Abstract
Lithium-ion batteries play an important role in our modern society as the main option to power portable electronic devices and electric vehicles. The growing demand for these batteries encourages the development of more efficient recycling processes, aiming to decrease the environmental impact of the spent batteries and recover their valuable components. In this paper, a combined hydrometallurgical-electrodialytic method is proposed for processing battery waste. In the combined technique, the amount of leaching solution is reduced as acid is generated via electrolysis. At the same time, the use of ion-exchange membranes and the possibility of electroplating allows for a selective separation of the target metals. Experiments were performed using LiCoO2, which is one of the most used cathodes in lithium-ion batteries. First, 0.1 M HCl solution was used in batch extractions to study the kinetics of LiCoO2 dissolution, reaching an extraction of 30% and 69% of cobalt and lithium, respectively. Secondly, hydrometallurgical extraction experiments were carried out in three-compartment electrodialytic cells, enhanced with cation-exchange membranes. Experiments yielded to a selective recovery in the catholyte of 62% of lithium and 33% of cobalt, 80% of the latter electrodeposited at the cathode.
1. Introduction
Nowadays, lithium-ion batteries (LIBs) are the most consumed energy storage and supply devices for portable electronics. Their characteristics (high energy density, long cycle life, high roundtrip efficiency, wide range of operating temperature, high reliability, safety, fast recharge and low self-discharge rate) make them the best option for numerous applications. LIBs play a vital role in the development of electric vehicles and in the storage of energy from renewable sources [1,2], which will contribute to decreasing the global carbon emissions. The increasing demand for LIBs from the sectors of portable electronics and transportation comes together with an increasing interest for recycling of spent batteries [2,3,4].
The main limitations for the implementation of proper recycling procedures are the lack of standards for the physical formats and the frequent appearance of new batteries with different chemistries [1]. There are normally three different shapes: prismatic, pouch (or polymer) and cylindrical cells. With respect to the chemical composition, LIBs consist of a cathode, an anode, an organic electrolyte and a separator. In most of the LIBs currently commercialized, the anode is made of graphite cast over copper foil. Nowadays, alternative anodes are under study, such as Li4Ti5O12, graphite nanotubes or nanoparticles, or compounds of Si and Sn. The cathode, supported on an aluminum foil, is usually built from transition metal oxides, such as LiMn2O4 (LCO), LiFePO4 (LFP), LiMn2O4 (LMO), Li(NiMnCo)O2 (NMC) and (NiCoAl)O2 (NCA) [5]. Most of the toxic and valuable metals are found in the cathode, which represent approximately 25–30% of the total battery mass.
According to the European Union, some components of LIBs, such as cobalt, phosphorous and natural graphite, are classified as “Critical Raw Materials” (CRMs) due to their high supply risk and economic importance [6]. Hence, recycling technologies for any CRM-containing waste are in the spotlight of the EU development plan. Nowadays, most battery recycling technologies focus on the recovery of Co, as it is considered the bottleneck in the battery industry, leaving the recuperation of Li in the background [1]. However, the increase in the LIBs market may place Li on the list of critical materials by 2030 [1,7,8]. Indeed, the increasing demand of Li for batteries may soon produce a supply shortage, which promotes the investigation of Li-free alternatives such as sodium or aluminum ion batteries [9]. In addition to the supply chain risk, some of the materials used in LIBs represent a threat to the environment, for example, the emissions of fluoride gas from battery fires or the aqueous contamination from the metals in the cathode. This environmental threat also motivates the development of recycling initiatives [10,11].
Regarding the currently existing LIBs recycling technologies, they can be classified into pyrometallurgical, hydrometallurgical, biometallurgical and combined techniques. Pyrometallurgical processes use high temperature smelting to recover cobalt, copper and nickel alloys [2]. Hydrometallurgical processes use chemical acidic leaching to dissolve the metal containing components, followed by chemical separation and recovery [12]. In biometallurgical processes, microbial activity promotes the production of inorganic and organic acids to leach metals from spent LIBs [13]. The main limitation of these techniques is that lithium is lost in slag, and they have high energy and chemical reagents consumption [3,14]. Up to 50 companies around the world recycle lithium-ion battery at some scale, most of them located in China, South Korea, Europe and North America [4]. Some important companies, such as Umicore, Duesenfeld, Toxco and Recupyl have developed industrial-scale recycling processes applying the aforementioned techniques [15,16].
