Beneficial Effects of Melatonin in the Ovarian Transport Medium on In Vitro Embryo Production of Iberian Red Deer (Cervus elaphus hispanicus)

 , , , , and
, , , , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Recovery of Oocytes and In Vitro Maturation
2.2. Collection of Follicular Fluid and Melatonin ELISA Assay
2.3. In Vitro Fertilization
2.4. In Vitro Embryo Culture
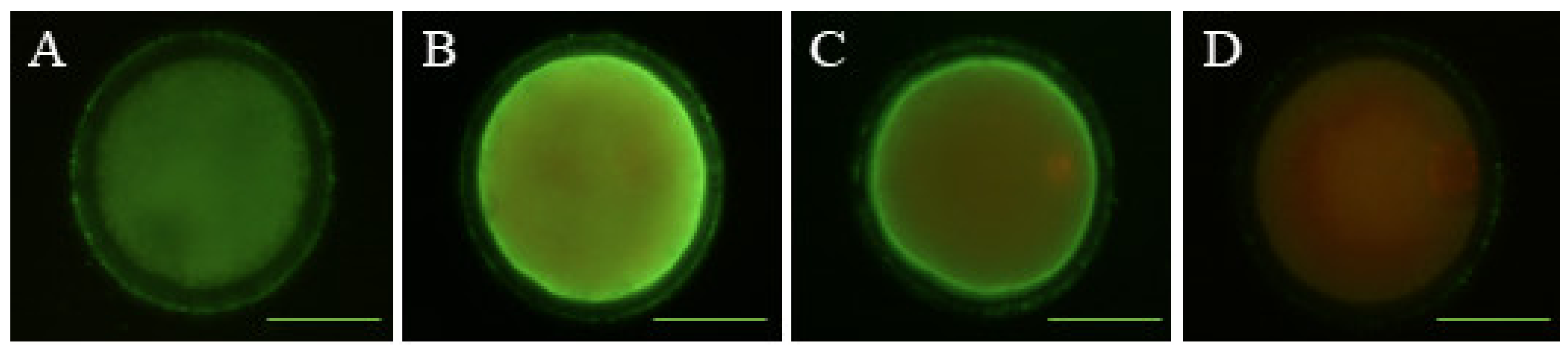
2.5. Determination of Early Apoptosis
2.6. Detection of DNA Fragmentation
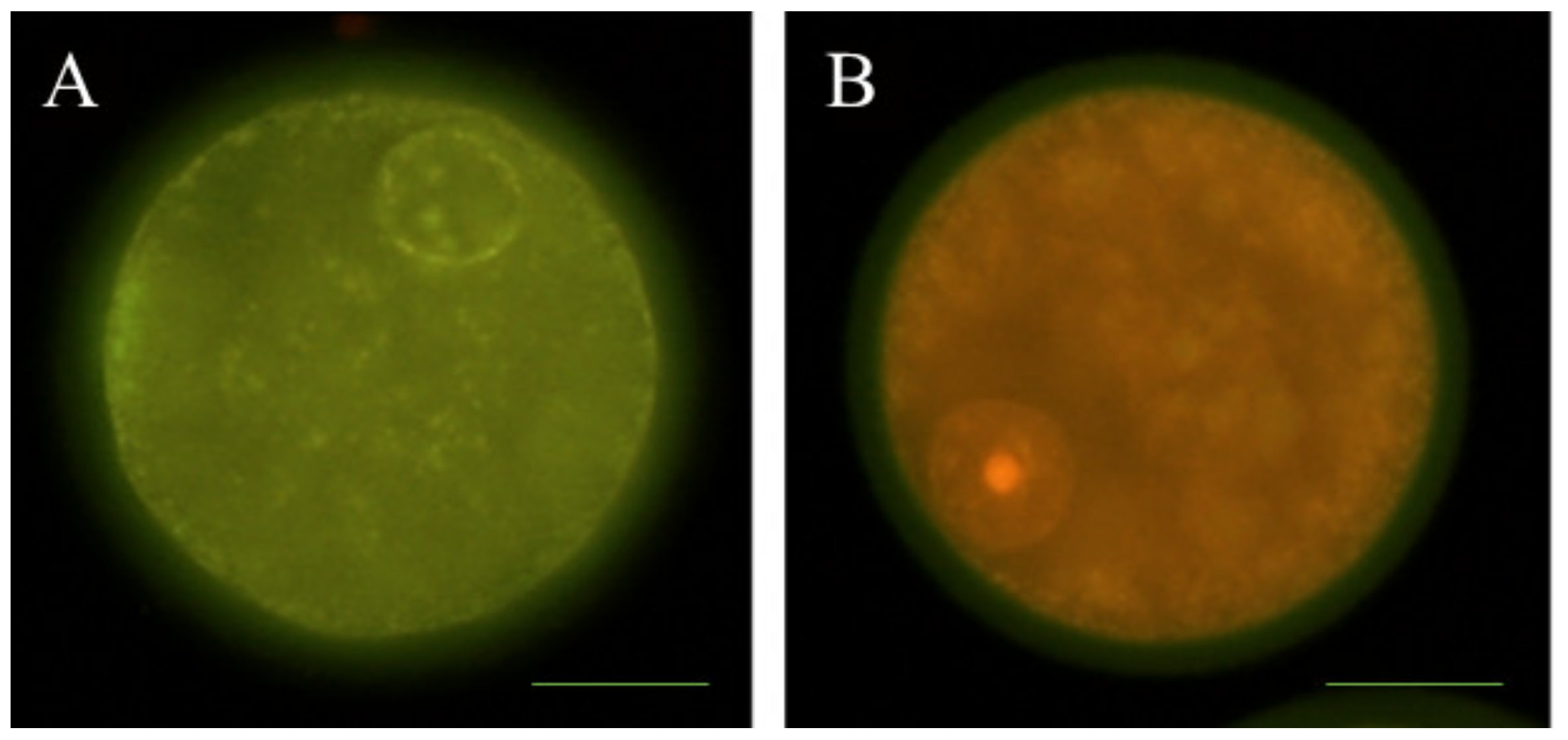
2.7. Determination of Intracellular ROS and GSH Levels
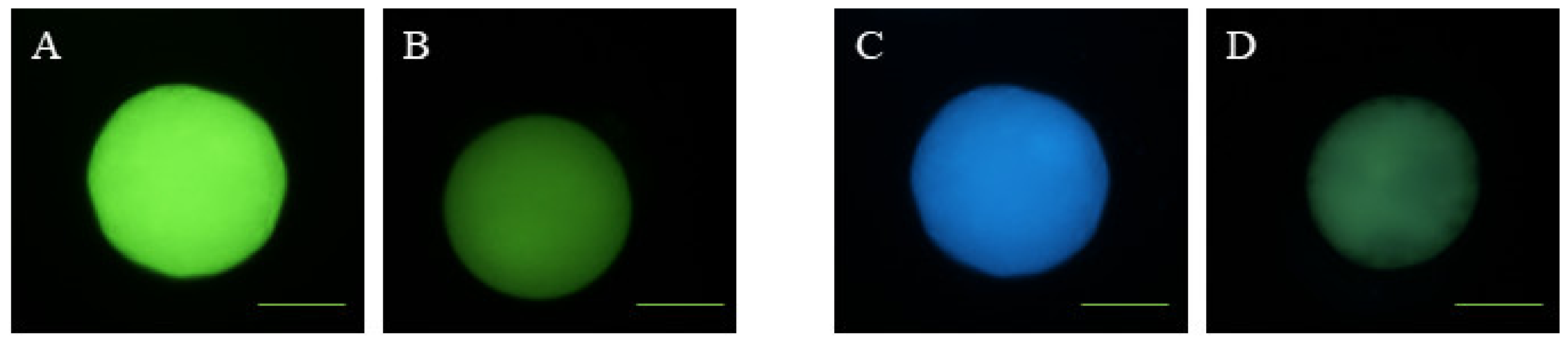
2.8. Mitochondrial Membrane Potential Analysis
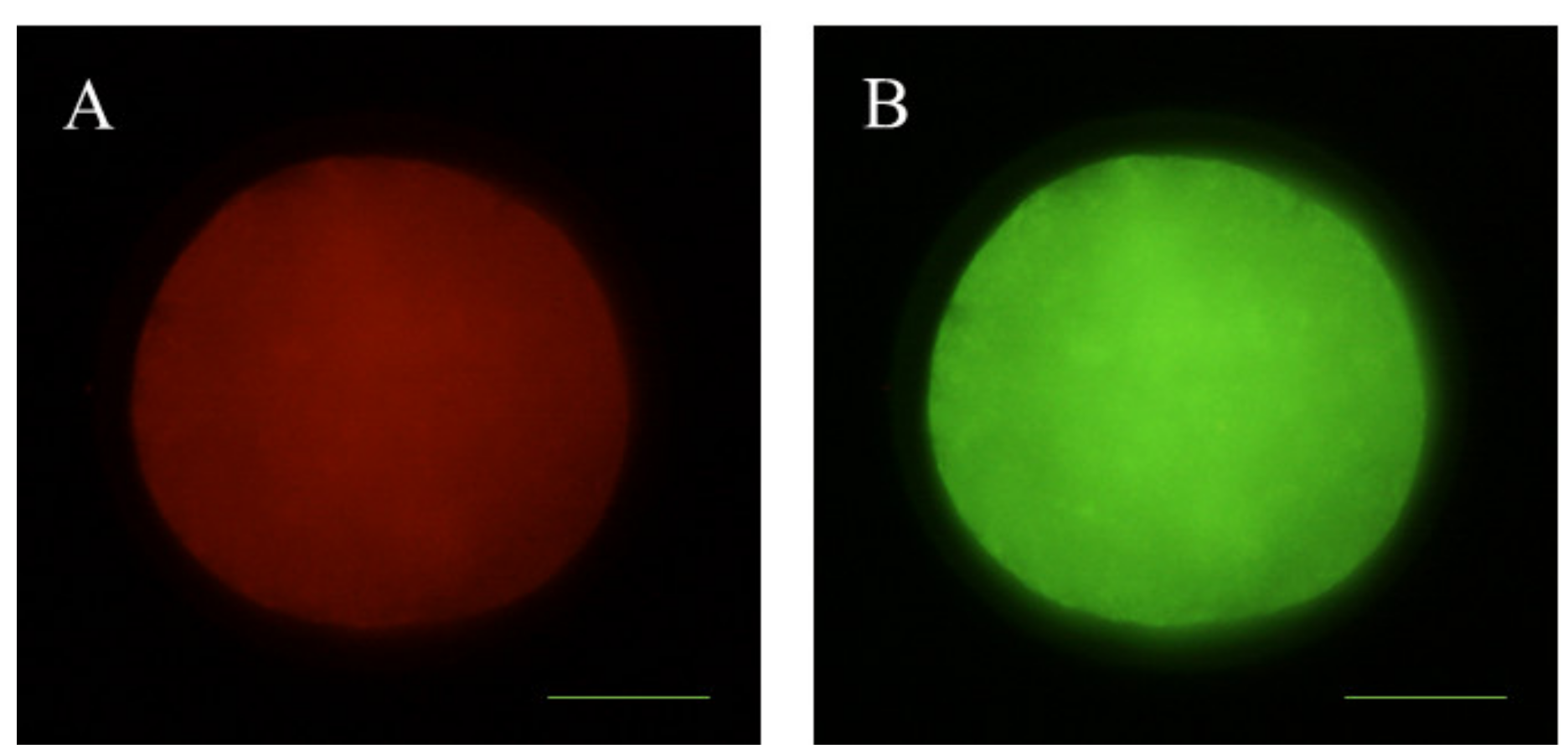
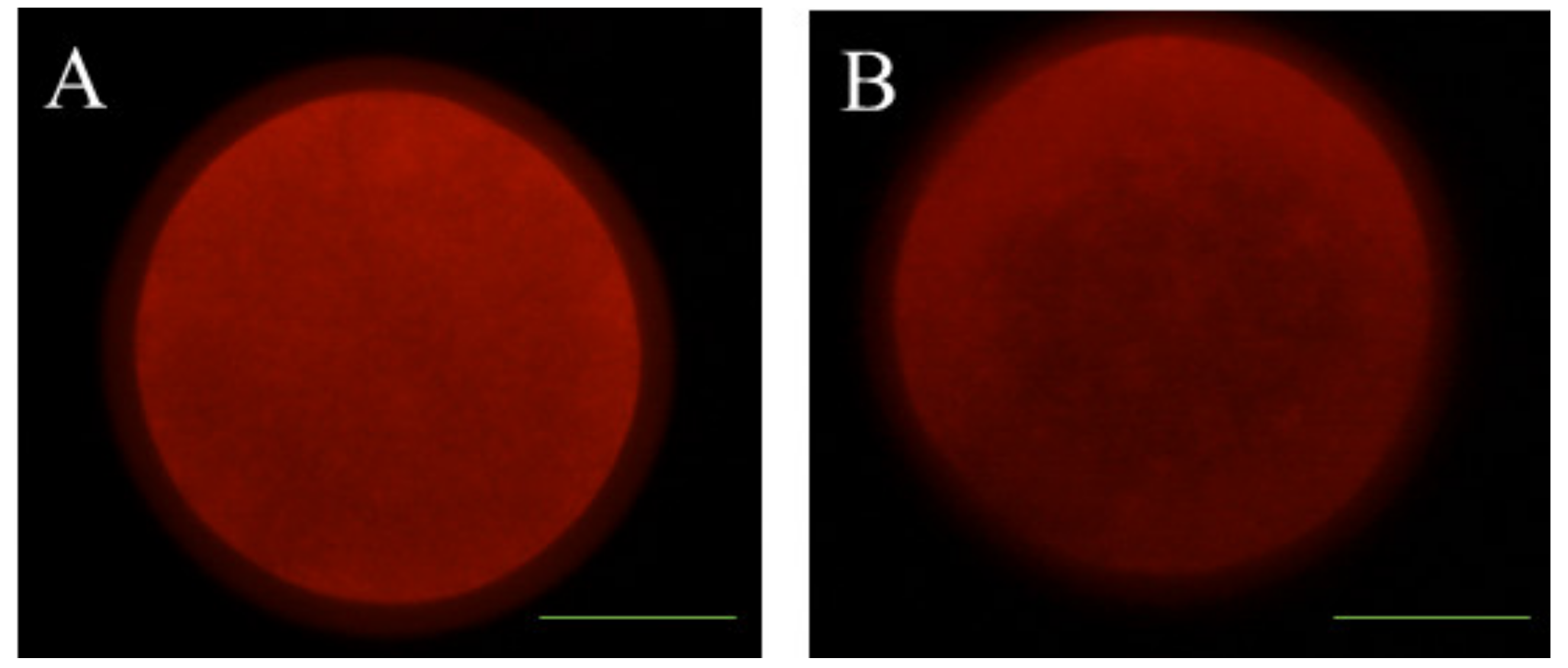
2.9. Examination of Mitochondrial Distribution
2.10. In Vitro Maturation Assessment
2.11. Transcript Quantification by qPCR
2.12. Statistical Analysis
3. Results
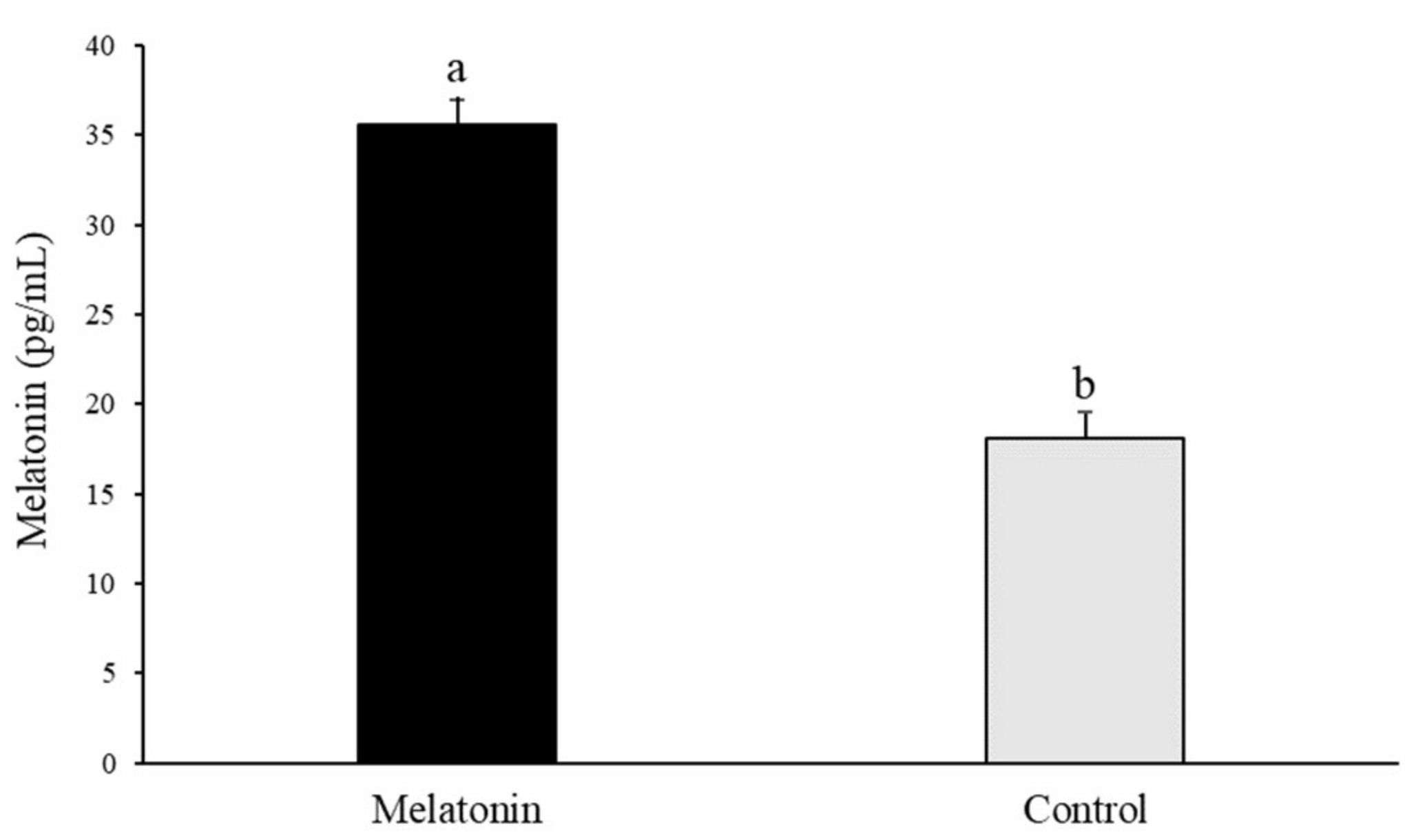
3.1. Melatonin Addition to the Ovary Storage Medium Increases Melatonin Levels in Follicular Fluid
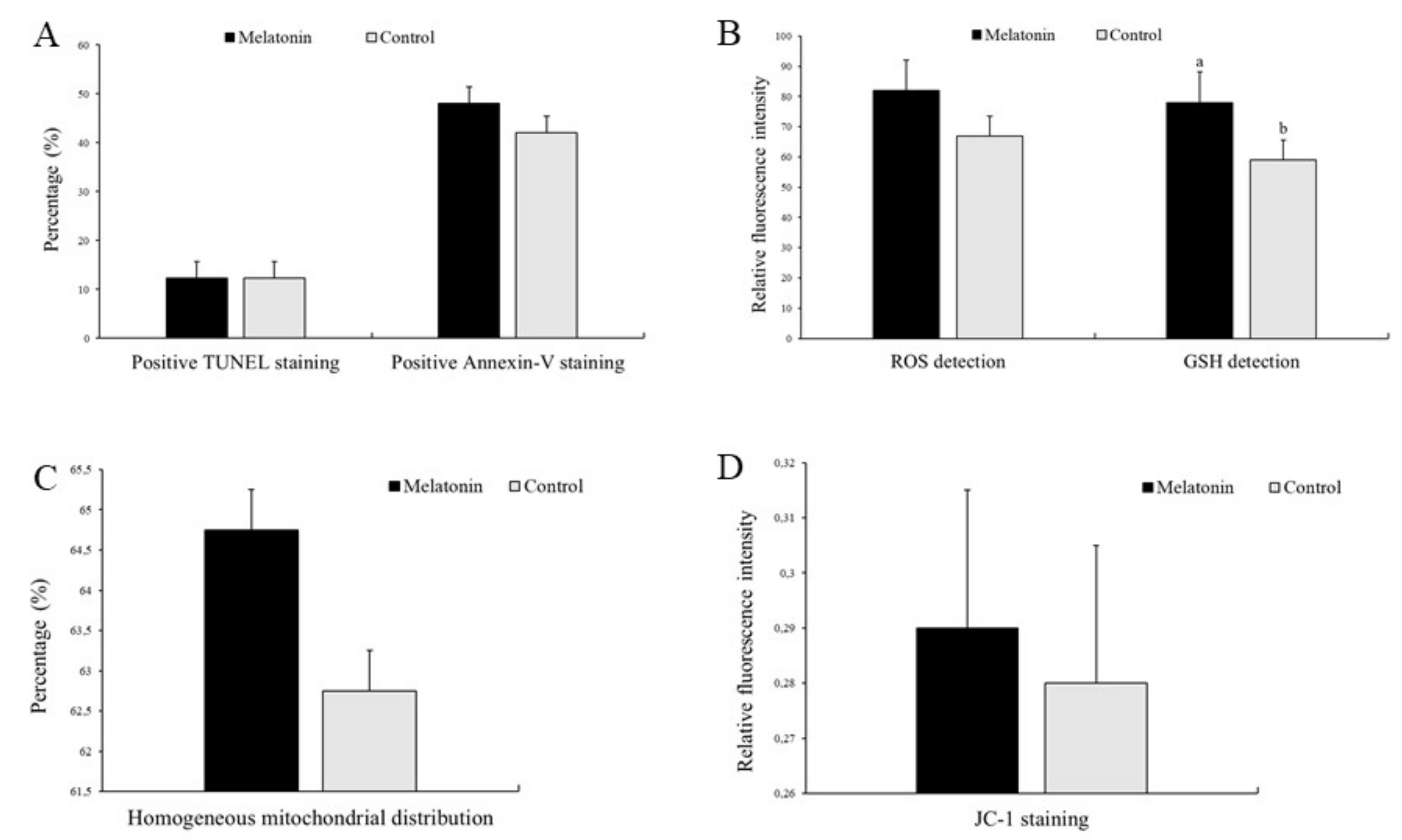
3.2. Effects of Melatonin on Oocyte Quality
3.3. Effects of Melatonin on the Meiotic Competence and Embryo Production
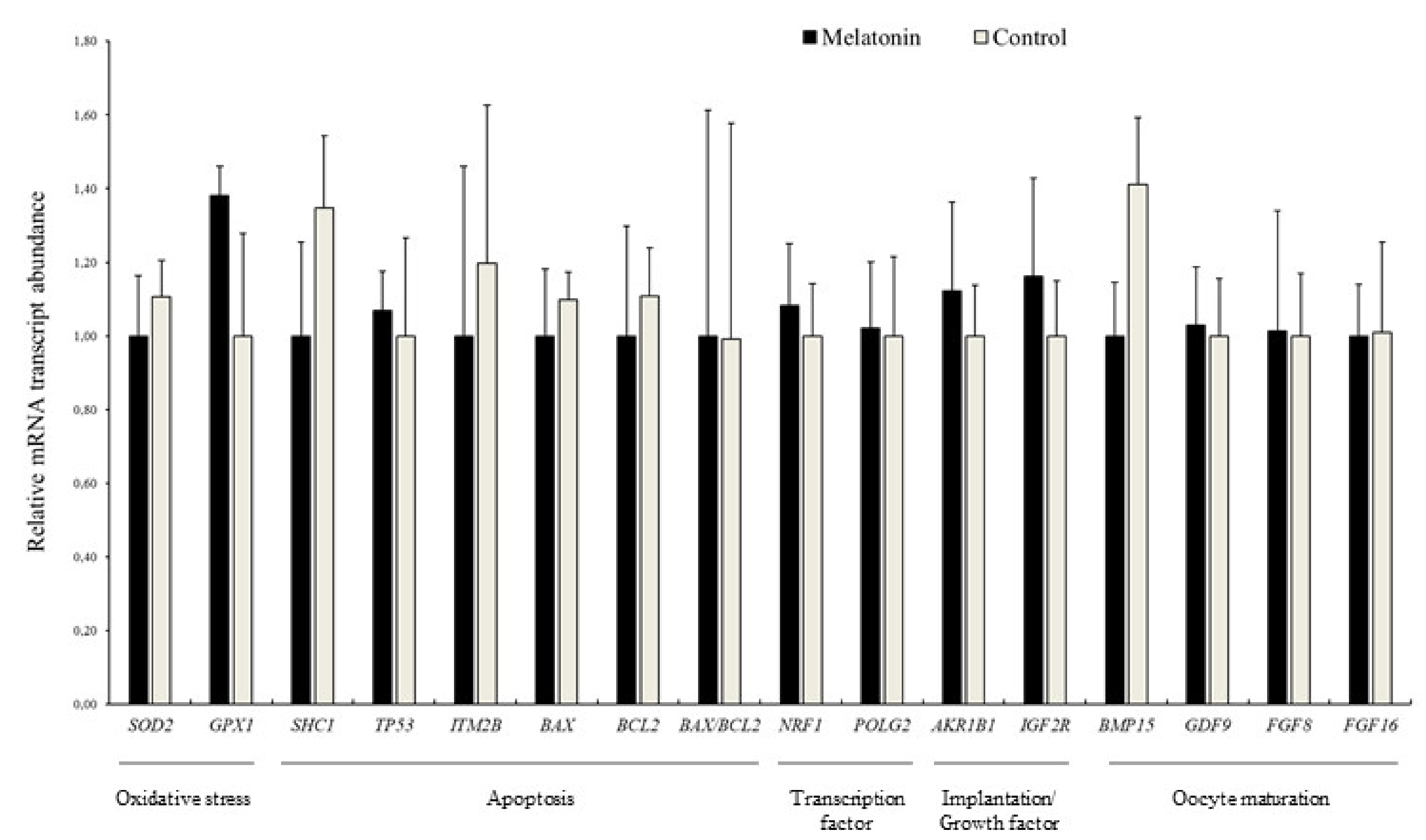
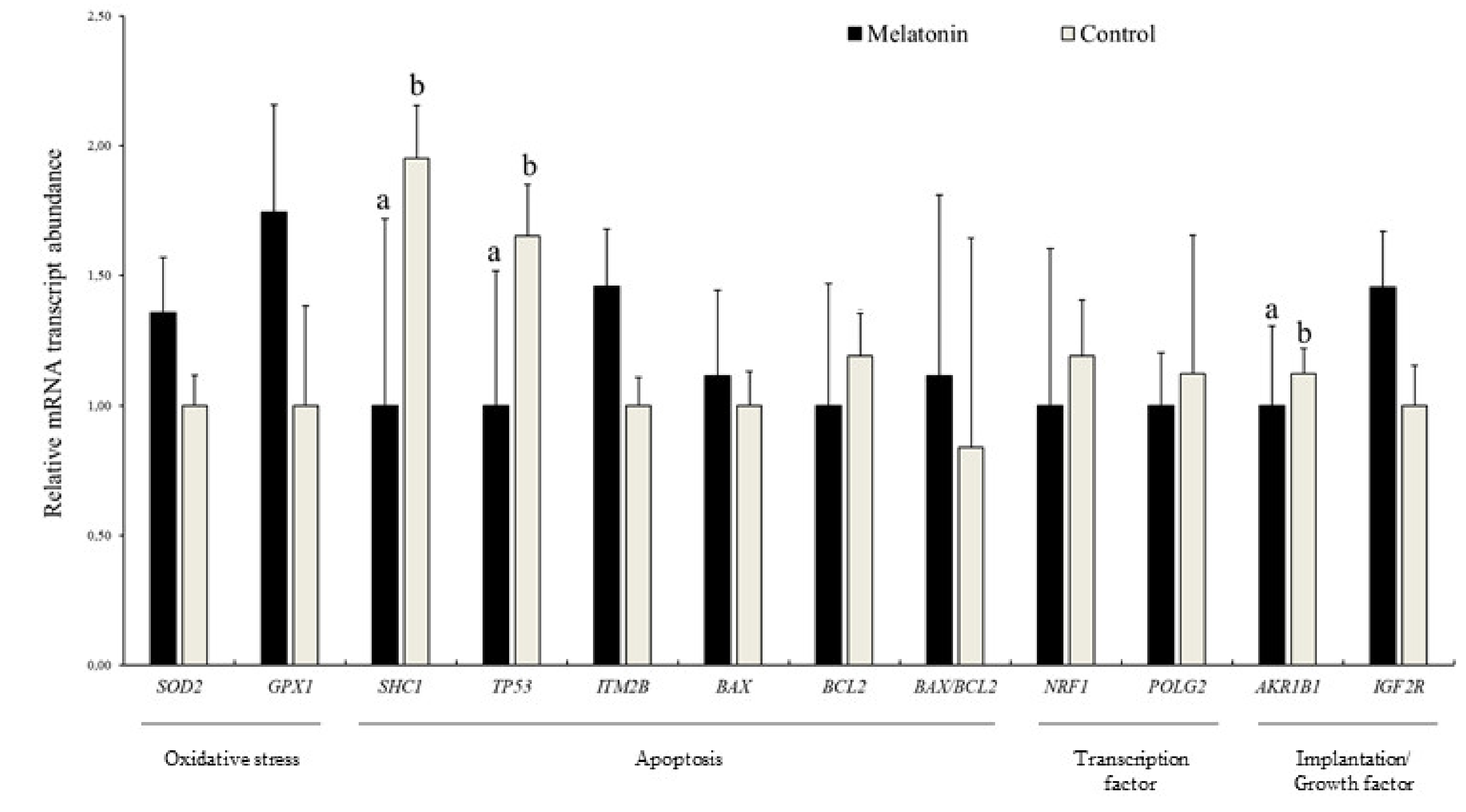
3.4. Effects of Melatonin on the Relative Abundance of mRNA Transcripts in Blastocysts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Berg, D.; Thongphakdee, A. In vitro Culture of Deer Embryos. In Comparative Embryo Culture; Springer: Berlin, Germany, 2006; pp. 191–208. ISBN 9781493995653. [Google Scholar]
- García-Álvarez, O.; Maroto-Morales, A.; Berlinguer, F.; Fernández-Santos, M.R.; Esteso, M.C.; Mermillod, P.; Ortiz, J.A.; Ramon, M.; Pérez-Guzmán, M.D.; Garde, J.J.; et al. Effect of storage temperature during transport of ovaries on in vitro embryo production in Iberian red deer (Cervus elaphus hispanicus). Theriogenology 2011, 75, 65–72. [Google Scholar]
- Martinez, J.G.; Carranza, J.; Fernandez-Garcia, J.L.; Sanchez-Prieto, C.B. Genetic Variation of Red Deer Populations under Hunting Exploitation in Southwestern Spain. J. Wildl. Manage. 2002, 66, 1273. [Google Scholar]
- Garde, J.J.; Martínez-Pastor, F.; Gomendio, M.; Malo, A.F.; Soler, A.J.; Fernández-Santos, M.R.; Esteso, M.C.; García, A.J.; Anel, L.; Roldán, E.R.S. The application of reproductive technologies to natural populations of red deer. Reprod. Domest. Anim. 2006, 41, 93–102. [Google Scholar] [PubMed]
