DMPC Phospholipid Bilayer as a Potential Interface for Human Cystatin C Oligomerization: Analysis of Protein-Liposome Interactions Using NMR Spectroscopy

 , ,
, ,  ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
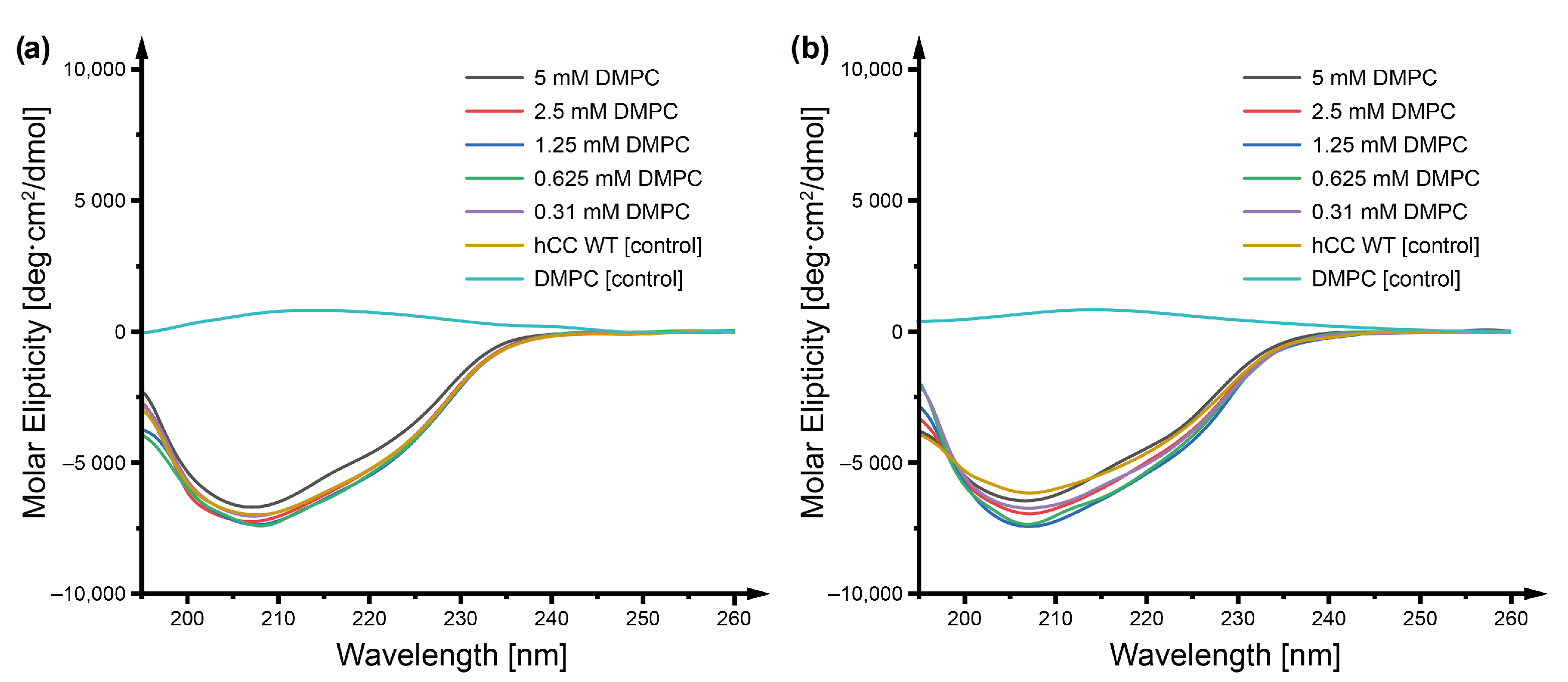
2.1. Circular Dichroism
2.2. Size Exclusion Chromatography
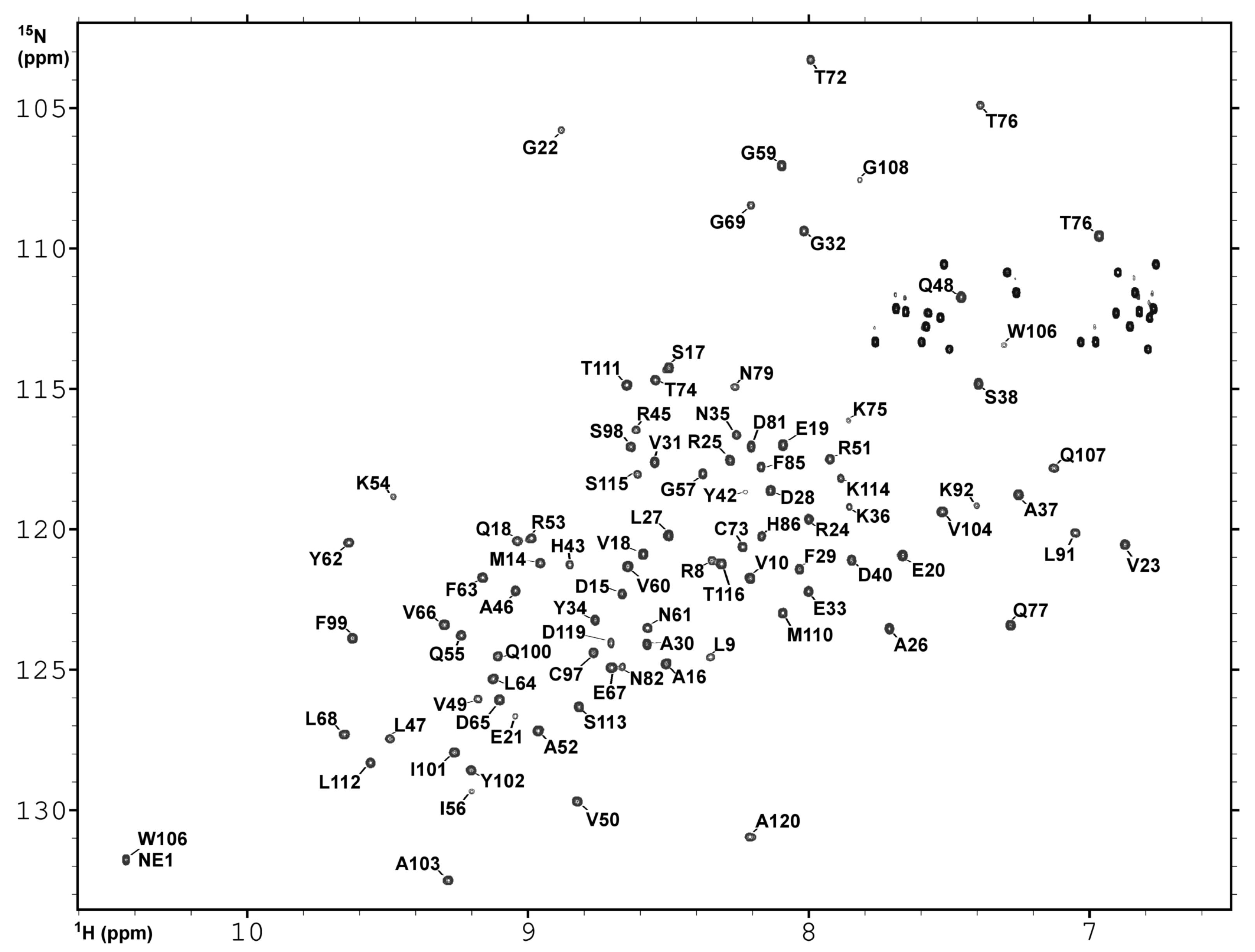
2.3. NMR Spectroscopy and Assignment of the H and N Backbone Resonances
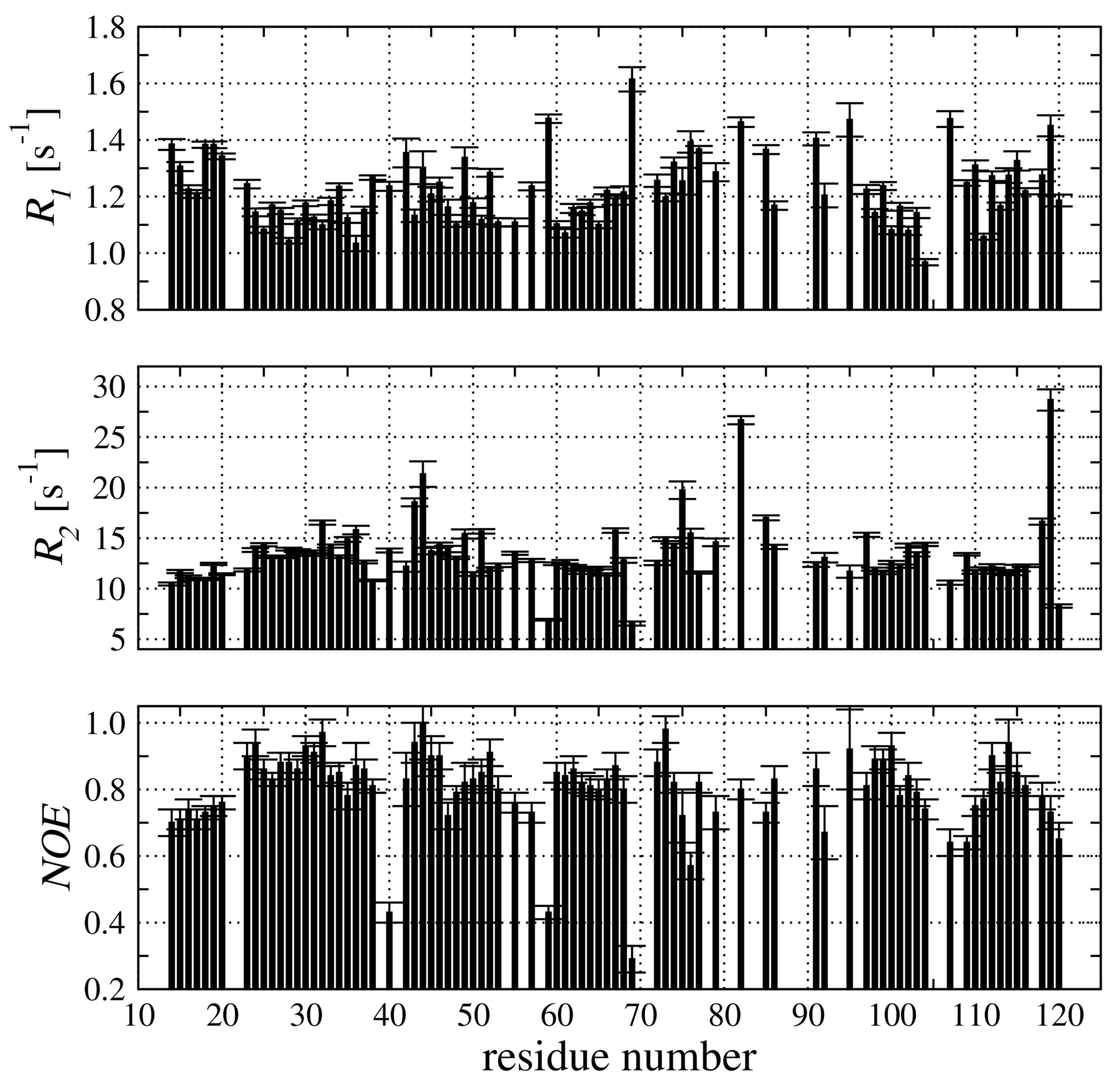
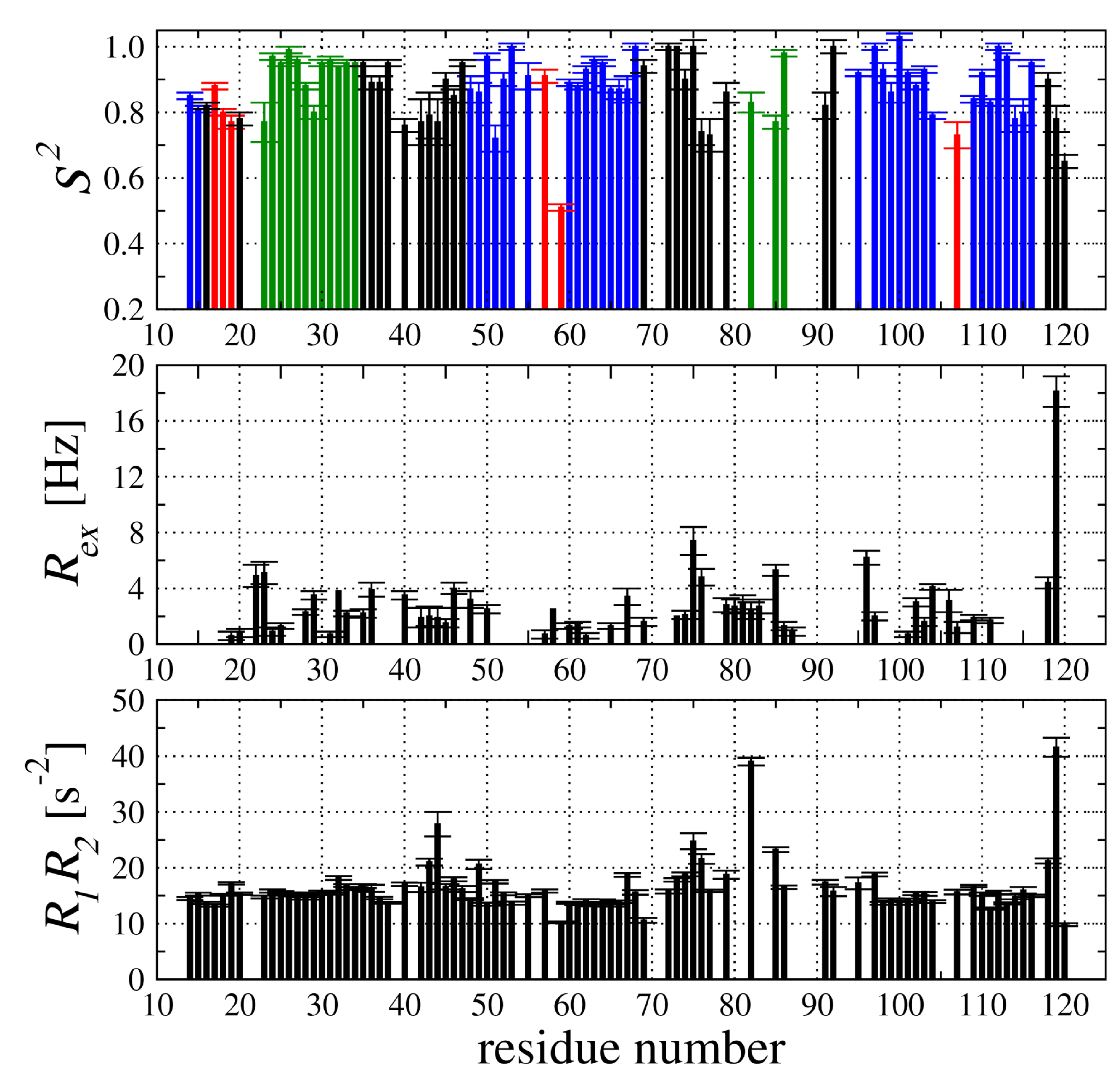
2.4. Backbone Dynamic of hCC V57G Based on the N Relaxation Data
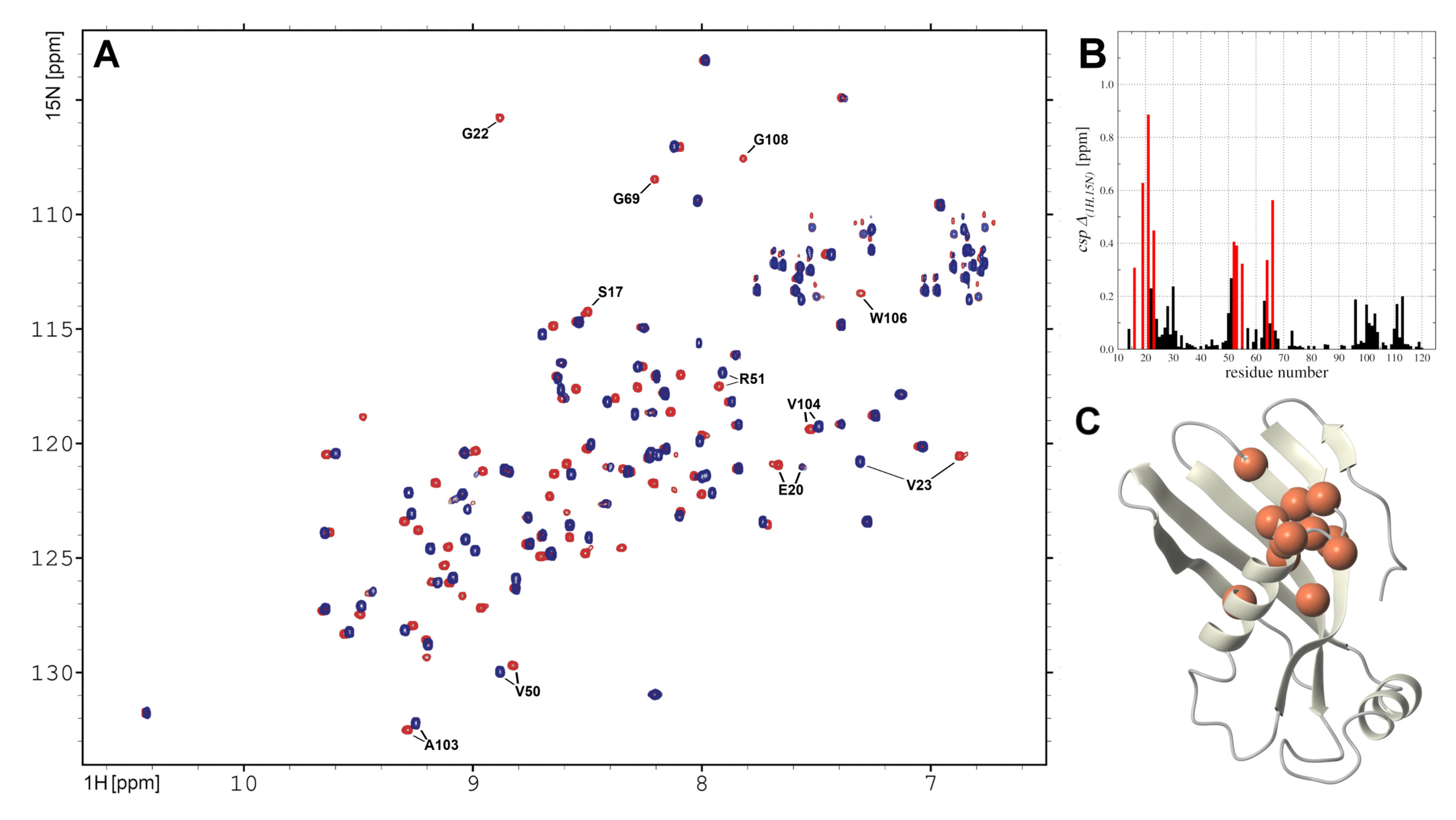
2.5. hCC V57G Interactions with DMPC Liposome
3. Conclusions
4. Materials and Methods
4.1. Expression and Purification of Labeled and Unlabeled Proteins
4.2. Liposome Sample Preparation
4.3. Circular Dichroism Spectroscopy
4.4. Analytical Size Exclusion Chromatography
4.5. NMR Sspectroscopy
4.6. N Relaxation Measurements
4.7. Analysis of N Relaxation Data with a Model-Free Approach
4.8. Translational Diffusion Measurement with PGSE-NMR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CD | Circular dichroism |
| hCC | human cystatine C |
| hCC V57G | Val57→Gly mutant of human cystatine C |
| DMPC | dimyristoylphosphocholine |
| DPC | dodecylphosphocholine |
| SDS | sodium dodecyl sulfate |
References
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Sakono, M.; Zako, T. Amyloid oligomers: Formation and toxicity of Aβ oligomers. FEBS J. 2010, 277, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, M.F.; Tempra, C.; Scollo, F.; Milardi, D.; La Rosa, C. Amyloid growth and membrane damage: Current themes and emerging perspectives from theory and experiments on Aβ and hIAPP. Biochim. Biophys. Acta (BBA) Biomembr. 