Large-Scale Hybridisation as an Extinction Threat to the Suweon Treefrog (Hylidae: Dryophytes suweonensis)



Simple Summary
Abstract
1. Introduction
2. Materials and Methods
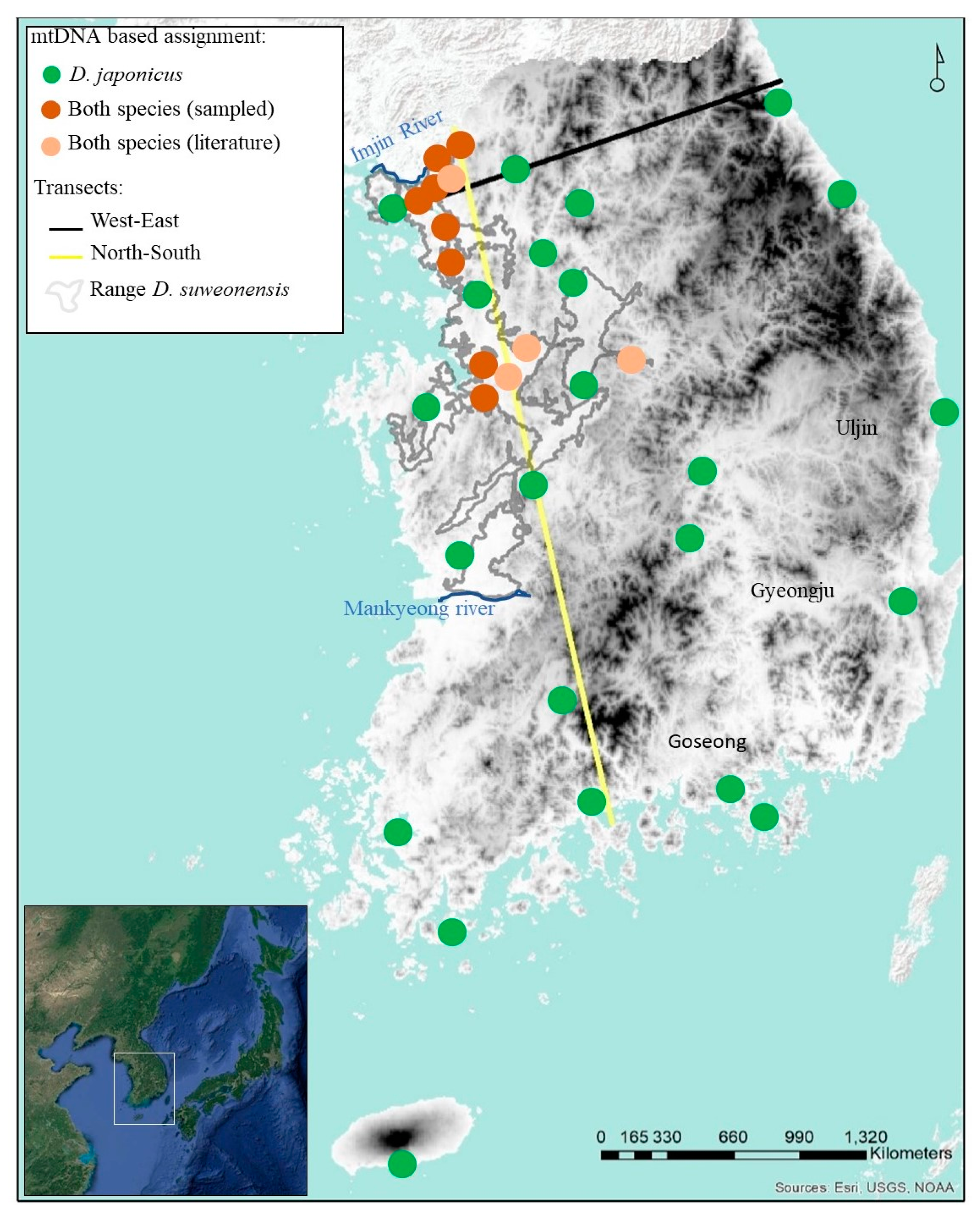
2.1. Field Work
2.2. Molecular Work
2.3. mtDNA Analyses
2.4. Microsatellites analysis
2.5. Identification of Potential Hybrids
3. Results
3.1. mtDNA: Estimation of Genetic Variables
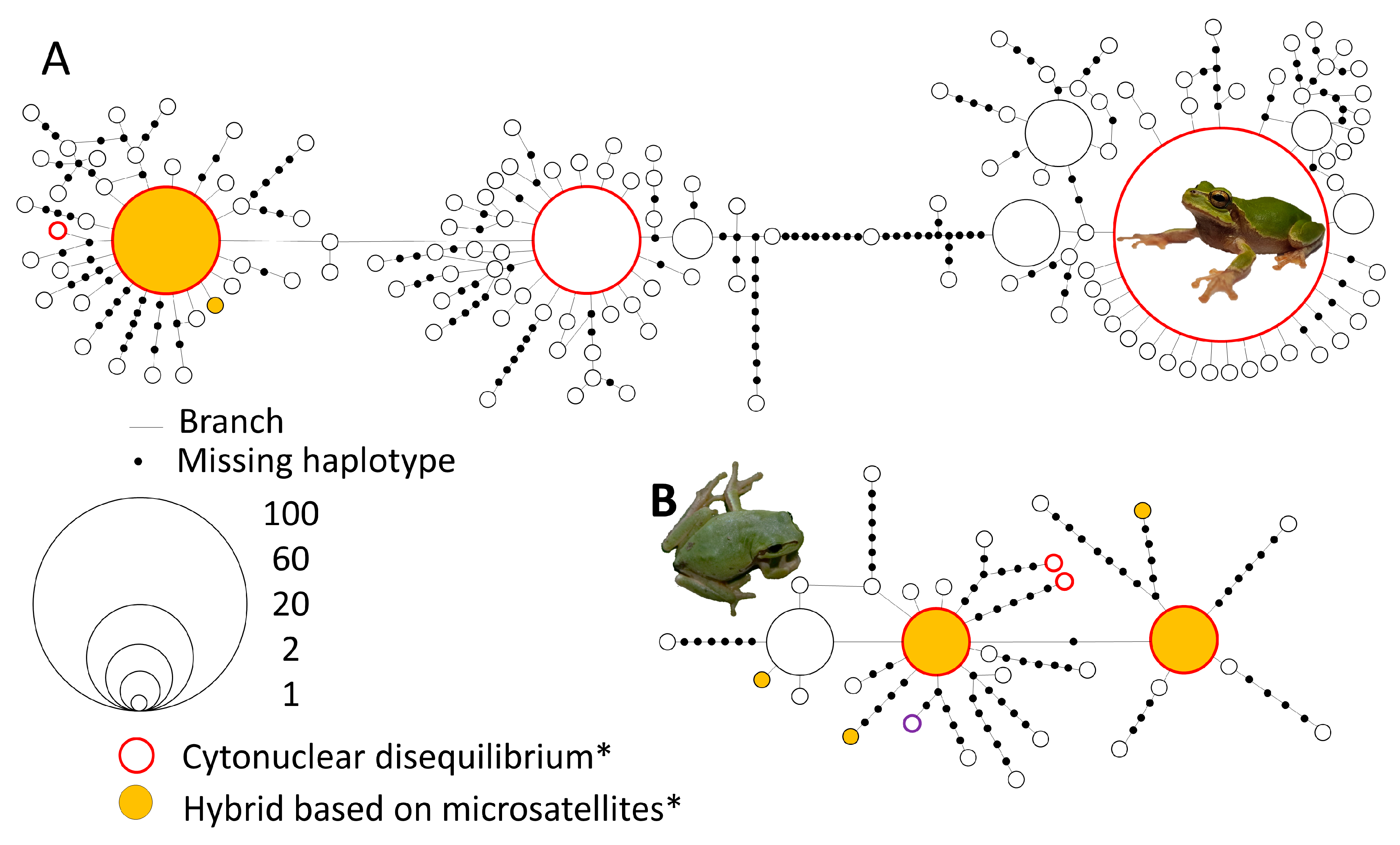
3.2. mtDNA Haplotype Network
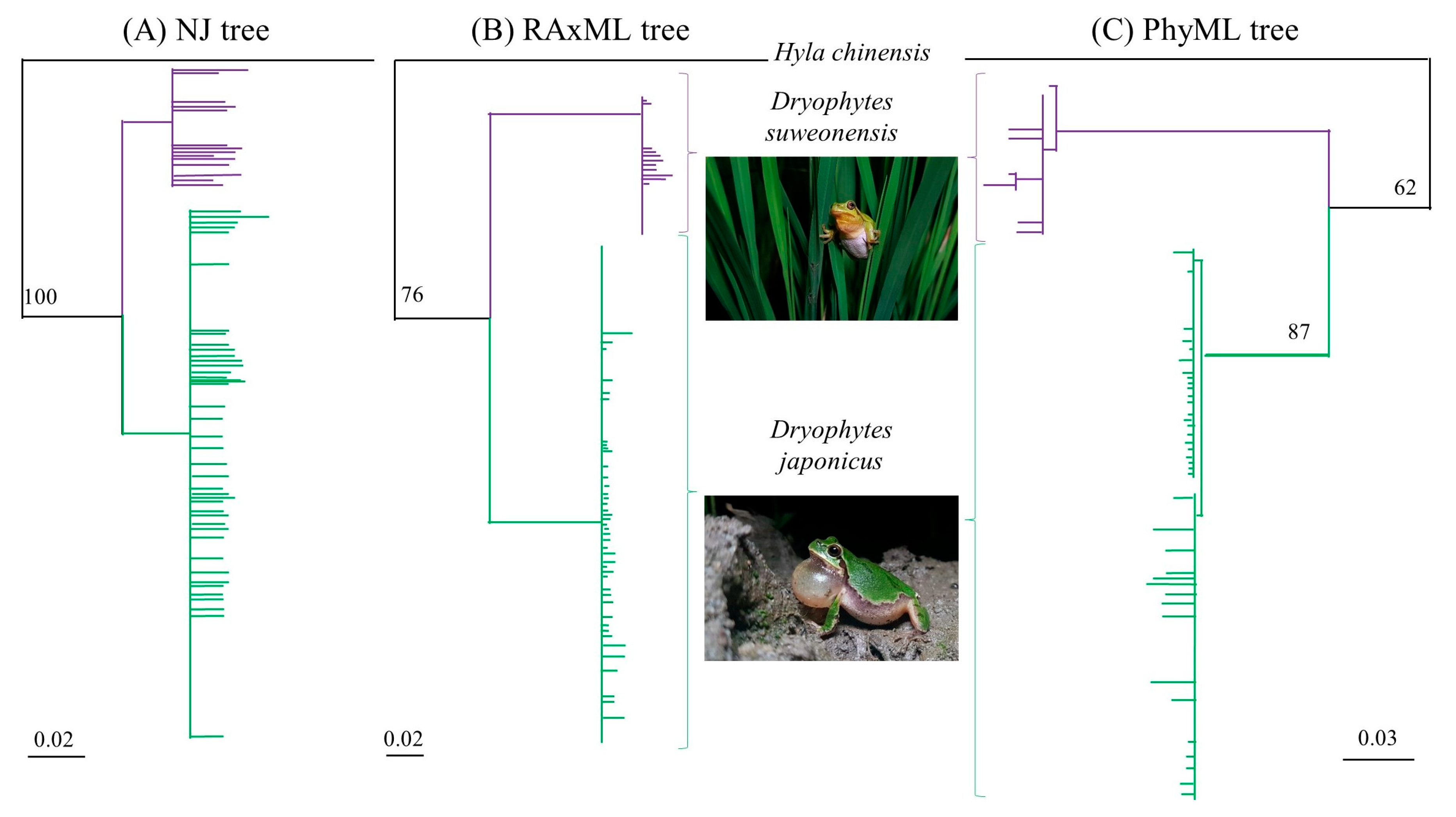
3.3. mtDNA Analyses
3.4. Microsatellites Analysis
3.5. Population Structure
3.6. Identification of Hybrids
4. Discussion
4.1. Genetic Structure
4.2. Hybridisation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Forester, D.J.; Machlist, G.E. Modeling human factors that affect the loss of biodiversity. Conserv. Biol. 1996, 10, 1253–1263. [Google Scholar] [CrossRef]
- Liu, J.; Ouyang, Z.; Tan, Y.; Yang, J.; Zhang, H. Changes in human population structure: Implications for biodiversity conservation. Popul. Environ. 1999, 21, 45–58. [Google Scholar] [CrossRef]