- Herrick, J. Assisted reproductive technologies for endangered species conservation: developing sophisticated protocols with limited access to animals with unique reproductive mechanisms. Biol. Reprod. 2019, 100, 1158–1170. [Google Scholar] [PubMed]
- Paramio, M.T.; Izquierdo, D. Current status of in vitro embryo production in sheep and goats. Reprod. Domest. Anim. 2014, 49, 37–48. [Google Scholar]
- Wani, N.A.; Wani, G.M.; Khan, M.Z.; Salahudin, S. Effect of oocyte harvesting techniques on in vitro maturation and in vitro fertilization in sheep. Small Rumin. Res. 2000, 36, 63–67. [Google Scholar]
- Sirard, M.A.; Desrosier, S.; Assidi, M. In vivo and in vitro effects of FSH on oocyte maturation and developmental competence. Theriogenology 2007, 68. [Google Scholar]
- Sánchez-Ajofrín, I.; Iniesta-Cuerda, M.; Sánchez-Calabuig, M.J.; Peris-Frau, P.; Martín-Maestro, A.; Ortiz, J.A.; del Rocío Fernández-Santos, M.; Garde, J.J.; Gutiérrez-Adán, A.; Soler, A.J. Oxygen tension during in vitro oocyte maturation and fertilization affects embryo quality in sheep and deer. Anim. Reprod. Sci. 2020, 213, 106279. [Google Scholar]
- Wongsrikeao, P.; Otoi, T.; Karja, N.W.K.; Agung, B.; Nii, M.; Nagai, T. Effects of ovary storage time and temperature on DNA fragmentation and development of porcine oocytes. J. Reprod. Dev. 2005, 51, 87–97. [Google Scholar]
- Guibert, E.E.; Petrenko, A.Y.; Balaban, C.L.; Somov, A.Y.; Rodriguez, J.V.; Fuller, B.J. Organ preservation: Current concepts and new strategies for the next decade. Transfus. Med. Hemotherapy 2011, 38, 125–142. [Google Scholar]
- Combelles, C.M.H.; Gupta, S.; Agarwal, A. Could oxidative stress influence the in-vitro maturation of oocytes? Reprod. Biomed. Online 2009, 18, 864–880. [Google Scholar] [PubMed]
- Liang, L.F.; Qi, S.T.; Xian, Y.X.; Huang, L.; Sun, X.F.; Wang, W.H. Protective effect of antioxidants on the pre-maturation aging of mouse oocytes. Sci. Rep. 2017, 7, 1–10. [Google Scholar]
- Torres-Osorio, V.; Urrego, R.; Echeverri-Zuluaga, J.J.; López-Herrera, A. Oxidative stress and antioxidant use during in vitro mammal embryo production: Review. Rev. Mex. Ciencias Pecu. 2019, 10, 433–459. [Google Scholar]
- Zhau, X.; Zuo, L. Characterization of oxygen radical formation mechanism at early cardiac ischemia. Cell Death Dis. 2013, 4, e787-7. [Google Scholar]
- Reynolds, L.P.; Grazul-Bilska, A.T.; Redmer, D.A. Angiogenesis in the female reproductive organs: Pathological implications. Int. J. Exp. Pathol. 2002, 83, 151–164. [Google Scholar]