2018, 1860, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Vahdat Shariat Panahi, A.; Hultman, P.; Öllinger, K.; Westermark, G.T.; Lundmark, K. Lipid membranes accelerate amyloid formation in the mouse model of AA amyloidosis. Amyloid 2019, 26, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, S.M.; Lashuel, H.A. Amyloidogenic protein–membrane interactions: Mechanistic insight from model systems. Angew. Chem. Int. Ed. 2010, 49, 5628–5654. [Google Scholar] [CrossRef]
- Harper, J.D.; Wong, S.S.; Lieber, C.M.; Lansbury, P.T., Jr. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem. Biol. 1997, 4, 119–125. [Google Scholar] [CrossRef]
- Ionescu-Zanetti, C.; Khurana, R.; Gillespie, J.R.; Petrick, J.S.; Trabachino, L.C.; Minert, L.J.; Carter, S.A.; Fink, A.L. Monitoring the assembly of Ig light-chain amyloid fibrils by atomic force microscopy. Proc. Natl. Acad. Sci. USA 1999, 96, 13175–13179. [Google Scholar] [CrossRef]
- Wahlbom, M.; Wang, X.; Lindström, V.; Carlemalm, E.; Jaskolski, M.; Grubb, A. Fibrillogenic oligomers of human cystatin C are formed by propagated domain swapping. J. Biol. Chem. 2007, 282, 18318–18326. [Google Scholar] [CrossRef]
- Ísleifur, Ó.; Grubb, A. Hereditary cystatin C amyloid angiopathy. Amyloid 2000, 7, 70–79. [Google Scholar]
- Deng, A.; Irizarry, M.C.; Nitsch, R.M.; Growdon, J.H.; Rebeck, G.W. Elevation of Cystatin C in Susceptible Neurons in Alzheimer’s Disease. Am. J. Pathol. 2001, 159, 1061–1068. [Google Scholar] [CrossRef]
- Sikorska, E.; Wyrzykowski, D.; Szutkowski, K.; Greber, K.; Lubecka, E.A.; Zhukov, I. Thermodynamics, size, and dynamics of zwitterionic dodecylphosphocholine and anionic sodium dodecyl sulfate mixed micelles. J. Therm. Anal. Calorim. 2016, 123, 511–523. [Google Scholar] [CrossRef]
