on 21 October 2021).
- Rocha, G.B.; Freire, R.O.; Simas, A.M.; Stewart, J.J.P. RM1: A reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I. J. Comput. Chem. 2006, 27, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- The Protein Data Bank. Available online: http://www.rcsb.org/ (accessed on 1 October 2020).
- Bystrov, V.S.; Kopyl, S.A.; Zelenovskiy, P.; Zhulyabina, O.A.; Tverdislov, V.; Salehli, F.; Ghermani, N.E.; Shur, V.; Kholkin, A.L. Investigation of physical properties of diphenylalanine peptide nanotubes having different chiralities and embedded water molecules. Ferroelectrics 2018, 525, 168–177. [Google Scholar] [CrossRef]












| Type of α-Turn | Number | Mean Chirality Value | Standard Deviation |
|---|---|---|---|
| F1 | 46 | 0.08628 | 0.01473 |
| F2 | 8 | 0.06922 | 0.013 |
| g1 | 5 | −0.03274 | 0.00846 |
| g2 | 2 | −0.04165 | 0.00864 |
| g3 | 4 | 0.02838 | 0.02813 |
| g4 | 4 | −0.06686 | 0.00568 |
| g5 | 3 | 0.10221 | 0.00191 |
| g6 | 3 | −0.00376 | 0.00415 |
| g7 | 2 | 0.09378 | 0.02175 |
| Other | 2 | 0.0323 | 0.05972 |
| Type of β-Turn | Number | Mean Chirality Value | Standard Deviation |
|---|---|---|---|
| AD | 481 | 0.05041 | 0.01634 |
| Pd | 61 | 0.02132 | 0.01371 |
| Pa | 36 | 0.00392 | 0.02101 |
| ad | 60 | −0.05916 | 0.00556 |
| AB1 | 17 | 0.00647 | 0.01820 |
| AZ | 16 | 0.04043 | 0.01255 |
| AB2 | 3 | 0.04561 | 0.00151 |
| pD | 30 | −0.01805 | 0.02312 |
| AG | 6 | 0.06884 | 0.00086 |
| BcisP | 10 | 0.03700 | 0.01090 |
| dD | 5 | 0.05961 | 0.01223 |
| PcisD | 10 | 0.00591 | 0.01596 |
| dN | 6 | 0.05235 | 0.00206 |
| Dd | 5 | −0.06710 | 0.00107 |
| cisDA | 6 | 0.06506 | 0.00022 |
| pG | 7 | 0.00086 | 0.02898 |
| cisDP | 3 | 0.04490 | 0.02358 |
| other | 88 | 0.02855 | 0.03472 |
| Number of Residues | Number of Vectors | Number of Mixed Products | Addition to the Chirality Value at This Step | Total Chirality Value at This Step |
|---|---|---|---|---|
| 1 | -- | -- | 0 | 0 |
| 2 | 1 | -- | 0 | 0 |
| 3 | 2 | -- | 0 | 0 |
| 4 | 3 | 1 | 0.0554 | 0.0554 |
| 5 | 4 | 2 | −0.0631 | −0.0077 |
| 6 | 5 | 3 | 0.0585 | 0.0508 |
| i | RM1 RHF | Amber | ||||||
|---|---|---|---|---|---|---|---|---|
| Di | Dx | Dy | Dz | Di | Dx | Dy | Dz | |
| 1 | 2.730 | 2.625 | −0.697 | 0.282 | 2.915 | 2.740 | −0.955 | −0.282 |
| 2 | 3.400 | 2.884 | 0.360 | −1.765 | 2.937 | 2.232 | 1.075 | −1.578 |
| 3 | 2.488 | 1.645 | 1.503 | 1.106 | 2.624 | 0.984 | 1.348 | 2.025 |
| 4 | 2.615 | 1.710 | 1.869 | 0.650 | 2.671 | 2.086 | 1.603 | 0.463 |
| 5 | 2.558 | −0.760 | 2.203 | 1.054 | 2.844 | −1.325 | 1.618 | 1.928 |
| 6 | 2.449 | −0.956 | 2.224 | 0.370 | 2.564 | −0.554 | 2.429 | −0.608 |
| 7 | 2.997 | −1.096 | 0.851 | −2.656 | 2.372 | −1.185 | −0.194 | −2.046 |
| 8 | 2.258 | −1.456 | 0.790 | −1.534 | 1.631 | −0.631 | 0.534 | −1.406 |
| 9 | 2.436 | −1.265 | −1.928 | −0.785 | 2.691 | −2.137 | −1.636 | −0.026 |
| 10 | 2.611 | −1.789 | −1.795 | 0.630 | 1.504 | −1.081 | −0.999 | 0.310 |
