The Influence of the Mixed DPC:SDS Micelle on the Structure and Oligomerization Process of the Human Cystatin C

 , , , ,
, , , ,  and
and
Abstract
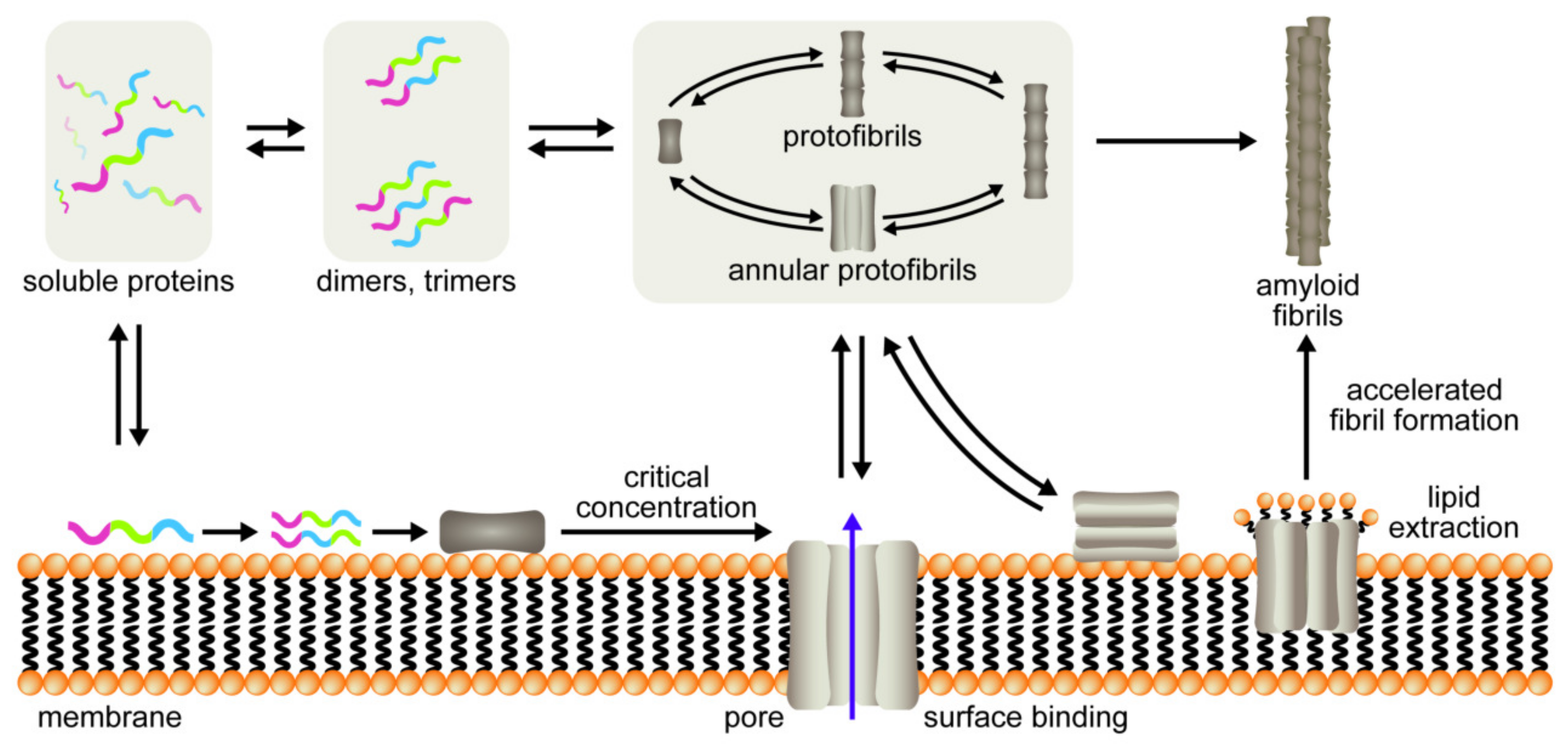
1. Introduction
2. Results
2.1. Selection of hCC Variants—What Are the Differences between hCC Variants’ Properties?
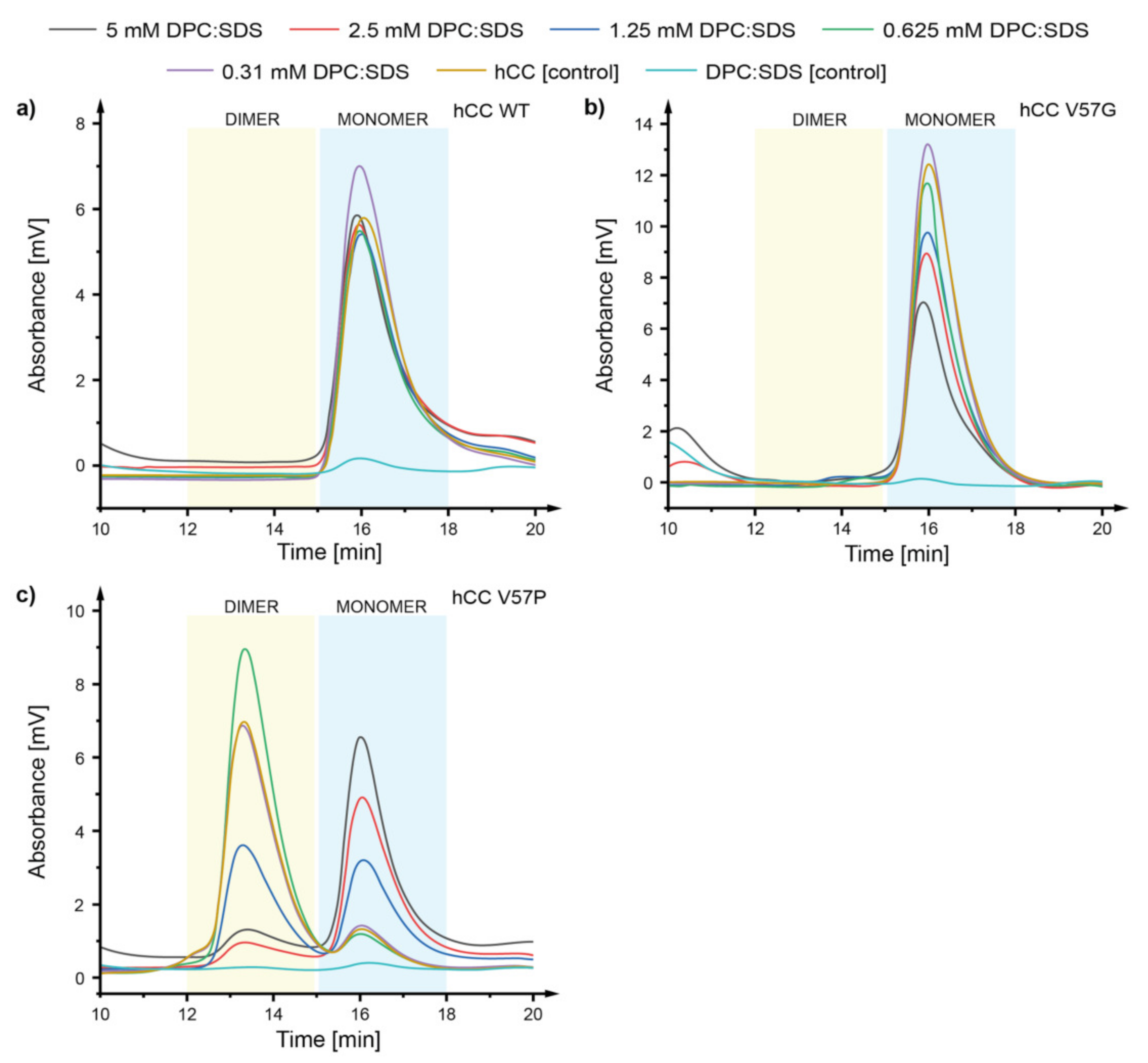
2.2. Size Exclusion Chromatography—How Does the hCC Monomer-Dimer Equilibrium Change in a Micellar Environment?
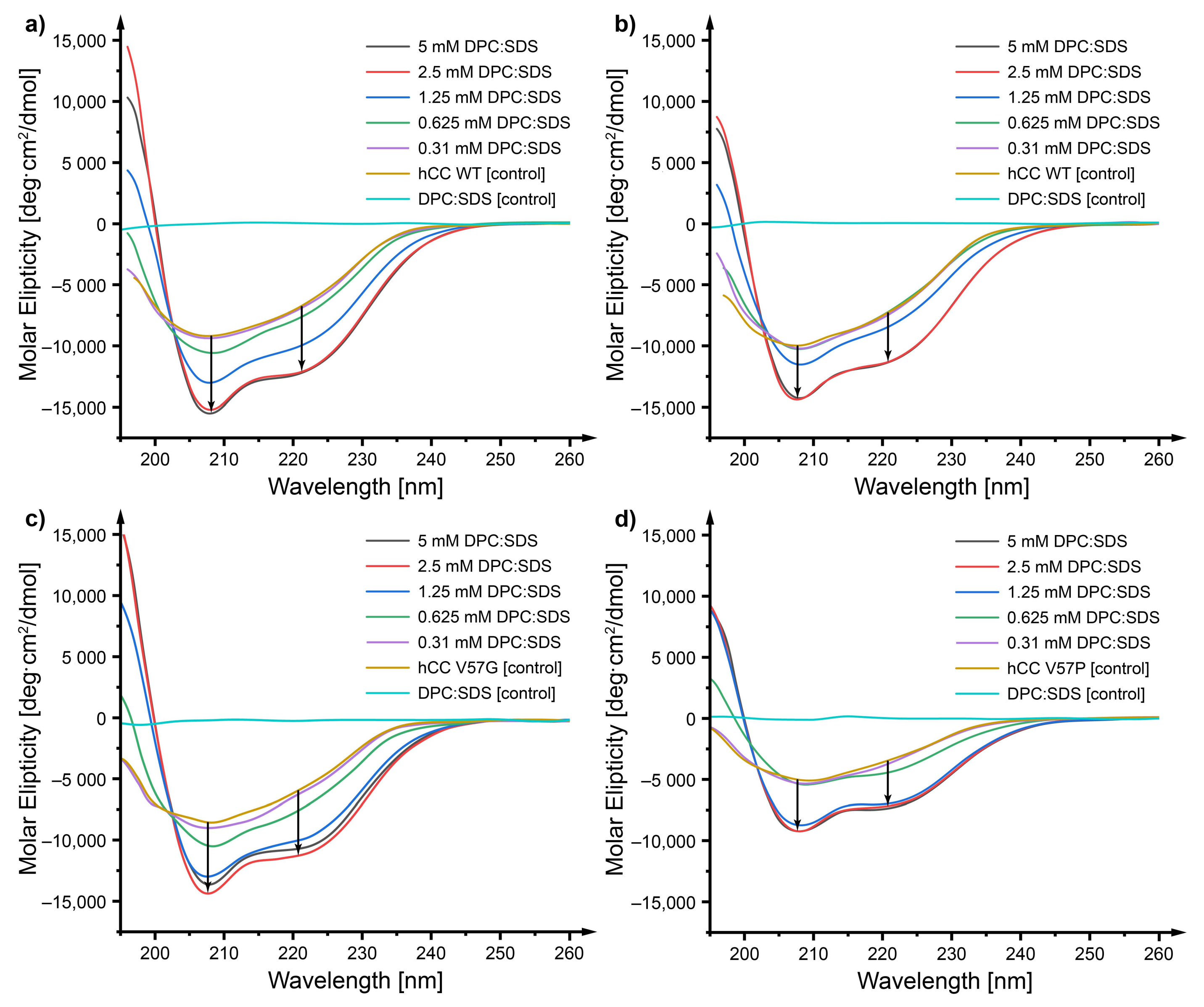
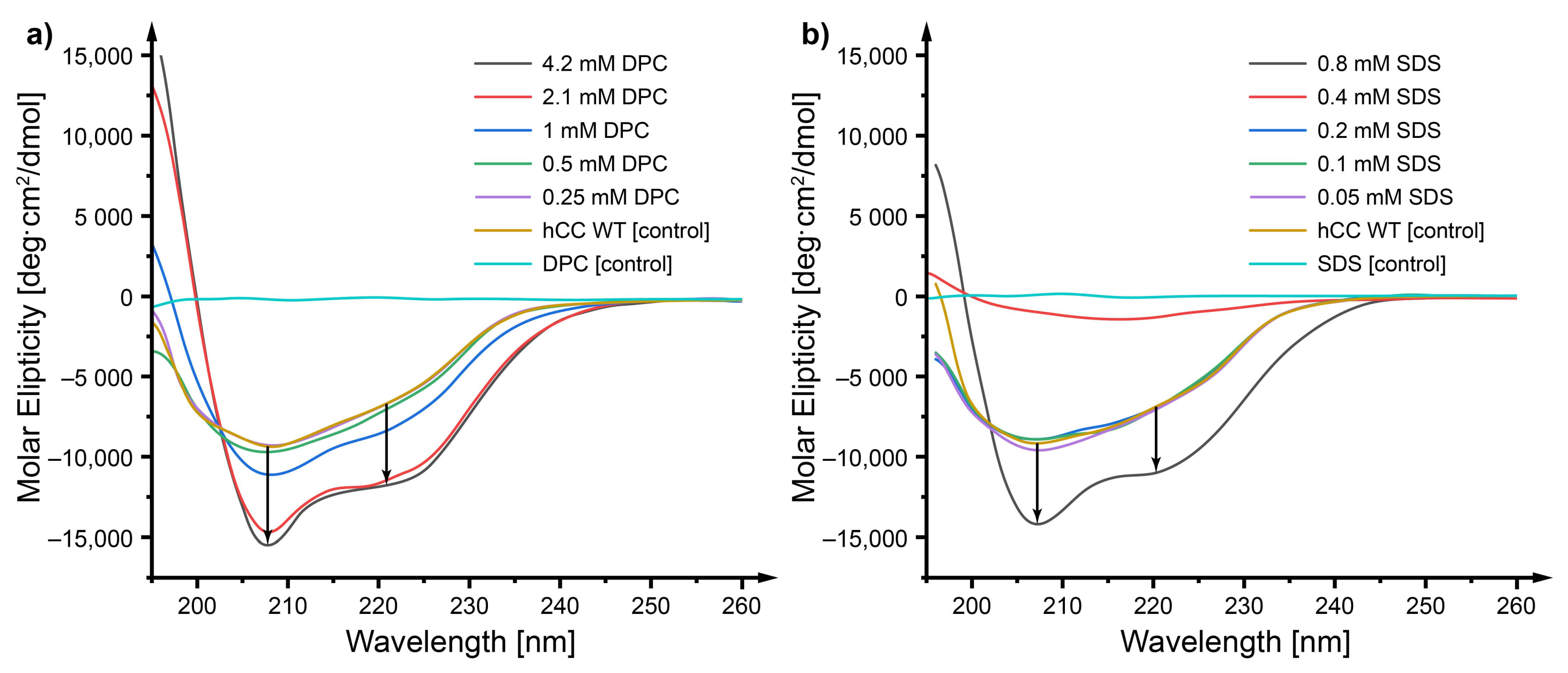
2.3. Circular Dichroism—Does a Micellar Environment Influence the Secondary Structure of hCC?
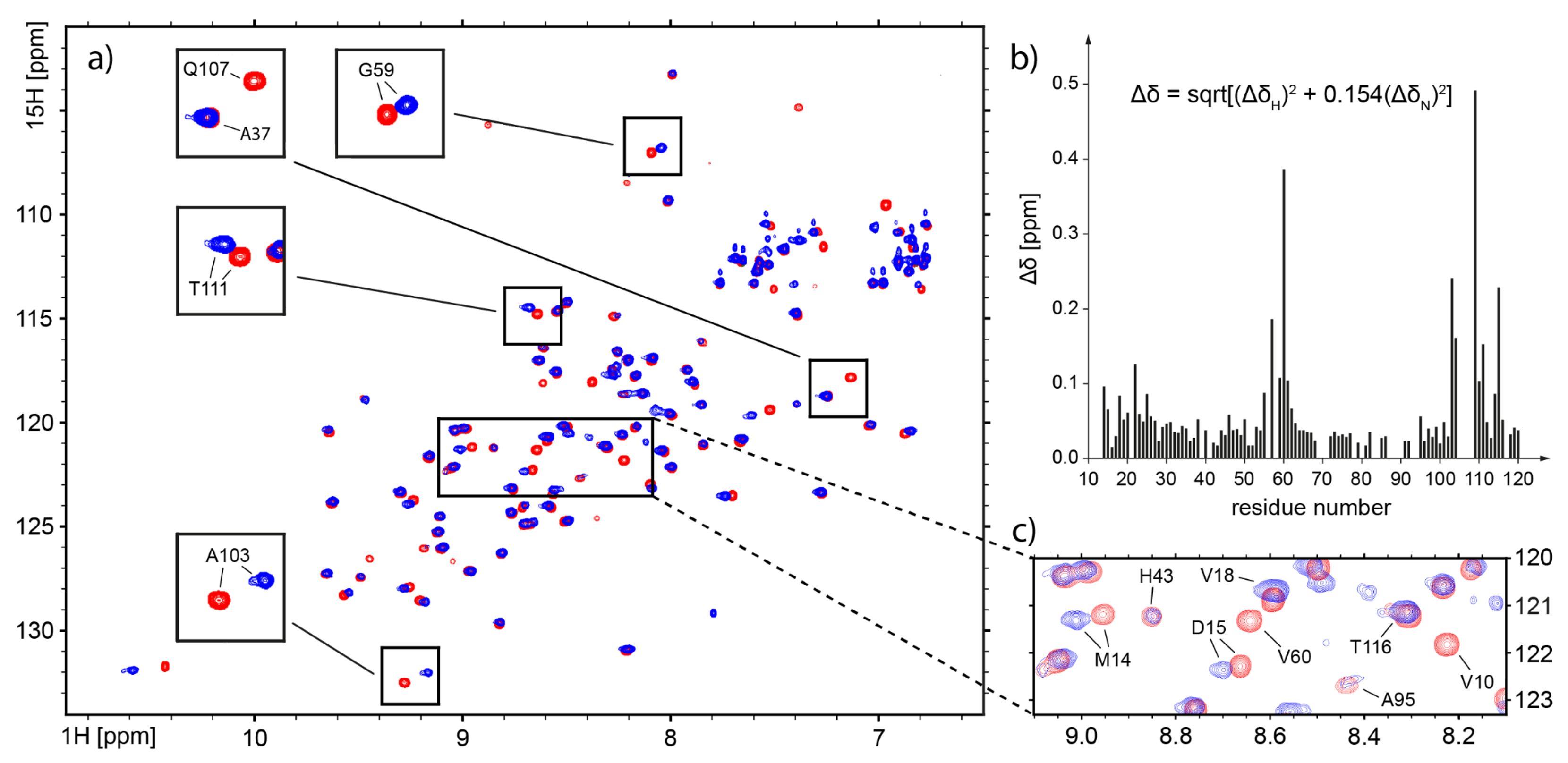
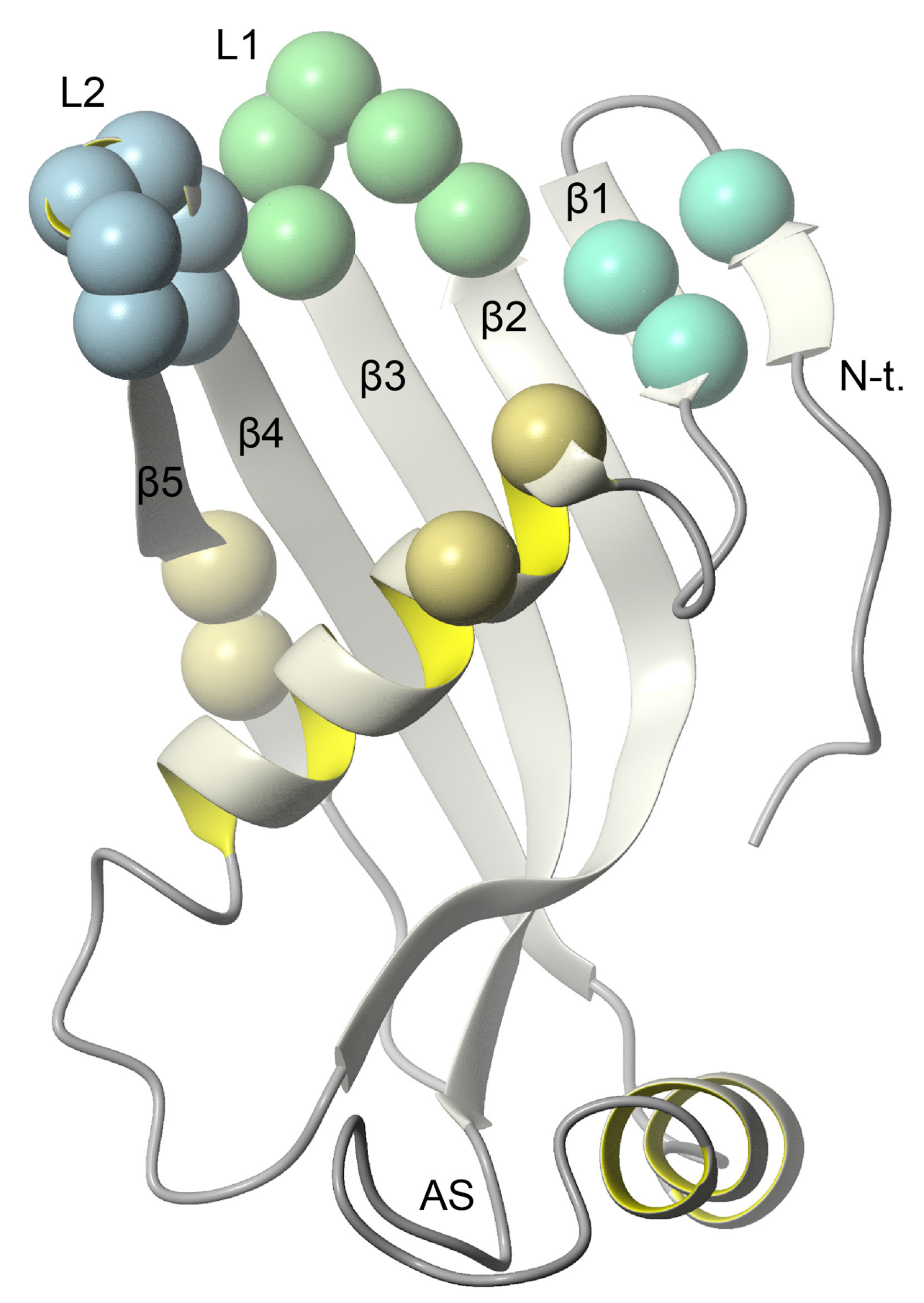
2.4. Nuclear Magnetic Resonance—Which Fragments in the hCC Structure Bind to the Micelle?
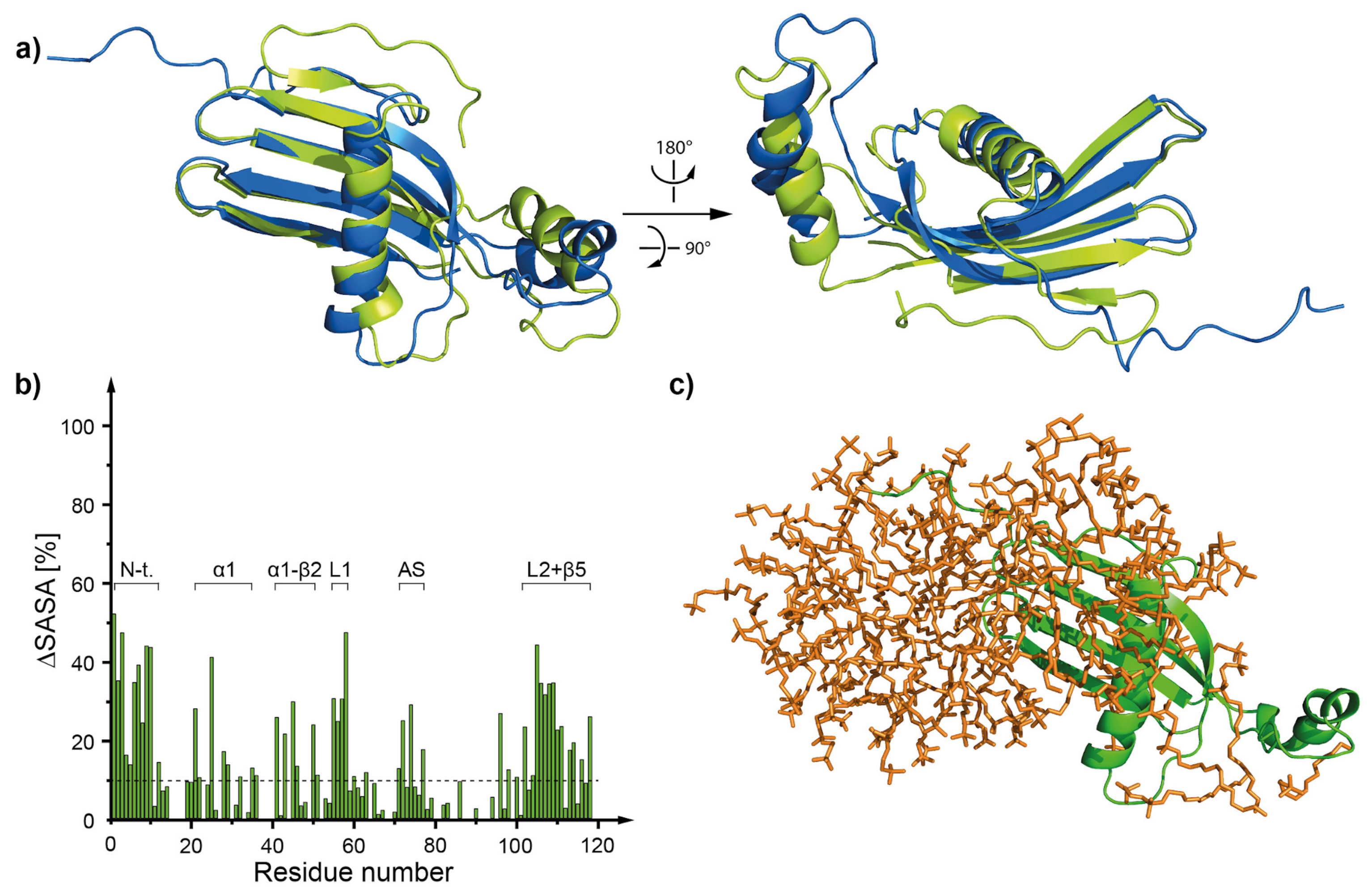
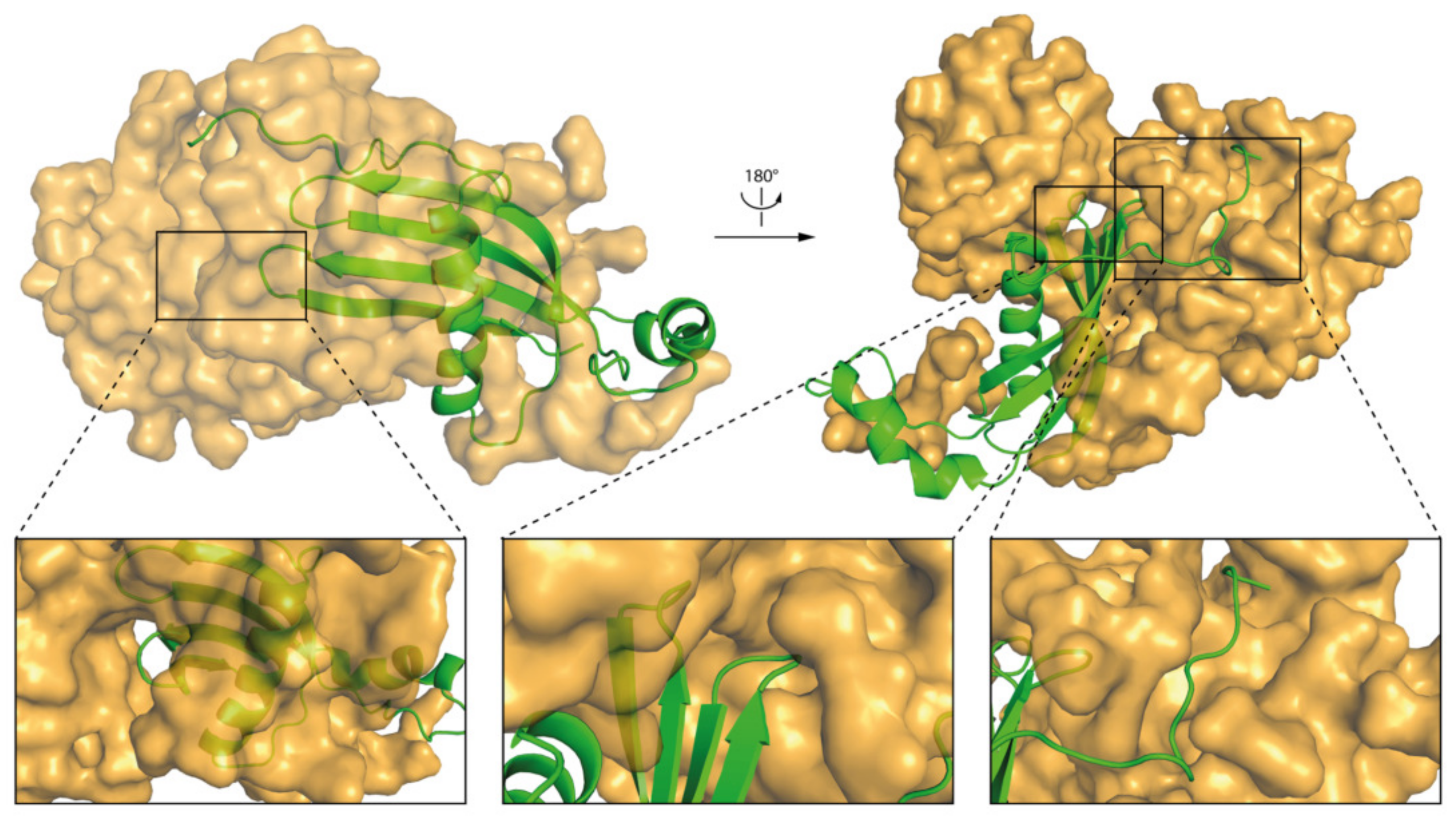
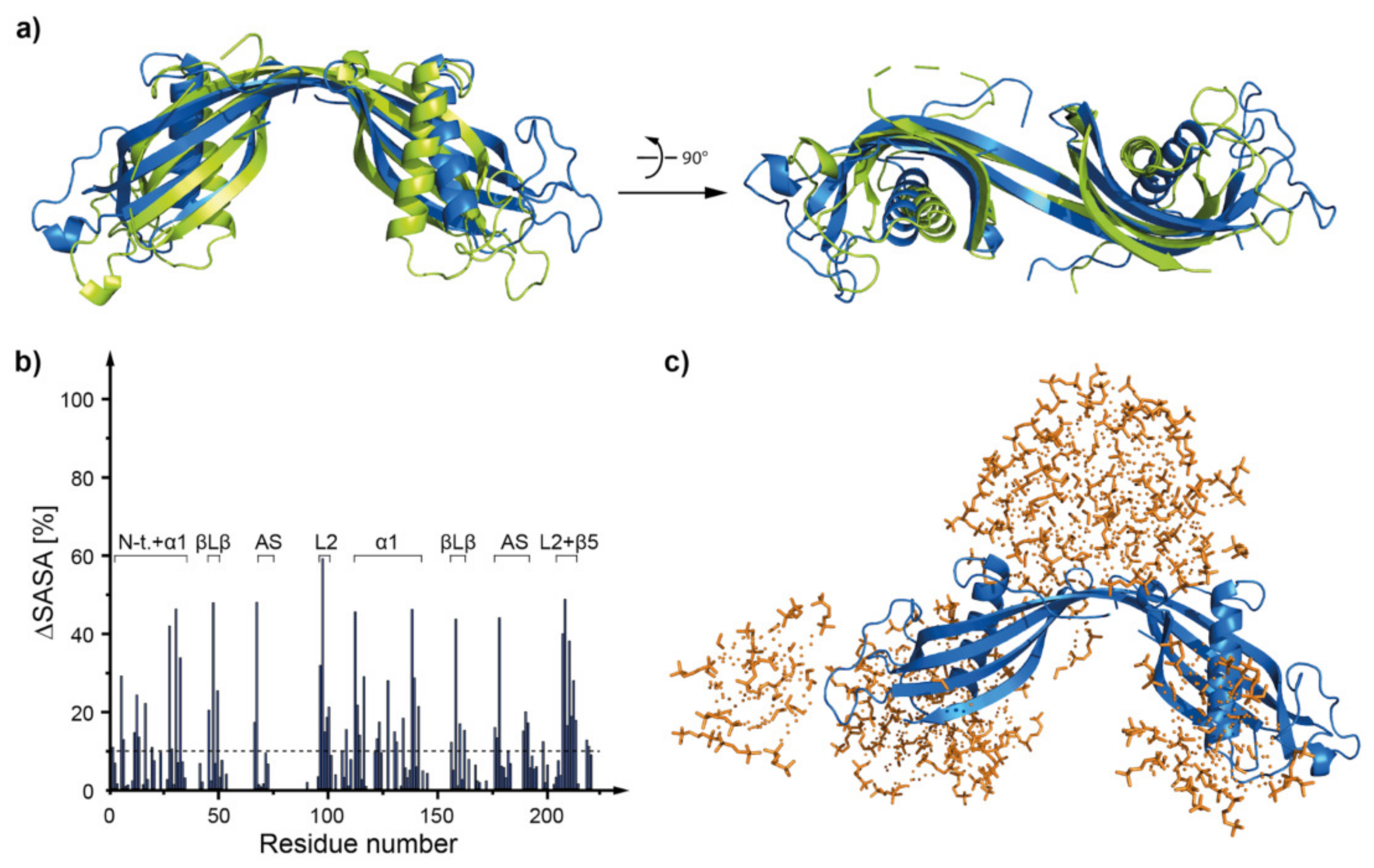
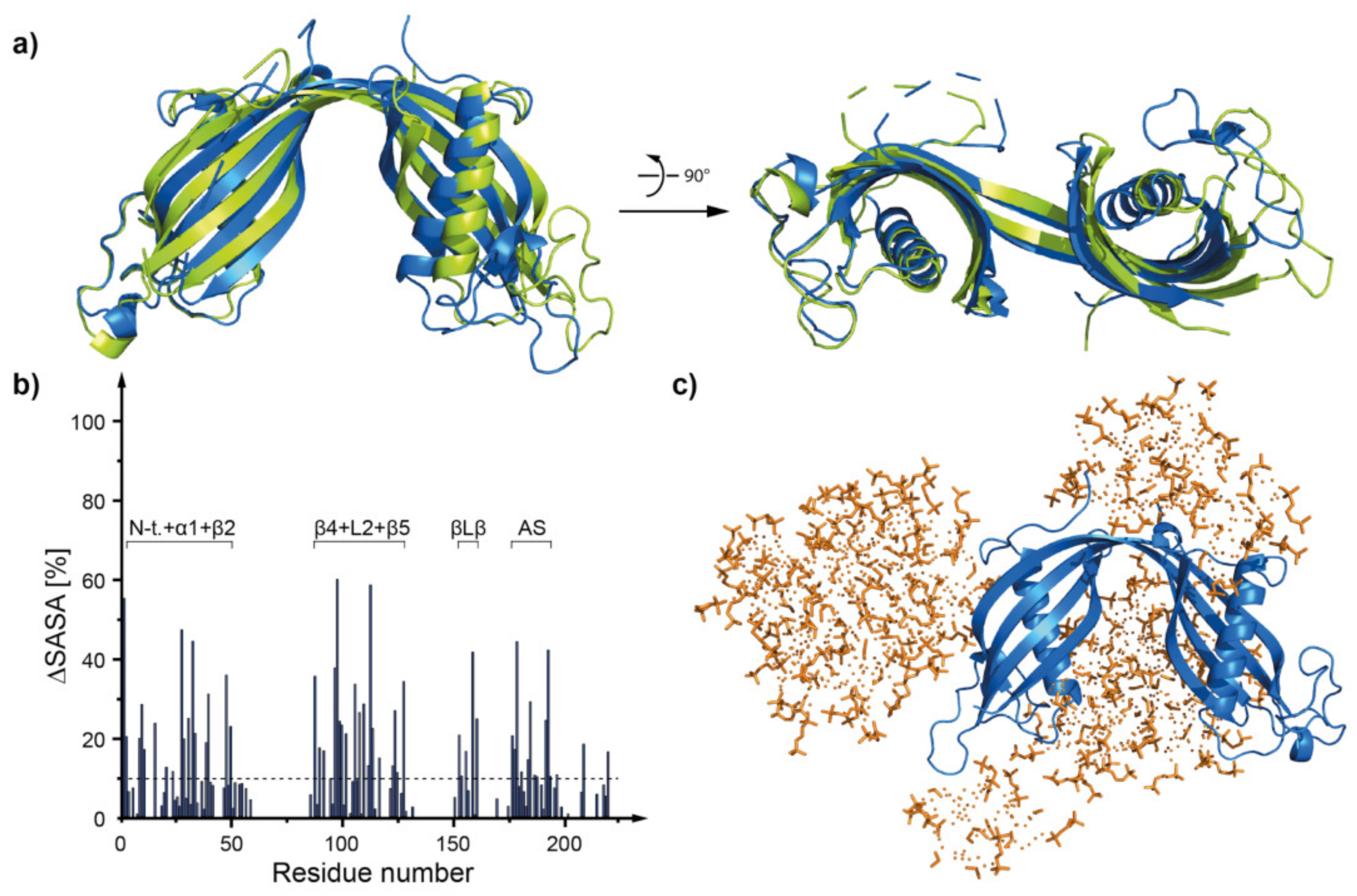
2.5. Molecular Dynamics—What Kind of Global and Local Structural Changes in the hCC Structure May Occur in a Virtual Micelle Environment?
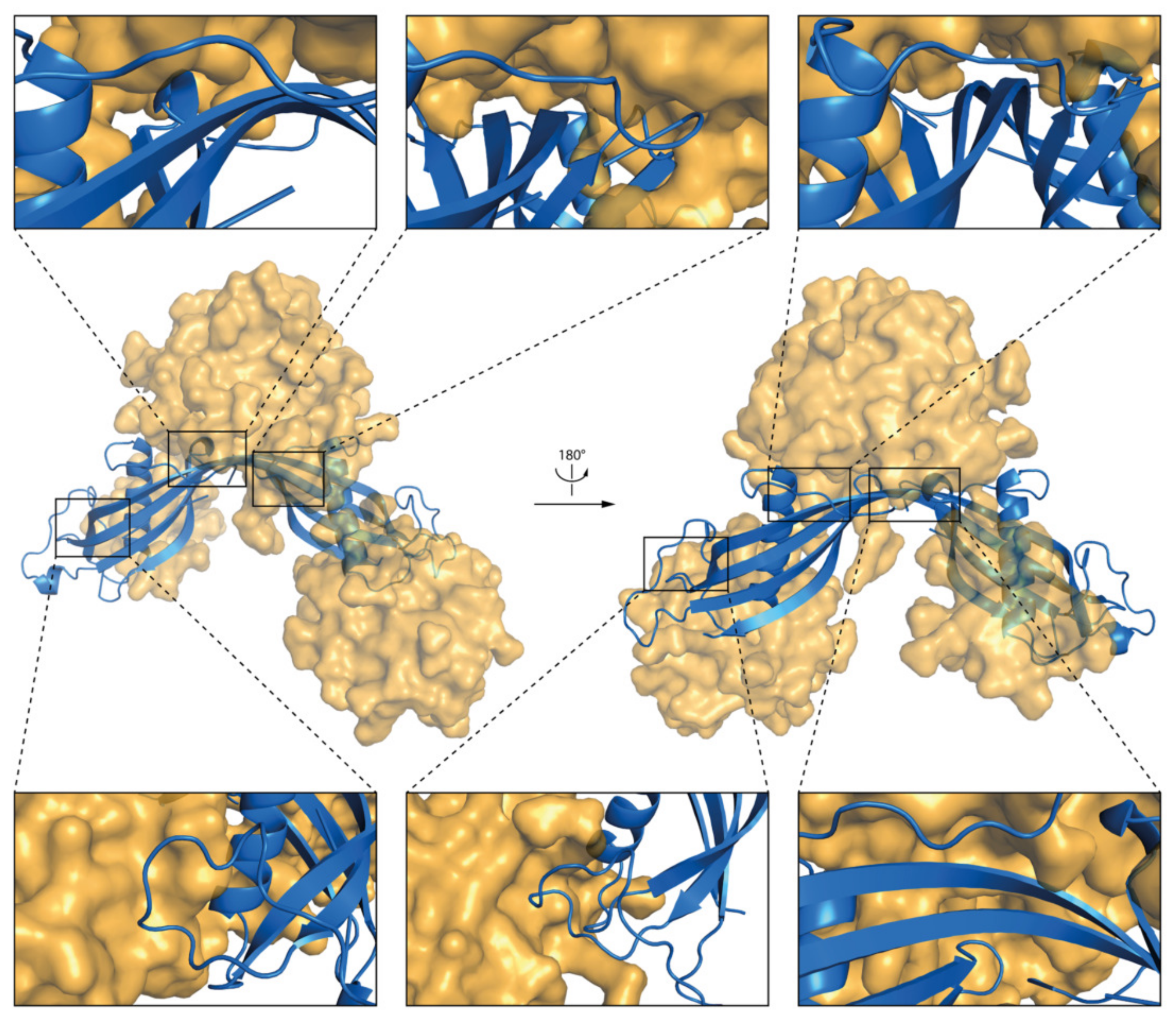
2.5.1. DPC:SDS Mixed Micelle—hCC Monomer (WT and hCC V57G)
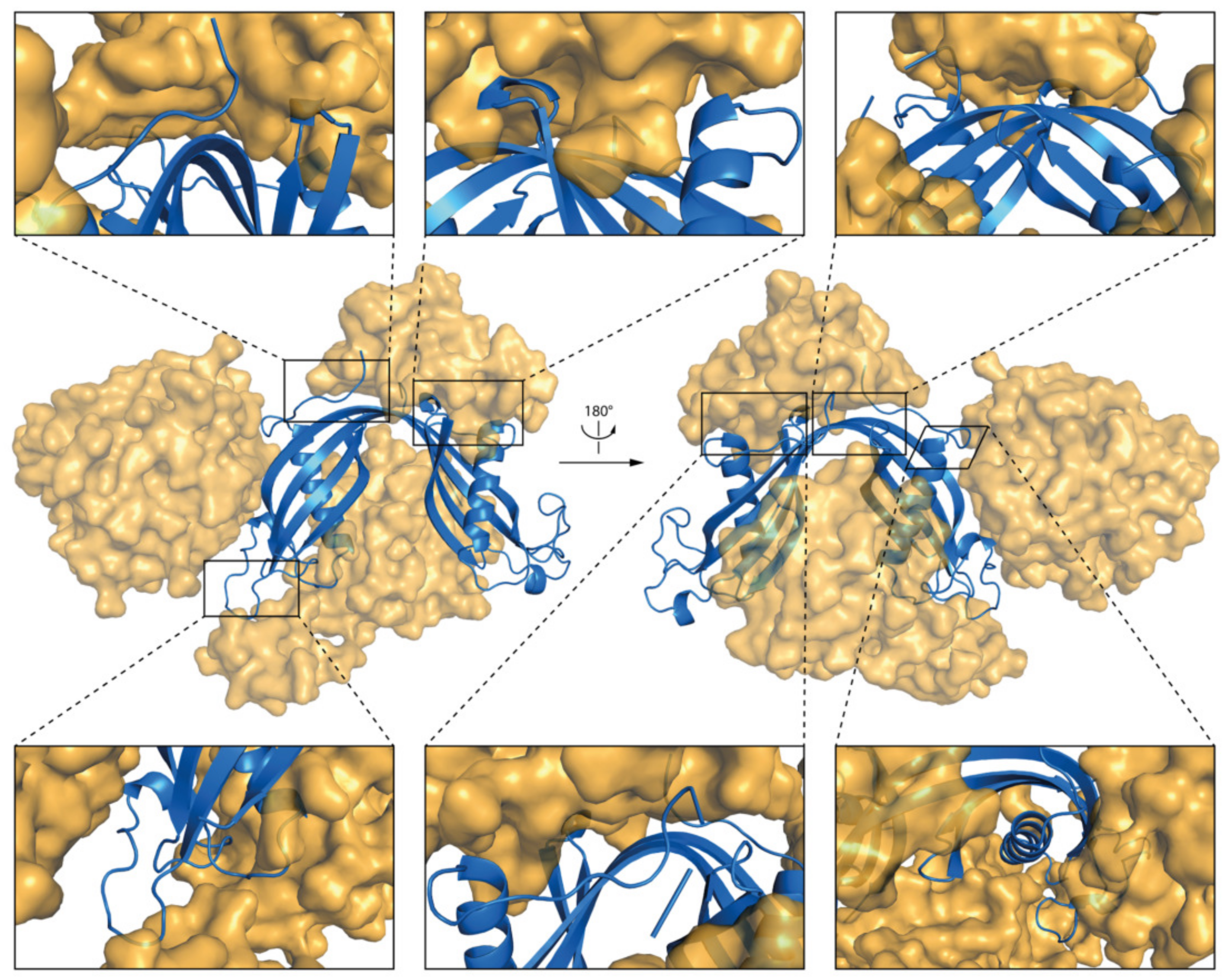
2.5.2. DPC:SDS Mixed Micelle—hCC Dimer (WT and V57P)
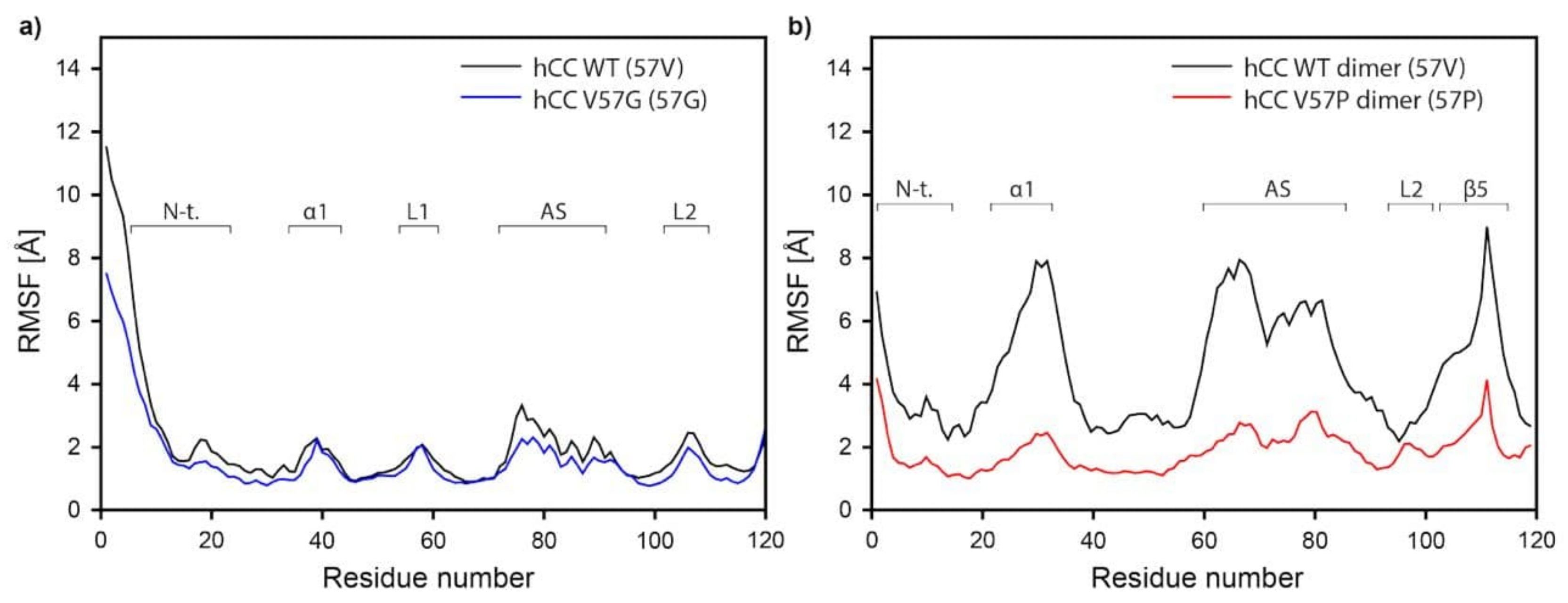
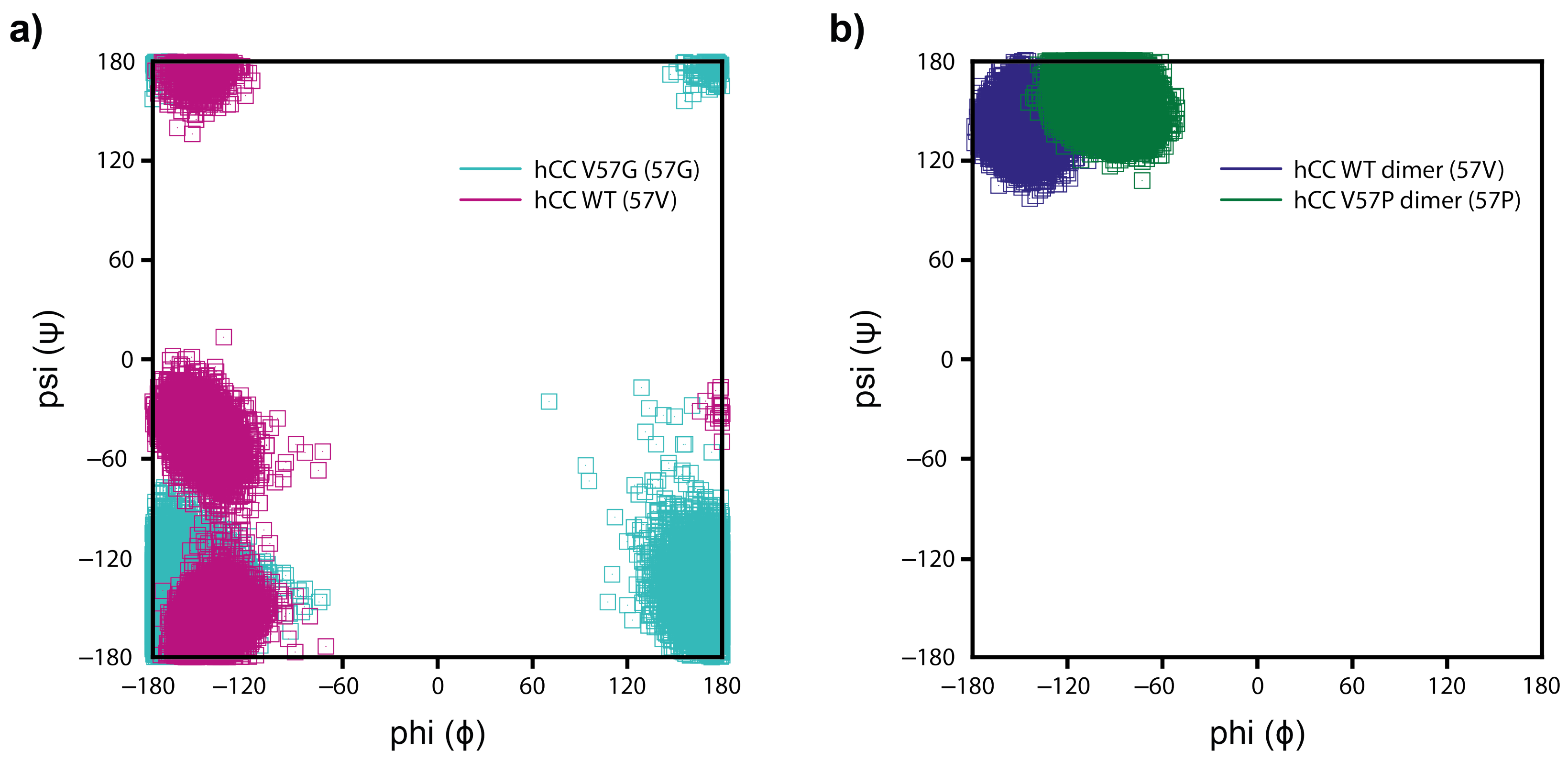
2.5.3. Molecular Dynamics—Fluctuations
3. Discussion
3.1. Size Exclusion Chromatography—The DPC:SDS Micelle Reverses the hCC Dimerization Process
3.2. Circular Dichroism—The Micelle Influences the Level of Order of the hCC Secondary Structure
3.3. Nuclear Magnetic Resonance—Micelle Binds to the Flexible Parts of hCC Structure
3.4. Molecular Dynamics—Micelle Causes Structural Displacement in the Flexible Parts of hCC Structure
4. Conclusions
5. Materials and Methods