- Sampaio, J.L.; Gerl, M.J.; Klose, C.; Ejsing, C.S.; Beug, H.; Simons, K.; Shevchenko, A. Membrane lipidome of an epithelial cell line. Proc. Natl. Acad. Sci. USA 2011, 108, 1903–1907. [Google Scholar] [CrossRef] [PubMed]
- Chipot, C.; Dehez, F.; Schnell, J.R.; Zitzmann, N.; Pebay-Peyroula, E.; Catoire, L.J.; Miroux, B.; Kunji, E.R.; Veglia, G.; Cross, T.A.; et al. Perturbations of native membrane protein structure in alkyl phosphocholine detergents: A critical assessment of NMR and biophysical studies. Chem. Rev. 2018, 118, 3559–3607. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Xue, D. The ins and outs of phospholipid asymmetry in the plasma membrane: Roles in health and disease. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Tun, T.N.; Jenkins, A.T.A. An electrochemical impedance study of the effect of pathogenic bacterial toxins on tethered bilayer lipid membrane. Electrochem. Commun. 2010, 12, 1411–1415. [Google Scholar] [CrossRef]
- Valincius, G.; Heinrich, F.; Budvytyte, R.; Vanderah, D.J.; McGillivray, D.J.; Sokolov, Y.; Hall, J.E.; Lösche, M. Soluble Amyloid β-Oligomers Affect Dielectric Membrane Properties by Bilayer Insertion and Domain Formation: Implications for Cell Toxicity. Biophys. J. 2008, 95, 4845–4861. [Google Scholar] [CrossRef]
- Hane, F.; Drolle, E.; Gaikwad, R.; Faught, E.; Leonenko, Z. Amyloid-β aggregation on model lipid membranes: An atomic force microscopy study. J. Alzheimers Dis. 2011, 26, 485–494. [Google Scholar] [CrossRef]