- Shirley, M.K.; Arthurs, O.J.; Seunarine, K.K.; Cole, T.J.; Eaton, S.; Williams, J.E.; Clark, C.A.; Wells, J.C.K. Metabolic rate of major organs and tissues in young adult South Asian women. Eur. J. Clin. Nutr. 2019, 73, 1164–1171. [Google Scholar]
- Wang, S.; He, G.; Chen, M.; Zuo, T.; Xu, W.; Liu, X. The Role of Antioxidant Enzymes in the Ovaries. Oxid. Med. Cell. Longev. 2017. [Google Scholar] [CrossRef]
- Tamura, H.; Nakamura, Y.; Korkmaz, A.; Manchester, L.C.; Tan, D.X.; Sugino, N.; Reiter, R.J. Melatonin and the ovary: physiological and pathophysiological implications. Fertil. Steril. 2009, 92, 328–343. [Google Scholar]
- Wang, F.; Tian, X.Z.; Zhou, Y.H.; Tan, D.X.; Zhu, S.E.; Dai, Y.P.; Liu, G.S. Melatonin improves the quality of in vitro produced (IVP) bovine embryos: Implications for blastocyst development, cryotolerance, and modifications of relevant gene. expression. PLoS ONE 2014, 9, 1–7. [Google Scholar]
- El-Raey, M.; Geshi, M.; Somfai, T.; Kaneda, M.; Hirako, M.; Abdel-Ghaffar, A.E.; Sosa, G.A.; El-Roos, M.E.A.A.; Nagai, T. Evidence of melatonin synthesis in the cumulus oocyte complexes and its role in enhancing oocyte maturation in vitro in cattle. Mol. Reprod. Dev. 2011, 78, 250–262. [Google Scholar]
- Tian, X.; Wang, F.; Zhang, L.; He, C.; Ji, P.; Wang, J.; Zhang, Z.; Lv, D.; Abulizi, W.; Wang, X.; et al. Beneficial effects of melatonin on the in vitro maturation of sheep oocytes and its relation to melatonin receptors. Int. J. Mol. Sci. 2017, 18, 834. [Google Scholar]
- Ishizuka, B.; Kuribayashi, Y.; Murai, K.; Amemiya, A.; Itoh, M.T. The effect of melatonin on in vitro fertilization and embryo development in mice. J. Pineal Res. 2000, 28, 48–51. [Google Scholar] [PubMed]
- Rodriguez-Osorio, N.; Kim, I.J.; Wang, H.; Kaya, A.; Memili, E. Melatonin increases cleavage rate of porcine preimplantation embryos in vitro. J. Pineal Res. 2007, 43, 283–288. [Google Scholar] [PubMed]
- Choi, J.; Park, S.M.; Lee, E.; Kim, J.H.; Jeong, Y.I.; Lee, J.Y.; Park, S.W.; Kim, H.S.; Hossein, M.S.; Jeong, Y.W.; et al. Anti-apoptotic effect of melatonin on preimplantation development of porcine parthenogenetic embryos. Mol. Reprod. Dev. 2008, 75, 1127–1135. [Google Scholar] [PubMed]
- Kang, J.T.; Koo, O.J.; Kwon, D.K.; Park, H.J.; Jang, G.; Kang, S.K.; Lee, B.C. Effects of melatonin on in vitro maturation of porcine oocyte and expression of melatonin receptor RNA in cumulus and granulosa cells. J. Pineal Res. 2009, 46, 22–28. [Google Scholar] [PubMed]