5.1. Expression and Purification of Labeled and Unlabeled Proteins
5.2. hCC WT Dimerization
5.3. Micelle Sample Preparation
5.4. Analytical Size Exclusion Chromatography of hCC
5.5. Circular Dichroism Spectroscopy
5.6. NMR Spectroscopy
5.7. Molecular Dynamics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CD | Circular dichroism |
| hCC | human cystatine C |
| hCC V57G | Val57→Gly mutant of human cystatine C |
| hCC V57P | Val57→Pro mutant of human cystatine C |
| DMPC | dimyristoylphosphocholine |
| DPC | dodecylphosphocholine |
| SDS | sodium dodecyl sulfate |
| CMC | critical micelle concentration |
| SEC | size exclusion chromatography |
| RMSD | root mean square distance |
| SASA | solvent accessible surface area |
| DSS | sodium 2,2-dimethyl-2-silapentane-5-sulfonat |
| WT | wild-type |
References
- Merlini, G.; Bellotti, V. Molecular mechanisms of amyloidosis. N. Engl. J. Med. 2003, 349, 583–596. [Google Scholar] [CrossRef]
- Siddiqi, M.K.; Alam, P.; Chaturvedi, S.K.; Shahein, Y.E.; Khan, R.H. Mechanisms of protein aggregation and inhibition. Front. Biosci. 2017, 9, 1–20. [Google Scholar]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef]
- Dobson, C.M. The amyloid phenomenon and its links with human disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a023648. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Butterfield, S.M.; Lashuel, H.A. Amyloidogenic protein–membrane interactions: Mechanistic insight from model systems. Angew. Chem. Int. Ed. 2010, 49, 5628–5654. [Google Scholar] [CrossRef]
- Marin-Argany, M.; Lin, Y.; Misra, P.; Williams, A.; Wall, J.S.; Howell, K.G.; Elsbernd, L.R.; McClure, M.; Ramirez-Alvarado, M. Cell damage in light chain amyloidosis fibril internalization, toxicity and cell-mediated seeding. J. Biol. Chem. 2016, 291, 19813–19825. [Google Scholar] [CrossRef]