- Drolle, E.; Hane, F.; Lee, B.; Leonenko, Z. Atomic force microscopy to study molecular mechanisms of amyloid fibril formation and toxicity in Alzheimer’s disease. Drug Metab. Rev. 2014, 46, 207–223. [Google Scholar] [CrossRef]
- Pinilla, C.M.B.; Brandelli, A.; López-Caballero, M.E.; Montero, P.; del Carmen Gómez-Guillén, M. Structural features of myofibrillar fish protein interacting with phosphatidylcholine liposomes. Food Res. Int. 2020, 137, 109687. [Google Scholar] [CrossRef]
- Foteini, P.; Pippa, N.; Naziris, N.; Demetzos, C. Physicochemical study of the protein–liposome interactions: Influence of liposome composition and concentration on protein binding. J. Liposome Res. 2019, 29, 313–321. [Google Scholar] [CrossRef]
- Brender, J.R.; Ghosh, A.; Kotler, S.A.; Krishnamoorthy, J.; Bera, S.; Morris, V.; Sil, T.B.; Garai, K.; Reif, B.; Bhunia, A.; et al. Probing transient non-native states in amyloid beta fiber elongation by NMR. Chem. Commun. 2019, 55, 4483–4486. [Google Scholar] [CrossRef] [PubMed]
- Divakara, M.B.; Martinez, D.; Ravi, A.; Bhavana, V.; Ramana, V.; Habenstein, B.; Loquet, A.; Santosh, M.S. Molecular mechanisms for the destabilization of model membranes by islet amyloid polypeptide. Biophys. Chem. 2019, 245, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Fawzi, N.L.; Ying, J.; Ghirlando, R.; Torchia, D.A.; Clore, G.M. Atomic-resolution dynamics on the surface of amyloid-β protofibrils probed by solution NMR. Nature 2011, 480, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Maszota-Zieleniak, M.; Jurczak, P.; Orlikowska, M.; Zhukov, I.; Borek, D.; Otwinowski, Z.; Skowron, P.; Pietralik, Z.; Kozak, M.; Szymańska, A.; et al. NMR and crystallographic structural studies of the extremely stable monomeric variant of human cystatin C with single amino acid substitution. FEBS J. 2020, 287, 361–376. [Google Scholar] [CrossRef]