- Shi, J.M.; Tian, X.Z.; Zhou, G.; Bin Wang, L.; Gao, C.; Zhu, S.E.; Zeng, S.M.; Tian, J.H.; Liu, G.S. Melatonin exists in porcine follicular fluid and improves in vitro maturation and parthenogenetic development of porcine oocytes. J. Pineal Res. 2009, 47, 318–323. [Google Scholar]
- Mehaisen, G.M.K.; Saeed, A.M.; Gad, A.; Abass, A.O.; Arafa, M.; El-Sayed, A.; Fraidenraich, D. Antioxidant capacity of melatonin on preimplantation development of fresh and vitrified rabbit embryos: Morphological and molecular aspects. PLoS ONE 2015, 10, 1–16. [Google Scholar]
- Mohseni, M.; Mihandoost, E.; Shirazi, A.; Sepehrizadeh, Z.; Bazzaz, J.T.; Ghazi-Khansari, M. Melatonin may play a role in modulation of bax and bcl-2 expression levels to protect rat peripheral blood lymphocytes from gamma irradiation-induced apoptosis. Mutat Res. 2012, 738–739, 19–27. [Google Scholar]
- Goodarzi, A.; Zare Shahneh, A.; Kohram, H.; Sadeghi, M.; Moazenizadeh, M.H.; Fouladi-Nashta, A.; Dadashpour Davachi, N. Effect of melatonin supplementation in the long-term preservation of the sheep ovaries at different temperatures and subsequent in vitro embryo production. Theriogenology 2018, 106, 265–270. [Google Scholar]
- Levraut, J.; Iwase, H.; Shao, Z.H.; Vanden Hoek, T.L.; Schumacker, P.T. Cell death during ischemia: Relationship to mitochondrial depolarization and ROS generation. Am. J. Physiol. Hear. Circ. Physiol. 2003, 284, 549–558. [Google Scholar]
- Matsukawa, K.; Akagi, S.; Adachi, N.; Kubo, M.; Hirako, M.; Watanabe, S.; Takahashi, S. Effect of Ovary Storage on Development of Bovine Oocytes after Intracytoplasmic Sperm Injection, Parthenogenetic Activation, or Somatic Cell Nuclear Transfer. J. Mamm. Ova Res. 2007, 24, 114–119. [Google Scholar]
- Ribeiro, B.I.; Love, L.B.; Choi, Y.H.; Hinrichs, K. Transport of equine ovaries for assisted reproduction. Anim. Reprod. Sci. 2008, 108, 171–179. [Google Scholar] [PubMed]
- Piras, A.R.; Burrai, G.P.; Ariu, F.; Falchi, L.; Zedda, M.T.; Pau, S.; Gadau, S.D.; Antuofermo, E.; Bebbere, D.; Ledda, S.; et al. Structure of preantral follicles, oxidative status and developmental competence of in vitro matured oocytes after ovary storage at 4 °C in the domestic cat model. Reprod. Biol. Endocrinol. 2018, 16, 1–14. [Google Scholar]
- Barberino, R.S.; Silva, J.R.V.; Figueiredo, J.R.; Matos, M.H.T. Transport of Domestic and Wild Animal Ovaries: A Review of the Effects of Medium, Temperature, and Periods of Storage on Follicular Viability. Biopreserv. Biobank. 2019, 17, 84–90. [Google Scholar]