- Ciudad, S.; Puig, E.; Botzanowski, T.; Meigooni, M.; Arango, A.S.; Do, J.; Mayzel, M.; Bayoumi, M.; Chaignepain, S.; Maglia, G.; et al. Aβ (1-42) tetramer and octamer structures reveal edge conductivity pores as a mechanism for membrane damage. Nat. Commun. 2020, 11, 3014. [Google Scholar] [CrossRef]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Lansbury, P.T. Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q. Rev. Biophys. 2006, 39, 167–201. [Google Scholar] [CrossRef]
- Wahlbom, M.; Wang, X.; Lindström, V.; Carlemalm, E.; Jaskolski, M.; Grubb, A. Fibrillogenic oligomers of human cystatin C are formed by propagated domain swapping. J. Biol. Chem. 2007, 282, 18318–18326. [Google Scholar] [CrossRef]
- Grubb, A.O. Cystatin C-properties and use as diagnostic marker. Adv. Clin. Chem. 2001, 35, 63–99. [Google Scholar]
- Turk, V.; Stoka, V.; Turk, D. Cystatins: Biochemical and structural properties, and medical relevance. Front. Biosci. 2008, 13, 5406–5420. [Google Scholar] [CrossRef] [PubMed]
- Mussap, M.; Plebani, M. Biochemistry and clinical role of human cystatin C. Crit. Rev. Clin. Lab. Sci. 2004, 41, 467–550. [Google Scholar] [CrossRef] [PubMed]
- Pirttilä, T.J.; Lukasiuk, K.; Håkansson, K.; Grubb, A.; Abrahamson, M.; Pitkänen, A. Cystatin C modulates neurodegeneration and neurogenesis following status epilepticus in mouse. Neurobiol. Dis. 2005, 20, 241–253. [Google Scholar] [CrossRef]
- Nagai, A.; Terashima, M.; Sheikh, A.M.; Notsu, Y.; Shimode, K.; Yamaguchi, S.; Kobayashi, S.; Kim, S.U.; Masuda, J. Involvement of cystatin C in pathophysiology of CNS diseases. Front. Biosci. 2008, 13, 3470–3479. [Google Scholar] [CrossRef]
- Wada, Y.; Nagai, A.; Sheikh, A.; Mitaki, S.; Yamaguchi, S. Aggregation mechanism of cystatin c in neuronal cell and its implication in neurodegeneration. J. Neurol. Sci. 2017, 381, 718. [Google Scholar] [CrossRef]