- Sreij, R.; Dargel, C.; Moleiro, L.H.; Monroy, F.; Hellweg, T. Aescin Incorporation and Nanodomain Formation in DMPC Model Membranes. Langmuir 2017, 33, 12351–12361. [Google Scholar] [CrossRef]
- Komljenović, I.; Marquardt, D.; Harroun, T.A.; Sternin, E. Location of chlorhexidine in DMPC model membranes: A neutron diffraction study. Chem. Phys. Lipids 2010, 163, 480–487. [Google Scholar] [CrossRef]
- Ezer, N.; Sahin, I.; Kazanci, N. Alliin interacts with DMPC model membranes to modify the membrane dynamics: FTIR and DSC Studies. Vib. Spectrosc. 2017, 89, 1–8. [Google Scholar] [CrossRef]
- Gagnon, M.C.; Strandberg, E.; Ulrich, A.S.; Paquin, J.F.; Auger, M. New insights into the influence of monofluorination on dimyristoylphosphatidylcholine membrane properties: A solid-state NMR study. Biochim. Biophys. Acta (BBA) Biomembr. 2018, 1860, 654–663. [Google Scholar] [CrossRef]
- Strandberg, E.; Grau-Campistany, A.; Wadhwani, P.; Bürck, J.; Rabanal, F.; Ulrich, A.S. Helix Fraying and Lipid-Dependent Structure of a Short Amphipathic Membrane-Bound Peptide Revealed by Solid-State NMR. J. Phys. Chem. B 2018, 122, 6236–6250. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Murase, O.; Sugishita, K.; Yoneyama, S.; Akada, K.; Ueha, M.; Nakamura, A.; Kobayashi, S. Optical characterization of liposomes by right angle light scattering and turbidity measurement. Biochim. Biophys. Acta (BBA) Biomembr. 2000, 1467, 219–226. [Google Scholar] [CrossRef]
- Szymańska, A.; Radulska, A.; Czaplewska, P.; Grubb, A.; Grzonka, Z.; Rodziewicz-Motowidło, S. Governing the monomer-dimer ratio of human cystatin c by single amino acid substitution in the hinge region. Acta Biochim. Pol. 2009, 56. [Google Scholar] [CrossRef]