- Takahashi, Y.; First, N.L. In vitro development of bovine ane-cell embryos: Influence of glucose, lactate, pyruvate, amino acids and vitamins. Theriogelology 1992, 37, 963–978. [Google Scholar]
- Bermejo-Álvarez, P.; Rizos, D.; Rath, D.; Lonergan, P.; Gutiérrez-Adán, A. Can Bovine In Vitro-Matured Oocytes Selectively Process X- or Y-Sorted Sperm Differentially?1. Biol. Reprod. 2008, 79, 594–597. [Google Scholar]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar]
- Ferreira, R.M.; Chiaratti, M.R.; Macabelli, C.H.; Rodrigues, C.A.; Ferraz, M.L.; Watanabe, Y.F.; Smith, L.C.; Meirelles, F.V.; Baruselli, P.S. The Infertility of Repeat-Breeder Cows During Summer Is Associated with Decreased Mitochondrial DNA and Increased Expression of Mitochondrial and Apoptotic Genes in Oocytes1. Biol. Reprod. 2016, 94, 1–10. [Google Scholar]
- Tellado, M.N.; Alvarez, G.M.; Dalvit, G.C.; Cetica, P.D. The Conditions of Ovary Storage Affect the Quality of Porcine Oocytes. Adv. Reprod. Sci. 2014, 02, 56–67. [Google Scholar]
- Costa, E.J.X.; Lopes, R.H.; Lamy-Freund, M.T. Permeability of pure lipid bilayers to melatonin. J. Pineal Res. 1995, 19, 123–126. [Google Scholar] [PubMed]
- Tamura, H.; Takasaki, A.; Taketani, T.; Tanabe, M.; Kizuka, F.; Lee, L.; Tamura, I.; Maekawa, R.; Aasada, H.; Yamagata, Y.; et al. The role of melatonin as an antioxidant in the follicle. J. Ovarian Res. 2012, 26, 5. [Google Scholar]
- Adriaens, I.; Jacquet, P.; Cortvrindt, R.; Janssen, K.; Smitz, J. Melatonin has dose-dependent effects on folliculogenesis, oocyte maturation capacity and steroidogenesis. Toxicology 2006, 228, 333–343. [Google Scholar] [PubMed]
- Nie, J.; Xiao, P.; Wang, X.; Yang, X.; Xu, H.; Lu, K.; Lu, S.; Liang, X. Melatonin prevents deterioration in quality by preserving epigenetic modifications of porcine oocytes after prolonged culture. Aging (Albany. NY). 2018, 10, 3897–3909. [Google Scholar] [PubMed]
- Ramis, M.; Esteban, S.; Miralles, A.; Tan, D.; Reiter, R. Protective Effects of Melatonin and Mitochondria-targeted Antioxidants Against Oxidative Stress: A Review. Curr. Med. Chem. 2015, 22, 2690–2711. [Google Scholar] [PubMed]
- Yang, M.; Tao, J.; Chai, M.; Wu, H.; Wang, J.; Li, G.; He, C.; Xie, L.; Ji, P.; Dai, Y.; et al. Melatonin improves the quality of inferior bovine oocytes and promoted their subsequent IVF embryo development: Mechanisms and results. Molecules 2017, 22, 12. [Google Scholar]