- Palsdottir, A.; Snorradottir, A.; Thorsteinsson, L. Hereditary cystatin C amyloid angiopathy: Genetic, clinical, and pathological aspects. Brain Pathol. 2006, 16, 55–59. [Google Scholar]
- Jensson, O.; Gudmundsson, G.; Arnason, A.; Blöndal, H.; Petursdottir, I.; Thorsteinsson, L.; Grubb, A.; Löfberg, H.; Cohen, D.; Frangione, B. Hereditary cystatin C (γ-trace) amyloid angiopathy of the CNS causing cerebral hemorrhage. Acta Neurol. Scand. 1987, 76, 102–114. [Google Scholar] [CrossRef]
- Levy, E. Cystatin C in Alzheimer’s disease. Front. Mol. Neurosci. 2012, 5, 79. [Google Scholar]
- Merz, G.S.; Benedikz, E.; Schwenk, V.; Johansen, T.E.; Vogel, L.K.; Rushbrook, J.I.; Wisniewski, H.M. Human cystatin C forms an inactive dimer during intracellular trafficking in transfected CHO cells. J. Cell. Physiol. 1997, 173, 423–432. [Google Scholar] [CrossRef]
- Serra-Batiste, M.; Ninot-Pedrosa, M.; Bayoumi, M.; Gairí, M.; Maglia, G.; Carulla, N. Aβ42 assembles into specific β-barrel pore-forming oligomers in membrane-mimicking environments. Proc. Natl. Acad. Sci. USA 2016, 113, 10866–10871. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Ng, W.J.; Bhattacharjya, S. NMR structure and localization of the host defense antimicrobial peptide thanatin in zwitterionic dodecylphosphocholine micelle: Implications in antimicrobial activity. Biochim. Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183432. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Sarker, M.; Huang, S.K.; Rainey, J.K. Apelin conformational and binding equilibria upon micelle interaction primarily depend on membrane-mimetic headgroup. Sci. Rep. 2017, 7, 15433. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Tamm, L.K. Solution NMR of SNAREs, complexin and α-synuclein in association with membrane-mimetics. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 105, 41–53. [Google Scholar] [CrossRef]