As an innovative recycling process, Villen-Guzman et al. [17] proposed the application of electrodialytic remediation (EDR) to LIB residues. This technique is based on the use of ion-exchange membranes for the selective separation of ions from liquid matrices by means of an applied electric potential. The EDR has been successfully used for the remediation of wet solid matrices and aqueous suspensions (e.g., polluted soil, treated timber waste, fly ash, wastewater sludge and harbor sediments [18,19,20,21,22]).
To the extent of our knowledge, there are no previous experimental studies evaluating the application of EDR to LIBs, apart from initial unpublished experiments carried out in our group. These previous experiments tested the recovery of Li and Co from LiCoO2 particles, placed in the central section of a standard three-compartment ED cell, using an anion-exchange membrane at the anode compartment and a cation-exchange membrane (CEM) at the cathode compartment. In those experiments, some limiting drawbacks were found, such as the difficulty to attain a proper stirring of the suspension within the cell, a significant deposition of particles at the surface of the ion-exchange membranes and the formation of chloride gas at the anode.
With the aim of tackling the aforementioned limitations, a novel experimental setup based on a hydrometallurgical–electrodialytic treatment is proposed in this paper. This experimental system basically consists of an electrodialytic cell using CEMs for the separation of the central compartment from both electrode compartments, while the dissolution of the LiCoO2 particles is carried out outside the electrodialytic cell, hindering the undissolved particles to reach the membranes and avoiding fouling. In order to achieve the proof of concept, the experiments presented here were carried out on new LiCoO2 particles.
2. Materials and Methods
2.1. Extraction Analysis
In order to study the kinetics of the dissolution of LiCoO2 particles, a number of extractions were carried out on LiCoO2 powder (Alfa Aesar, 97%) in HCl 0.1 M solution. Batch-extraction experiments were carried out in well-stirred 50 mL polypropylene vessels containing LiCoO2 powder suspended in the extracting solution using a liquid/solid ratio of L:S = 200, namely 125 mg of solid powder suspended in 25 mL of 0.1 M HCl solution. The vessels with the suspensions were stirred in a rotatory shaking table at room temperature, and they were withdrawn and analyzed at different times (up to 6 days) to obtain the transient concentration profile of and in the aqueous phase. With the aim of determining the initial amount of metals, microwave-assisted acid digestion was carried out. All the samples were filtered using 0.60 µm glass-fiber (Macherey-Nagel (MN) GF-3) and analyzed for Co and Li using atomic absorption spectrophotometry (Varian SpectrAA 1101).
In hydrometallurgical processes, different extracting agents (organic and inorganic acids) have been tested [3,17,23,24,25]. In this study, 0.1 M HCl was used to extract Co and Li from LiCoO2. The choice of the extracting agent was done to evaluate the capability of chloride ions as extraction agent, as it may act as a reducing agent of to .
2.2. Solid Surface Characterization
X-ray photoelectron spectroscopy (XPS) measurements were performed with a Physical Electronics PHI 5701 spectrometer with a multi-channel hemispherical electron analyzer. Samples of solid before and after extraction were mounted on a sample holder without adhesive tape and kept overnight at high vacuum in the preparation chamber before being transferred to the analysis chamber for testing. The photoelectron lines Cu 2p3/2, Ag 3d5/2 and Au 4f7/2 at 932.7, 368.3 and 84.0 eV were selected to calibrate the spectrometer energy scale. High-resolution spectra were collected in the constant pass energy mode at 29.35 eV operating at a given take-off angle of 45°. The adventitious carbon (C1 s at 284.8 eV) was used as charge reference. The PHI ACCESS ESCA-V8.0 software package was used for acquisition and data analysis.
2.3. Hydrometallurgical–Electrodialytic Experiments
The combined hydrometallurgical–electrodialytic experiments were carried out in duplicate, using cells electrically connected in series, i.e., the cathode of one of the ED cells was connected to the anode of the other one, assuring that both cells were submitted to the same electrical current. The experiments were performed at constant current of 50 mA by means of a DC power supply unit (Genesys RDK Lambda GEN 600-2.6), which corresponded to a current density of 1 mA cm−2 referred to the internal diameter of the ED cell. The experimental system is schematically presented in Figure 1.

Figure 1.