- Taube, M.; Pietralik, Z.; Szymanska, A.; Szutkowski, K.; Clemens, D.; Grubb, A.; Kozak, M. The domain swapping of human cystatin C induced by synchrotron radiation. Sci. Rep. 2019, 9, 8548. [Google Scholar] [CrossRef] [PubMed]
- Lipari, G.; Szabo, A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982, 104, 4546–4559. [Google Scholar] [CrossRef]
- Korzhnev, D.; Billeter, M.; Arseniev, A.; Orekhov, V. NMR studies of Brownian tumbling and internal motions in proteins. Prog. Nucl. Magn. Reson. Spectrosc. 2001, 3, 197–266. [Google Scholar] [CrossRef]
- Stone, M.J.; Fairbrother, W.J.; Palmer, A.G., III; Reizer, J.; Saier, M.H., Jr.; Wright, P.E. Backbone dynamics of the Bacillus subtilis glucose permease IIA domain determined from nitrogen-15 NMR relaxation measurements. Biochemistry 1992, 31, 4394–4406. [Google Scholar] [CrossRef]
- Tjandra, N.; Garrett, D.S.; Gronenborn, A.M.; Bax, A.; Clore, G.M. Defining long range order in NMR structure determination from the dependence of heteronuclear relaxation times on rotational diffusion anisotropy. Nat. Struct. Biol. 1997, 4, 443–449. [Google Scholar] [CrossRef]
- Clore, G.M.; Gronenborn, A.M.; Szabo, A.; Tjandra, N. Determining the magnitude of the fully asymmetric diffusion tensor from heteronuclear relaxation data in the absence of structural information. J. Am. Chem. Soc. 1998, 120, 4889–4890. [Google Scholar] [CrossRef]
- Kneller, J.M.; Lu, M.; Bracken, C. An effective method for the discrimination of motional anisotropy and chemical exchange. J. Am. Chem. Soc. 2002, 124, 1852–1853. [Google Scholar] [CrossRef]