- Krainer, G.; Hartmann, A.; Anandamurugan, A.; Gracia, P.; Keller, S.; Schlierf, M. Ultrafast protein folding in membrane-mimetic environments. J. Mol. Biol. 2018, 430, 554–564. [Google Scholar] [CrossRef]
- Langham, A.A.; Waring, A.J.; Kaznessis, Y. Comparison of interactions between beta-hairpin decapeptides and SDS/DPC micelles from experimental and simulation data. BMC Biochem. 2007, 8, 11. [Google Scholar] [CrossRef]
- Sikorska, E.; Wyrzykowski, D.; Szutkowski, K.; Greber, K.; Lubecka, E.A.; Zhukov, I. Thermodynamics, size, and dynamics of zwitterionic dodecylphosphocholine and anionic sodium dodecyl sulfate mixed micelles. J. Therm. Anal. Calorim. 2016, 123, 511–523. [Google Scholar] [CrossRef]
- Szymanska, A.; Jankowska, E.; Orlikowska, M.; Behrendt, I.; Czaplewska, P.; Rodziewicz-Motowidło, S. Influence of point mutations on the stability, dimerization, and oligomerization of human cystatin C and its L68Q variant. Front. Mol. Neurosci. 2012, 5, 82. [Google Scholar] [CrossRef]
- Rodziewicz-Motowidło, S.; Iwaszkiewicz, J.; Sosnowska, R.; Czaplewska, P.; Sobolewski, E.; Szymańska, A.; Stachowiak, K.; Liwo, A. The role of the Val57 amino-acid residue in the hinge loop of the human cystatin C. Conformational studies of the beta2-L1-beta3 segments of wild-type human cystatin C and its mutants. Biopolymers 2009, 91, 373–383. [Google Scholar] [CrossRef]
- Szymańska, A.; Radulska, A.; Czaplewska, P.; Grubb, A.; Grzonka, Z.; Rodziewicz-Motowidło, S. Governing the monomer-dimer ratio of human cystatin c by single amino acid substitution in the hinge region. Acta Biochim. Pol. 2009, 56. [Google Scholar] [CrossRef]
- Maszota-Zieleniak, M.; Jurczak, P.; Orlikowska, M.; Zhukov, I.; Borek, D.; Otwinowski, Z.; Skowron, P.; Pietralik, Z.; Kozak, M.; Szymańska, A.; et al. NMR and crystallographic structural studies of the extremely stable monomeric variant of human cystatin C with single amino acid substitution. FEBS J. 2020, 287, 361–376. [Google Scholar] [CrossRef]