Experimental set-up scheme.
Each ED cylindrical cell was built using three methacrylate compartments (4 cm length and 8 cm internal diameter). The central compartment of the electrodialytic cell was connected with an external vessel and a separatory funnel of 100 mL capacity. A volume of 350 mL of the suspension with the LiCoO2 particles in 0.1 M HCl (L:S = 200) was initially placed in the magnetically and well stirred vessel. The suspension was continuously pumped from the vessel to the central compartment, passing through the separatory funnel using a four-channel Watson Marlow 302S peristaltic pump. The conical shape of the separatory funnel helps the solid particles to not follow the liquid phase containing the extracted metals and therefore prevents the particles from reaching the central compartment of the ED cell. A fiberglass filter was added at the upper end of the separatory funnel to assure that no solid particles were dragged by the aqueous flow. The liquid phase continuously flowed back to the vessel from the central compartment. The flow rate was 0.2 mL s−1, optimized to assure the retention of the undissolved particles in the separatory funnel.
The central compartment was separated from both electrode compartments by means of CEMs (Neosepta CMX-fg standard grade; electrical resistance 1.8 , thickness 0.16 mm, stability pH 0–14). The commercial anode consisted of the base titanium material covered with mixed oxides, mainly Ir, to prevent their oxidation (Metakem GmbH), and the cathode was stainless steel. The catholyte was 0.1 M HCl, similar to the extracting solution. In turn, the anolyte was 0.1 M solution, as it could not be further oxidized at the anode. The use of CEMs to separate the anode and the central compartments hindered the passing of towards the anode and the corresponding oxidation to chlorine gas. This configuration allows the regeneration and reuse of the extracting solution, which is an advantage of this technique due to the continual addition of extracting agent solution not being necessary.
Liquid samples of 10 mL from the catholyte and the central compartments were withdrawn twice a day during the experimental time (6 days). With the aim of keeping the liquid volume constant, 10 mL of 0.1 M HCl solution was added to the central compartment after sampling. The metal concentration of all liquid samples was determined by an AAS Varian SpectrAA-110.
At the end of the experiment, the cells were disassembled and the cathodes were soaked in 1:1 solution for 24 h to determine the metal electrodeposited on them. Similarly, the CEMs were soaked in 1 M solution for 24 h to determine the amount of metal accumulated. The membranes used in these experiments are stable in strong acids, which allows low pH regeneration. With the aim of complete global mass balance, the fiberglass was also soaked in concentrated acid to extract the metals accumulated. Particles attached to the fiberglass used in the separator funnel were considered as part of the central compartment solution.
3. Results and Discussion
3.1. Extractions Eexperiments and Chemical Equilibrium Calculaitons
The time-transient evolution of the dissolved metal ( and ) from the solid matrix and of the solution pH are presented in Figure 2. The solid dissolution trend was approximately linear in the first few hours of the experiment, slowing down asymptotically as the particles were dissolved. It could be concluded that the kinetics of particle dissolution was moderately slow, reaching a plateau after approximately 3 days. Regarding the pH evolution, a slight increase from pH = 1 to pH = 1.4 was observed, which could be associated with the consumption of protons during the extraction process. A non-equimolar proportion between Li+ and Co2+ was also observed; that is, the concentration of Li+ in the equilibrium was approximately twice as high as the concentration of Co2+.
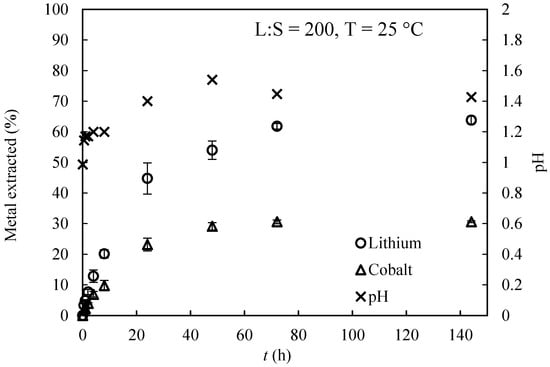
Figure 2.
Time-transient values for the percentage of extracted metal with respect to the amount in the initial solid.