- Abrahamson, M.; Dalbøge, H.; Olafsson, I.; Carlsen, S.; Grubb, A. Efficient production of native, biologically active human cystatin C by Escherichia coli. FEBS Lett. 1988, 236, 14–18. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Farrow, N.A.; Muhandiram, R.; Singer, A.U.; Pascal, S.M.; Kay, C.M.; Gish, G.; Shoelson, S.E.; Pawson, T.; Forman-Kay, J.D.; Kay, L.E. Backbone dynamics of a free and a phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 1994, 33, 5984–6003. [Google Scholar] [CrossRef] [PubMed]
- Kay, L.E.; Nicholson, L.K.; Delaglio, F.; Bax, A.; Torchia, D. Pulse sequences for removal of the effects of cross correlation between dipolar and chemical-shift anisotropy relaxation mechanisms on the measurement of heteronuclear T1 and T2 values in proteins. J. Magn. Reson. 1992, 97, 359–375. [Google Scholar] [CrossRef]
- d’Auvergne, E.J.; Gooley, P.R. Optimisation of NMR dynamic models I. Minimisation algorithms and their performance within the model-free and Brownian rotational diffusion spaces. J. Biomol. NMR 2008, 40, 107. [Google Scholar] [CrossRef]
- Peng, J.W.; Wagner, G. Frequency spectrum of NH bonds in eglin c from spectral density mapping at multiple fields. Biochemistry 1995, 34, 16733–16752. [Google Scholar] [CrossRef]
- Barbato, G.; Ikura, M.; Kay, L.E.; Pastor, R.W.; Bax, A. Backbone dynamics of calmodulin studied by nitrogen-15 relaxation using inverse detected two-dimensional NMR spectroscopy: The central helix is flexible. Biochemistry 1992, 31, 5269–5278. [Google Scholar] [CrossRef]
- Wu, D.; Chen, A.; Johnson, C.S. An improved diffusion-ordered spectroscopy experiment incorporating bipolar-gradient pulses. J. Magn. Reson. Ser. A 1995, 115, 260–264. [Google Scholar] [CrossRef]