- Yang, L.; Wang, Q.; Cui, M.; Li, Q.; Mu, S.; Zhao, Z. Effect of Melatonin on the In Vitro Maturation of Porcine Oocytes, Development of Parthenogenetically Activated Embryos, and Expression of Genes Related to the Oocyte Developmental Capability. Animals 2020, 10, 209. [Google Scholar]
- De Matos, D.G.; Furnus, C.C.; Moses, D.F.; Baldassarre, H. Effect of cysteamine on glutathione level and developmental capacity of bovine oocyte matured in vitro. Mol. Reprod. Dev. 1995, 42, 432–436. [Google Scholar]
- Gardiner, C.S.; Reed, D.J. Synthesis of glutathione in the preimplantation mouse embryo. Arch. Biochem. Biophys. 1995, 318, 30–36. [Google Scholar]
- Guérin, P.; El Mouatassim, S.; Ménézo, Y. Oxidative stress and protection against reactive oxygen species in the pre-implantation embryo and its surroundings. Hum. Reprod. Update 2001, 7, 175–189. [Google Scholar]
- Lee, C.S.; Koo, D.B.; Fang, N.; Lee, Y.; Shin, S.T.; Park, C.S.; Lee, K.K. Potent and stage-specific action of glutathione on the development of goat early embryos in vitro. Mol. Reprod. Dev. 2000, 57, 48–54. [Google Scholar] [PubMed]
- Gardiner, C.S.; Salmen, J.J.; Brandt, C.J.; Stover, S.K. Glutathione is present in reproductive tract secretions and improves development of mouse embryos after chemically induced glutathione depletion. Biol. Reprod. 1998, 59, 431–436. [Google Scholar] [PubMed]
- Kong, X.; Yang, S.; Gong, F.; Lu, C.; Zhang, S.; Lu, G.; Lin, G. The relationship between cell number, division behavior and developmental potential of cleavage stage human embryos: A time-lapse study. PLoS ONE 2016, 11, 1–11. [Google Scholar]
- Rodríguez-González, E.; López-Bejar, M.; Mertens, M.J.; Paramio, M.T. Effects on in vitro embryo development and intracellular glutathione content of the presence of thiol compounds during maturation of prepubertal goat oocytes. Mol. Reprod. Dev. 2003, 65, 446–453. [Google Scholar]
- Esfandiari, N.; Falcone, T.; Agarwal, A.; Attaran, M.; Nelson, D.R.; Sharma, R.K. Protein supplementation and the incidence of apoptosis and oxidative stress in mouse embryos. Obstet. Gynecol. 2005, 105, 653–660. [Google Scholar]
- Takahashi, M. Oxidative stress and redox regulation on in vitro development of mammalian embryos. J. Reprod. Dev. 2012, 58, 1–9. [Google Scholar]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shcgenerates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar]