- Berek, D. Size exclusion chromatography—A blessing and a curse of science and technology of synthetic polymers. J. Sep. Sci. 2010, 33, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Grubisic, Z.; Rempp, P.; Benoit, H. A universal calibration for gel permeation chromatography. J. Polym. Sci. Part B Polym. Lett. 1967, 5, 753–759. [Google Scholar] [CrossRef]
- Deep, S.; Ahluwalia, J.C. Interaction of bovine serum albumin with anionic surfactants. Phys. Chem. Chem. Phys. 2001, 3, 4583–4591. [Google Scholar] [CrossRef]
- Li, Y.; Cao, M.; Wang, Y. Alzheimer amyloid β (1- 40) peptide: Interactions with cationic gemini and single-chain surfactants. J. Phys. Chem. B 2006, 110, 18040–18045. [Google Scholar] [CrossRef]
- Österlund, N.; Kulkarni, Y.S.; Misiaszek, A.D.; Wallin, C.; Krüger, D.M.; Liao, Q.; Mashayekhy Rad, F.; Jarvet, J.; Strodel, B.; Wärmländer, S.K.; et al. Amyloid-β peptide interactions with amphiphilic surfactants: Electrostatic and hydrophobic effects. ACS Chem. Neurosci. 2018, 9, 1680–1692. [Google Scholar] [CrossRef] [PubMed]
- Sureshbabu, N.; Kirubagaran, R.; Jayakumar, R. Surfactant-induced conformational transition of amyloid β-peptide. Eur. Biophys. J. 2009, 38, 355. [Google Scholar] [CrossRef]
- Ausubel, F.; Brent, R.; Kingston, R.; Moore, D.; Seidman, J.; Smith, J.; Struhl, K. Analysis of proteins. Curr. Protoc. Mol. Biol. 1987, 2. [Google Scholar] [CrossRef]
- Hall, A.; Håkansson, K.; Mason, R.W.; Grubb, A.; Abrahamson, M. Structural basis for the biological specificity of cystatin C identification of leucine 9 in the N-terminal binding region as a selectivity-conferring residue in the inhibition of mammalian cysteine peptidases. J. Biol. Chem. 1995, 270, 5115–5121. [Google Scholar] [CrossRef]