With the aim of evaluating the equilibrium system, the software PHREEQC [26] was used to simulate the extraction experiments. The database “phreeqc.dat” adapted with the specific chemical system presented in Table 1 was used to simulate the chemical system. Equilibrium data for the solid phases were adapted from those reported in [25] while the /HCl/HClO equilibria was from [27]. According to Equation (1), Co3+, within the LiCoO2 particles, is reduced into soluble Co2+. Using HCl solution, and according to Equations (9) and (10), the reducing agent could be expected to be the chloride ions. However, the experimental results indicated that the reducing agent was water and not the chloride ions. The chemical equilibrium simulations carried out corroborated that the oxidation of water, Equation (11), was thermodynamically favored in acid media. This was also consistent with the fact that other acids, such as HNO3 and H2SO4, were also used in the literature achieving similar results, rather than HCl [25].

Table 1.
Chemical system.
Regarding the dissolution percentage of LiCoO2, equilibrium simulations predicted the total dissolution of the LiCoO2 particles and the complete re-precipitation of dissolved Co2+ as Co3O4 (s), together with the production of an aqueous phase rich in LiCl. However, in practice, the precipitation of Co3O4 (s) from dissolved Co2+ needs a precursor to take place [28]. This would explain the presence of Co2+ in the dissolution at the end of the experiments. Instead, the formation of Co3O4 is more likely to happen through the decomposition of Li0.5CoO2 as follows:
Furthermore, the plateau observed in the experimental data could be associated with kinetic mechanisms where (s) forms in the outer part of the (s) particles, acting as a passivating layer that stops the leaching process [29,30]. This behavior would explain why higher acid concentrations and the use of stronger reducing agents, such as H2O2, produce higher extraction yields through the elimination of that passive (s) outer layer.
For a better understanding of the chemical processes involved, the solid surface properties of the solid matrix before and after experiments were studied by XPS analysis. Figure 3 presents the results of the XPS analysis of the (s) particles, showing the binding energy of the particles (a) before the extraction and (b) 6 days after the extraction. The former presents a composition consistent with the (s) stoichiometry, while the latter presents no Li at the surface of the particles, indicating only the presence of cobalt oxides. These results are consistent with a dissolution mechanism that would take place with the existence of an external passive layer.
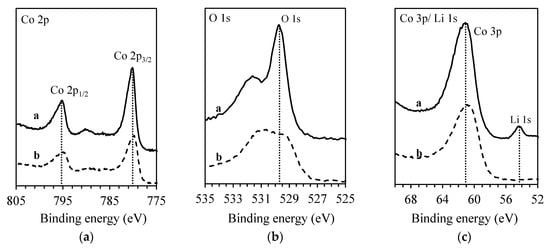
Figure 3.
XPS results for the solid sample before and after extraction. (a) Co 2p core level spectra; (b) Co 3p core level spectra; (c) Li 1s and O 1s core level spectra. The solid line: Initial LiCoO2 particles. The dashed line: Solid particles after extraction.
3.2. Extraction with ED
Figure 4 shows the results from the combined hydrometallurgical–electrodialytic experiment. The figure shows the time-transient percentage of dissolved metal ( and ) in the central and the catholyte compartments, indicating the initial amount in the solid particles. It can be observed that the total amount of and in the liquid phase increased during the first 2 days of treatment up to 25.53% 0.89% and 56.36% 0.96%, respectively. After that time, the concentration of aqueous remained constant, indicating that a point was reached from where it was not possible to extract more lithium nor dissolve more particles, as observed before in the extraction experiments. Despite there being no more extraction after 2 days, the dissolved was transported from the central compartment to the catholyte in the electrodialytic cell.
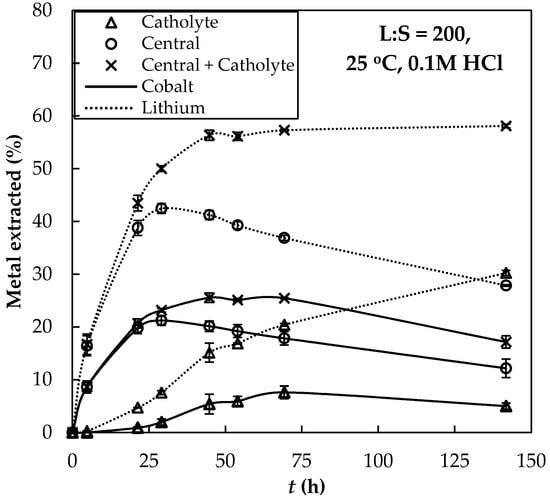
Figure 4.