- Favetta, L.A.; St. John, E.J.; King, W.A.; Betts, D.H. High levels of p66shcand intracellular ROS in permanently arrested early embryos. Free Radic. Biol. Med. 2007, 42, 1201–1210. [Google Scholar]
- Betts, D.H.; Bain, N.T.; Madan, P. The p66Shc adaptor protein controls oxidative stress response in early bovine embryos. PLoS ONE 2014, 9, 1. [Google Scholar]
- Soliman, M.A.; Abdel Rahman, A.M.; Lamming, D.A.; Birsoy, K.; Pawling, J.; Frigolet, M.E.; Lu, H.; Fantus, I.G.; Pasculescu, A.; Zheng, Y.; et al. The adaptor protein p66Shc inhibits mTOR-dependent anabolic metabolism. Sci. Signal. 2014, 7, 1–11. [Google Scholar]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [PubMed]
- Pan, B.; Yang, H.; Wu, Z.; Qazi, I.H.; Liu, G.; Han, H.; Meng, Q.; Zhou, G. Melatonin improves parthenogenetic development of vitrified–warmed mouse oocytes potentially by promoting G1/S cell cycle progression. Int. J. Mol. Sci. 2018, 19, 1–16. [Google Scholar]
- El-Sayed, A.; Hoelker, M.; Rings, F.; Salilew, D.; Jennen, D.; Tholen, E.; Sirard, M.-A.; Schellander, K.; Tesfaye, D. Large-scale transcriptional analysis of bovine embryo biopsies in relation to pregnancy success after transfer to recipients. Physiol. Genomics 2006, 28, 84–96. [Google Scholar] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Function | Primer Sequence (5′–3′) | Product Size (bp) | Accession No. |
|---|---|---|---|---|
| H2AFZ | Reference gene | F-ATTGCTGGTGGTGGTGTCAT | 147 | NM_001009270.1 |
| R-ACTGGAATCACCAACACTGGA | ||||
| SOD2 | Oxidative stress | F-GCTTACAGATTGCTGCTTGT | 101 | NM_201527.2 |
| R-AAGGTAATAAGCATGCTCCC | ||||
| GPX1 | F-GCAACCAGTTTGGGCATCA | 116 | NM_174076.3 | |
| R-CTCGCACTTTTCGAAGAGCATA | ||||
| SHC1 | Apoptosis | F-GTGAGGTCTGGGCAGAAGC | 335 | XM_024986737.1 |
| R-GGTTCGGACAAAAGGATCACC | ||||
| TP53 | F-GACTCTCGTGGTAACCTGCT | 91 | NM_001009403.1 | |
| R-AATTTTCTTCCTCAGTGCGGC | ||||
| ITM2Ba | F-GTCCCAGAGTTTGCAGATAGTGA | 104 | NM_001035093.1 | |
| R-GGAATCACATAGCACTTATCCAGGTT | ||||
| BAXb | F-GTTGTCGCCCTTTTCTACTTTGC | 89 | NM_173894.1 | |
| R-CAGCCCATGATGGTCCTGATC | ||||
| BCL2 | F-GGAGCTGGTGGTTGACTTTC | 518 | NM_001077486.2 | |
| R-CTAGGTGGTCATTCAGGTAAG | ||||
| NRF1c | Transcription factor | F-CTGTCGCCCAAGTGAATTATTCG | 67 | NM_001098002.2 |
| R-TGTAACGTGGCCCAGTTTTGT | ||||
| POLG2d | F-CTTCTGGGAAACTACGGGAGAAC | 84 | NM_001075191.1 | |
| R-GTAGCCTCTTGTTTACCAGATCCA | ||||
| AKR1B1 | Implantation | F-CGTGATCCCCAAGTCAGTGA | 152 | NM_001012519.1 |
| R-AATCCCTGTGGGAGGCACA | ||||