- Alvarez-Fernandez, M.; Barrett, A.J.; Gerhartz, B.; Dando, P.M.; Ni, J.; Abrahamson, M. Inhibition of mammalian legumain by some cystatins is due to a novel second reactive site. J. Biol. Chem. 1999, 274, 19195–19203. [Google Scholar] [CrossRef] [PubMed]
- Janowski, R.; Kozak, M.; Abrahamson, M.; Grubb, A.; Jaskolski, M. 3D domain-swapped human cystatin C with amyloidlike intermolecular β-sheets. Proteins Struct. Funct. Bioinform. 2005, 61, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Staniforth, R.A.; Giannini, S.; Higgins, L.D.; Conroy, M.J.; Hounslow, A.M.; Jerala, R.; Craven, C.J.; Waltho, J.P. Three-dimensional domain swapping in the folded and molten-globule states of cystatins, an amyloid-forming structural superfamily. EMBO J. 2001, 20, 4774–4781. [Google Scholar] [CrossRef] [PubMed]
- Pujols, J.; Peña-Díaz, S.; Lázaro, D.F.; Peccati, F.; Pinheiro, F.; González, D.; Carija, A.; Navarro, S.; Conde-Giménez, M.; García, J.; et al. Small molecule inhibits α-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2018, 115, 10481–10486. [Google Scholar] [CrossRef]
- Abrahamson, M.; Dalbøge, H.; Olafsson, I.; Carlsen, S.; Grubb, A. Efficient production of native, biologically active human cystatin C by Escherichia coli. FEBS Lett. 1988, 236, 14–18. [Google Scholar] [CrossRef]
- Ekiel, I.; Abrahamson, M. Folding-related dimerization of human cystatin C. J. Biol. Chem. 1996, 271, 1314–1321. [Google Scholar] [CrossRef]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef]
- Ayed, A.; Mulder, F.A.; Yi, G.S.; Lu, Y.; Kay, L.E.; Arrowsmith, C.H. Latent and active p53 are identical in conformation. Nat. Struct. Biol. 2001, 8, 756–760. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System. 2002. Available online: https://www.pymol.org (accessed on 1 December 2019).