Time-transient values for percentage of extracted metal in the different compartments with respect to the amount in the initial solid. —: Cobalt; ···: Lithium. Error bars represent two times the standard deviation of the duplicated texts.
In the case of cobalt, the total concentration in the liquid phase decreased with the treatment as it got electrodeposited at the surface of the cathode. Electrodeposited was dissolved in HNO3 after the experiment, and it corresponded to 11% of the total Co in the system (initially contained in the particles), and to 80% of the extracted cobalt. Therefore, when a certain concentration of is reached at the catholyte, there may be competition between the electrolytic reduction of water to produce hydrogen gas and the reduction of Co2+ into metallic shown in Equation (8). Although the hydrogen gas evolution seems thermodynamically more favorable at the working pH values, overpotentials may promote the electrodeposition of into . The pH values at the central and the cathode compartments were both approximately constant at pH ≈ 1.2 during the experiments. The average value of the electric potential drop measured at the electrodes of each cell increased from 2.53 to 2.94 V.
The results suggest a switch between the hydrometallurgical and the electrodialytic mechanisms in controlling the recuperation rate of the target metals. In order to make the technique energetically efficient, it would be important to optimize the applied electric current to produce the ED separation at the same rate as the extraction of metals from the solid particles. In the experiments presented here, the applied electric current was set from the dissolution rate observed in the linear part at the beginning of the extraction experiments shown in Section 3.1.
Figure 5 shows the percentage of Co and Li (referred to the total amount of these chemical species in the initial solid samples) in the different components of the experimental set-up at the end of the experiment. It can be observed that after 6 days of treatment, 33% of Co and 62% of Li was extracted. This indicates again that in the absence of reducing agents, the dissolution of the LiCoO2 (s) particles is limited by certain competitive mechanisms, such as the precipitation of Co3O4 (s). Figure 5 also shows that 16% of Co and 30% of Li was transported to the catholyte compartment. Furthermore, approximately 68% of the cobalt in the catholyte compartment was found electrodeposited on the cathode surface. Figure 5 shows that small fractions of Co (0.5%) and Li (3.9%) were found in the anolyte compartment, as CEMs do not hinder diffusion of the cations. It can also be observed certain amount of Co (4.4%) and Li (0.9%) accumulated within the structure of the CEMs. Cobalt had a bigger tendency to accumulate within the membranes, as expected from divalent cations. No metal was found on the anode electrode.

Figure 5.
Percentage of cobalt and lithium, referred to the total amount of these chemical species in the initial solid samples, in the different parts of the electrodialytic cell at the end of the experiment.
The results indicate that the combined hydrometallurgical-electrodialytic cell allows the extraction of Li and Co from the particles and the separation of the metals in a catholyte solution. Furthermore, it would be possible to selectively separate the metals, as cobalt metal can be obtained via electrodeposition at the cathode surface. The experiments indicated that the dissolution of the solid particles limits the time efficiency of the technique. Future work is planned to evaluate the performance of this kind of set-up in combination with reducing agents and optimizing the applied electrical current to the dissolution of the solid particles.
4. Conclusions
This paper presents an innovative experimental technique that combines hydrometallurgical extraction with electrodialytic cells for the selective recovery of Li and Co from lithium-ion battery waste. The experiments carried out in this paper were focused on LiCoO2, but the technique has the potential to be applied to different kinds of cathode scraps from spent batteries.
The results indicate that it is possible to produce a selective separation of the target metals without the addition of a continuous inlet of extracting agents. In the hydrometallurgical–electrodialytic procedure, the electrolysis reactions would provide the necessary acid media for the dissolution of the metals, while the suggested combination of cation-exchange membranes keep the extracting agents within a closed well-stirred fluid.
Author Contributions
Conceptualization, all authors; methodology, all authors; software, J.M.P.-G. and C.V.-A.; validation, J.M.P.-G., M.V.-G. and C.V.-A.; formal analysis, J.M.R.-M.; investigation, M.M.C.-G. and M.V.-G.; resources, all authors; data curation, J.M.P.-G. and M.V.-G.; writing—original draft preparation, M.M.C.-G., J.M.P.-G. and M.V.-G.; writing—review and editing, all authors; visualization, C.G.-L. and C.V.-A.; supervision, J.M.P.-G. and M.V.-G.; project administration, J.M.P.-G.; funding acquisition, C.G.-L. and J.M.P.-G. All authors have read and agreed to the published version of the manuscript.