| IGF2R | Growth factor | F-GCTGCGGTGTGCCAAGTGAAAAAG | 201 | NM_174352.2 |
| R-AGCCCCTCTGCCGTTGTTACCT | ||||
| GJA1 | GAP junctions | F-TGCCTTTCGTTGTAACACTCA | 143 | NM_174068.2 |
| R-AGAACACATGAGCCAGGTACA | ||||
| BMP15e | Oocyte maturation | F-CTACGACTCCGCTTCGTGTGT | 69 | NM_001031752.1 |
| R-AGTGCCATGCCACCAGAAC | ||||
| GDF9f | F-GAAGTGGGACAACTGGATTGTG | 71 | NM_174681.2 | |
| R-CCCTGGGACAGTCCCCTTTA | ||||
| FGF8g | F-GGAGATCGTGCTGGAGAACAA | 66 | NM_001206678.1 | |
| R-GCCATGTACCAGCCCTCGTA | ||||
| FGF16h | F-CGCTTCGGAATTCTGGAGTT | 62 | NM_001192777.1 | |
| R-TCCACTCCCCGGATGCT |
| Treatment | n | Cleaved Embryo at 48 hpi (%) | Expanded Blastocyst (%) | |
|---|---|---|---|---|
| Total | Cleaved | |||
| Melatonin | 157 | 40.08 ± 2.51 a | 14.06 ± 2.42 a | 35.62 ± 9.58 |
| Control | 212 | 31.85 ± 2.45 b | 6.63 ± 2.36 b | 26.17 ± 9.33 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Ajofrín, I.; Iniesta-Cuerda, M.; Peris-Frau, P.; Martín-Maestro, A.; Medina-Chávez, D.-A.; Maside, C.; Fernández-Santos, M.R.; Ortiz, J.A.; Montoro, V.; Garde, J.J.; et al. Beneficial Effects of Melatonin in the Ovarian Transport Medium on In Vitro Embryo Production of Iberian Red Deer (Cervus elaphus hispanicus). Animals 2020, 10, 763. https://doi.org/10.3390/ani10050763
Sánchez-Ajofrín I, Iniesta-Cuerda M, Peris-Frau P, Martín-Maestro A, Medina-Chávez D-A, Maside C, Fernández-Santos MR, Ortiz JA, Montoro V, Garde JJ, et al. Beneficial Effects of Melatonin in the Ovarian Transport Medium on In Vitro Embryo Production of Iberian Red Deer (Cervus elaphus hispanicus). Animals. 2020; 10(5):763. https://doi.org/10.3390/ani10050763
Chicago/Turabian StyleSánchez-Ajofrín, Irene, María Iniesta-Cuerda, Patricia Peris-Frau, Alicia Martín-Maestro, Daniela-Alejandra Medina-Chávez, Carolina Maside, María Rocío Fernández-Santos, José Antonio Ortiz, Vidal Montoro, José Julián Garde, and et al. 2020. "Beneficial Effects of Melatonin in the Ovarian Transport Medium on In Vitro Embryo Production of Iberian Red Deer (Cervus elaphus hispanicus)" Animals 10, no. 5: 763. https://doi.org/10.3390/ani10050763
APA StyleSánchez-Ajofrín, I., Iniesta-Cuerda, M., Peris-Frau, P., Martín-Maestro, A., Medina-Chávez, D.-A., Maside, C., Fernández-Santos, M. R., Ortiz, J. A., Montoro, V., Garde, J. J., & Soler, A. J. (2020). Beneficial Effects of Melatonin in the Ovarian Transport Medium on In Vitro Embryo Production of Iberian Red Deer (Cervus elaphus hispanicus). Animals, 10(5), 763. https://doi.org/10.3390/ani10050763