| 11 | 1.887 | −0.942 | −0.771 | −1.442 | 1.165 | −1.135 | −0.254 | 0.068 |
| 12 | 2.201 | −0.996 | −1.873 | 0.586 | 1.771 | −0.819 | −1.533 | −0.338 |
| Dsum | 30.630 | −0.396 | 3.433 | −3.504 | 27.692 | 2.111 | 3.036 | −2.138 |
| Dcoil | 9.995 | 6.624 | 2.820 | −6.934 | 6.3004 | 4.2597 | 2.0522 | −4.164 |
| Dav | 2.553 | 2.308 | ||||||
| i | RM1 RHF | Amber | ||||||
|---|---|---|---|---|---|---|---|---|
| Di | Dx | Dy | Dz | Di | Dx | Dy | Dz | |
| 1 | 2.992 | −2.037 | 2.152 | 0.414 | 3.041 | −2.109 | 1.870 | 1.142 |
| 2 | 3.676 | −3.516 | −0.872 | −0.630 | 2.980 | −2.724 | −0.862 | −0.846 |
| 3 | 2.864 | −2.084 | −1.822 | −0.735 | 2.727 | −1.612 | −1.660 | −1.443 |
| 4 | 2.943 | 0.259 | −2.654 | 1.244 | 2.797 | −0.790 | −2.055 | 1.726 |
| 5 | 2.746 | 0.852 | −2.590 | −0.325 | 2.780 | 1.098 | −2.542 | 0.245 |
| 6 | 2.821 | 1.415 | −2.392 | −0.483 | 3.192 | 1.285 | −2.357 | −1.726 |
| 7 | 3.781 | 1.444 | −3.251 | 1.282 | 3.691 | 1.197 | −2.894 | 1.953 |
| 8 | 2.503 | 1.317 | 2.048 | 0.578 | 2.387 | 2.070 | 1.060 | −0.539 |
| 9 | 2.888 | 1.720 | 1.513 | 1.758 | 2.483 | 1.376 | 1.563 | 1.353 |
| 10 | 3.282 | −1.128 | 2.525 | 1.766 | 2.732 | −0.748 | 2.323 | 1.230 |
| 11 | 3.762 | −3.239 | 1.798 | 0.657 | 3.072 | −2.448 | 1.774 | 0.547 |
| 12 | 2.667 | −2.229 | 0.793 | −1.231 | 2.836 | −2.774 | 0.589 | 0.034 |
| Dsum | 36.925 | −7.224 | −2.752 | 4.294 | 34.718 | −6.179 | −3.191 | 3.675 |
| Dcoil | 9.599 | −6.395 | −5.676 | 4.362 | 8.234 | −4.333 | −5.081 | 4.818 |
| Dav | 3.077 | 2.893 | ||||||
| Type of PNT | L-F | D-F | ||
|---|---|---|---|---|
| Calculating method | RM1 RHF | Amber | RM1 RHF | Amber |
| ctotal, Debye3 | 20.266 | 18.171 | −19.647 | −26.204 |
| cnorm | 1.219 | 1.479 | −0.674 | −1.082 |
| Chirality sign | positive | positive | negative | negative |
| Chirality symbol | D | D | L | L |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidorova, A.; Bystrov, V.; Lutsenko, A.; Shpigun, D.; Belova, E.; Likhachev, I. Quantitative Assessment of Chirality of Protein Secondary Structures and Phenylalanine Peptide Nanotubes. Nanomaterials 2021, 11, 3299. https://doi.org/10.3390/nano11123299
Sidorova A, Bystrov V, Lutsenko A, Shpigun D, Belova E, Likhachev I. Quantitative Assessment of Chirality of Protein Secondary Structures and Phenylalanine Peptide Nanotubes. Nanomaterials. 2021; 11(12):3299. https://doi.org/10.3390/nano11123299
Chicago/Turabian StyleSidorova, Alla, Vladimir Bystrov, Aleksey Lutsenko, Denis Shpigun, Ekaterina Belova, and Ilya Likhachev. 2021. "Quantitative Assessment of Chirality of Protein Secondary Structures and Phenylalanine Peptide Nanotubes" Nanomaterials 11, no. 12: 3299. https://doi.org/10.3390/nano11123299
APA StyleSidorova, A., Bystrov, V., Lutsenko, A., Shpigun, D., Belova, E., & Likhachev, I. (2021). Quantitative Assessment of Chirality of Protein Secondary Structures and Phenylalanine Peptide Nanotubes. Nanomaterials, 11(12), 3299. https://doi.org/10.3390/nano11123299