Funding
This work has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 778045. Financial support from E3TECH Excellence Network under project CTQ2017-90659-REDT (MCIUN, Spain) is acknowledged. Paz-Garcia acknowledges the financial support from the program “Proyectos I+D+i en el marco del Programa Operativo FEDER Andalucía 2014–2020”, No. UMA18-FEDERJA-279.
Acknowledgments
Villen-Guzman acknowledges the postdoctoral fellowship obtained from the University of Malaga. Cerrillo-Gonzalez acknowledges the FPU grant obtained from the Spanish Ministry of Education.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zubi, G.; Dufo-López, R.; Carvalho, M.; Pasaoglu, G. The lithium-ion battery: State of the art and future perspectives. Renew. Sustain. Energy Rev. 2018, 89, 292–308. [Google Scholar] [CrossRef]
- Heelan, J.; Gratz, E.; Zheng, Z.; Wang, Y.; Chen, M.; Apelian, D. Current and prospective Li-Ion battery recycling and recovery processes. JOM 2016, 68, 2632–2638. [Google Scholar] [CrossRef]
- Zheng, X.; Zhu, Z.; Lin, X.; Zhang, Y.; He, Y.; Cao, H.; Sun, Z. A mini-review on metal recycling from spent lithium ion batteries. Engineering 2018, 4, 361–370. [Google Scholar] [CrossRef]
- Melin, H.E. State of the Art in Reuse and Recycling of Lithium-Ion Batteries—A Research Review; The Swedish Energy Agency: London, UK, 2019. [Google Scholar]
- Nitta, N.; Wu, F.; Lee, J.T.; Yushin, G. Li-ion battery materials: Present and future. Mater. Today 2015, 18, 252–264. [Google Scholar] [CrossRef]
- Mathieux, F.; Ardente, F.; Bobba, S.; Nuss, P.; Blengini, G.A.; Dias, P.A.; Blagoeva, D.; De Matos, C.T.; Wittmer, D.; Pavel, C.; et al. Critical Raw Materials and the Circular Economy; Background report; Publications Office of the European Union: Luxembourg, 2017. [Google Scholar] [CrossRef]
- Swain, B. Recovery and recycling of lithium: A review. Sep. Purif. Technol. 2017, 172, 388–403. [Google Scholar] [CrossRef]
- Graedel, T.E.; Allwood, J.; Birat, J.P.; Buchert, M.; Hagelüken, C.; Reck, B.K.; Sibley, S.F.; Sonnemann, G. Recycling Rates of Metals: A Status Report; United Nations Environment Programme: Paris, France, 2011; ISBN 978-92-807-3161-3. [Google Scholar]
- Navarro-Suárez, A.; Casado, N.; Mecerreyes, D.; Rojo, T.; Castillo-Martínez, E.; Carretero-González, J. Hybrid biopolymer electrodes for lithium- and sodium-ion batteries in organic electrolytes. Sustain. Energy Fuels 2018, 2, 836–842. [Google Scholar] [CrossRef]
- Larcher, D.; Tarascon, J.-M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2014, 7, 19–29. [Google Scholar] [CrossRef]
- Larsson, F.; Andersson, P.; Blomqvist, P.; Mellander, B.-E. Toxic fluoride gas emissions from lithium-ion battery fires. Sci. Rep. 2017, 7, 10018. [Google Scholar] [CrossRef]
- Chen, L.; Tang, X.; Zhang, Y.; Li, L.; Zeng, Z.; Zhang, Y. Process for the recovery of cobalt oxalate from spent lithium-ion batteries. Hydrometallurgy 2011, 108, 80–86. [Google Scholar] [CrossRef]
- Zeng, X.; Li, J.; Singh, N. Recycling of Spent Lithium-Ion Battery: A Critical Review. Crit. Rev. Environ. Sci. Technol. 2014, 44, 1129–1165. [Google Scholar] [CrossRef]
- Meshram, P.; Pandey, B.; Mankhand, T. Extraction of lithium from primary and secondary sources by pre-treatment, leaching and separation: A comprehensive review. Hydrometallurgy 2014, 150, 192–208. [Google Scholar] [CrossRef]