- Koradi, R.; Billeter, M.; Wuthrich, K. MOLMOL: Programa para la visualización y el análisis de estructuras macromoleculares. J. Mol. Graph. Model. 1996, 14, 51–55. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Whole hCC | Protein (Monomer or Dimer) 1 | Protein (Monomer) 2 |
|---|---|---|---|
| hCC-1 | 6.27 ± 2.71 Å | 2.05 ± 0.89 Å | 1.38 ± 0.60 Å |
| hCC-2 | 2.85 ± 1.23 Å | 2.72 ± 1.18 Å | 2.49 ± 1.07 Å |
| hCC-3 | 4.57 ± 1.98 Å | 2.74 ± 1.18 Å | 1.73 ± 0.75 Å |
| V57G-1 | 4.00 ± 1.73 Å | 2.56 ± 1.10 Å | 1.48 ± 0.64 Å |
| V57G-2 | 3.64 ± 1.58 Å | 1.84 ± 0.80 Å | 1.56 ± 0.68 Å |
| V57G-3 | 2.52 ± 1.09 Å | 2.48 ± 1.07 Å | 1.53 ± 0.66 Å |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurczak, P.; Sikorska, E.; Czaplewska, P.; Rodziewicz-Motowidlo, S.; Zhukov, I.; Szymanska, A. The Influence of the Mixed DPC:SDS Micelle on the Structure and Oligomerization Process of the Human Cystatin C. Membranes 2021, 11, 17. https://doi.org/10.3390/membranes11010017
Jurczak P, Sikorska E, Czaplewska P, Rodziewicz-Motowidlo S, Zhukov I, Szymanska A. The Influence of the Mixed DPC:SDS Micelle on the Structure and Oligomerization Process of the Human Cystatin C. Membranes. 2021; 11(1):17. https://doi.org/10.3390/membranes11010017
Chicago/Turabian StyleJurczak, Przemyslaw, Emilia Sikorska, Paulina Czaplewska, Sylwia Rodziewicz-Motowidlo, Igor Zhukov, and Aneta Szymanska. 2021. "The Influence of the Mixed DPC:SDS Micelle on the Structure and Oligomerization Process of the Human Cystatin C" Membranes 11, no. 1: 17. https://doi.org/10.3390/membranes11010017
APA StyleJurczak, P., Sikorska, E., Czaplewska, P., Rodziewicz-Motowidlo, S., Zhukov, I., & Szymanska, A. (2021). The Influence of the Mixed DPC:SDS Micelle on the Structure and Oligomerization Process of the Human Cystatin C. Membranes, 11(1), 17. https://doi.org/10.3390/membranes11010017