- Hwang, T.L.; Shaka, A. Water suppression that works. Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson. Ser. A 1995, 112, 275–279. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Tanner, J.E. Spin diffusion measurements: Spin echoes in the presence of a time-dependent field gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurczak, P.; Szutkowski, K.; Lach, S.; Jurga, S.; Czaplewska, P.; Szymanska, A.; Zhukov, I. DMPC Phospholipid Bilayer as a Potential Interface for Human Cystatin C Oligomerization: Analysis of Protein-Liposome Interactions Using NMR Spectroscopy. Membranes 2021, 11, 13. https://doi.org/10.3390/membranes11010013
Jurczak P, Szutkowski K, Lach S, Jurga S, Czaplewska P, Szymanska A, Zhukov I. DMPC Phospholipid Bilayer as a Potential Interface for Human Cystatin C Oligomerization: Analysis of Protein-Liposome Interactions Using NMR Spectroscopy. Membranes. 2021; 11(1):13. https://doi.org/10.3390/membranes11010013
Chicago/Turabian StyleJurczak, Przemyslaw, Kosma Szutkowski, Slawomir Lach, Stefan Jurga, Paulina Czaplewska, Aneta Szymanska, and Igor Zhukov. 2021. "DMPC Phospholipid Bilayer as a Potential Interface for Human Cystatin C Oligomerization: Analysis of Protein-Liposome Interactions Using NMR Spectroscopy" Membranes 11, no. 1: 13. https://doi.org/10.3390/membranes11010013
APA StyleJurczak, P., Szutkowski, K., Lach, S., Jurga, S., Czaplewska, P., Szymanska, A., & Zhukov, I. (2021). DMPC Phospholipid Bilayer as a Potential Interface for Human Cystatin C Oligomerization: Analysis of Protein-Liposome Interactions Using NMR Spectroscopy. Membranes, 11(1), 13. https://doi.org/10.3390/membranes11010013