- Foster, M.; Isely, P.; Standridge, C.R.; Hasan, M. Feasibility assessment of remanufacturing, repurposing, and recycling of end of vehicle application lithium-ion batteries. J. Ind. Eng. Manag. 2014, 7, 698–715. [Google Scholar] [CrossRef]
- Choubey, P.K.; Chung, K.-S.; Kim, M.-S.; Lee, J.-C.L.; Srivastava, R.R. Advance review on the exploitation of the prominent energy-storage element Lithium. Part II: From sea water and spent lithium ion batteries (LIBs). Miner. Eng. 2017, 110, 104–121. [Google Scholar] [CrossRef]
- Villen-Guzman, M.; Arhoun, B.; Vereda-Alonso, C.; Gomez-Lahoz, C.; Rodriguez-Maroto, J.M.; Paz-Garcia, J.M. Electrodialytic processes in solid matrices. New insights into battery recycling. A review. J. Chem. Technol. Biotechnol. 2019, 94, 1727–1738. [Google Scholar] [CrossRef]
- Velizarova, E.; Ribeiro, A.B.; Ottosen, L.M. A comparative study on Cu, Cr and as removal from CCA-treated wood waste by dialytic and electrodialytic processes. J. Hazard. Mater. 2002, 94, 147–160. [Google Scholar] [CrossRef]
- Pedersen, A.J.; Ottosen, L.M.; Villumsen, A.; Damø, A.J. Electrodialytic removal of heavy metals from different fly ashes. J. Hazard. Mater. 2003, 100, 65–78. [Google Scholar] [CrossRef]
- Nystroem, G.M.; Ottosen, L.M.; Villumsen, A. Acidification of harbor sediment and removal of heavy metals induced by water splitting in electrodialytic remediation. Sep. Sci. Technol. 2005, 40, 2245–2264. [Google Scholar] [CrossRef]
- Ottosen, L.M.; Lepkova, K.; Kubal, M. Comparison of electrodialytic removal of Cu from spiked kaolinite, spiked soil and industrially polluted soil. J. Hazard. Mater. 2006, 137, 113–120. [Google Scholar] [CrossRef]
- Guedes, P.; Couto, N.; Ottosen, L.M.; Ribeiro, A.B. Phosphorus recovery from sewage sludge ash through an electrodialytic process. Waste Manag. 2014, 34, 886–892. [Google Scholar] [CrossRef]
- Huang, B.; Pan, Z.; Su, X.; An, L. Recycling of lithium-ion batteries: Recent advances and perspectives. J. Power Sources 2018, 399, 274–286. [Google Scholar] [CrossRef]
- Lee, C.K.; Rhee, K.-I. Preparation of LiCoO2 from spent lithium-ion batteries. J. Power Sources 2002, 109, 17–21. [Google Scholar] [CrossRef]
- Takacova, Z.; Havlik, T.; Kukurugya, F.; Orac, D. Cobalt and lithium recovery from active mass of spent Li-ion batteries: Theoretical and experimental approach. Hydrometallurgy 2016, 163, 9–17. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input and Examples for PHREEQC Version 3: A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations. In U.S. Geological Survey Techniques and Methods, Book 6, Chapter A43; U.S. Geological Survey: Reston, VA, USA, 2014. [Google Scholar] [CrossRef]
- Stumm, W.; Morgan, J.J. Oxidation and Reduction; Equilibria and Microbial Mediation. In Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters, 3rd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2012; pp. 425–515. [Google Scholar]
- Nakamura, T.; Kajiyama, A. Synthesis of LiCoO2 particles with uniform size distribution using hydrothermally precipitated Co3O4 fine particles. Solid State Ionics 1999, 123, 95–101. [Google Scholar] [CrossRef]
- Shuva, M.A.H.; Kurny, A.S.W. Dissolution Kinetics of Cathode of Spent Lithium Ion Battery in Hydrochloric Acid Solutions. J. Inst. Eng. (India) Ser. D 2013, 94, 13–16. [Google Scholar] [CrossRef]
- Li, L.; Bian, Y.; Zhang, X.; Guan, Y.; Fan, E.; Wu, F.; Chen, R. Process for recycling mixed-cathode materials from spent lithium-ion batteries and kinetics of leaching. Waste Manag. 2018, 71, 362–371. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).