- Pereira, H.M.; Leadley, P.W.; Proença, V.; Alkemade, R.; Scharlemann, J.P.; Fernandez-Manjarrés, J.F.; Araújo, M.B.; Balvanera, P.; Biggs, R.; Cheung, W.W.; et al. Scenarios for global biodiversity in the 21st century. Science 2010, 330, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Rhymer, J.M.; Simberloff, D. Extinction by hybridization and introgression. Annu. Rev. Ecol. Syst. 1996, 27, 83–109. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Leary, R.F.; Spruell, P.; Wenburg, J.K. The problems with hybrids: Setting conservation guidelines. Trends Ecol. Evol. 2001, 16, 613–622. [Google Scholar] [CrossRef]
- Olden, J.D.; Douglas, M.E.; Douglas, M.R. The human dimensions of biotic homogenization. Conserv. Biol. 2005, 19, 2036–2038. [Google Scholar] [CrossRef]
- Laikre, L.; Schwartz, M.K.; Waples, R.S.; Ryman, N.; Group, G.W. Compromising genetic diversity in the wild: Unmonitored large-scale release of plants and animals. Trends Ecol. Evol. 2010, 25, 520–529. [Google Scholar] [CrossRef]
- Casas, F.; Mougeot, F.; Sánchez-Barbudo, I.; Dávila, J.; Viñuela, J. Fitness consequences of anthropogenic hybridization in wild red-legged partridge (Alectoris rufa, Phasianidae) populations. Biol. Invasions 2012, 14, 295–305. [Google Scholar] [CrossRef]
- Seehausen, O. Conservation: Losing biodiversity by reverse speciation. Curr. Biol. 2006, 16, R334–R337. [Google Scholar] [CrossRef]
- Garrick, R.C.; Banusiewicz, J.D.; Burgess, S.; Hyseni, C.; Symula, R.E. Extending phylogeography to account for lineage fusion. J. Biogeogr. 2019, 46, 268–278. [Google Scholar] [CrossRef]
- Vonlanthen, P.; Bittner, D.; Hudson, A.G.; Young, K.A.; Müller, R.; Lundsgaard-Hansen, B.; Roy, D.; Di Piazza, S.; Largiadèr, C.R.; Seehausen, O. Eutrophication causes speciation reversal in whitefish adaptive radiations. Nature 2012, 482, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Marques, D.A.; Meier, J.I.; Seehausen, O. A combinatorial view on speciation and adaptive radiation. Trends Ecol. Evol. 2019, 34, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.H. The role of hybridization in evolution. Mol. Ecol. 2001, 10, 551–568. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.B.; Bernatchez, L. Evolutionary change in human-altered environments. Mol. Ecol. 2008, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.M. A taxonomist’s experience with hybrids in the wild. Science 1935, 81, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Rhymer, J.M.; Williams, M.J.; Braun, M.J. Mitochondrial analysis of gene flow between New Zealand mallards (Anas platyrhynchos) and grey ducks (A. superciliosa). Auk 1994, 111, 970–978. [Google Scholar] [CrossRef]
- Holsbeek, G.; Jooris, R. Potential impact of genome exclusion by alien species in the hybridogenetic water frogs (Pelophylax esculentus complex). Biol. Invasions 2010, 12, 1–13. [Google Scholar] [CrossRef]
- Arano, B.; Llorente, G.; Garcia-Paris, M.; Herrero, P. Species translocation menaces Iberian waterfrogs. Conserv. Biol. 1995, 9, 196–198. [Google Scholar] [CrossRef]
- Kuramoto, M. Mating calls of treefrogs (genus Hyla) in the far east, with description of a new species from Korea. Copeia 1980, 1, 100–108. [Google Scholar] [CrossRef]
- Duellman, W.E.; Marion, A.B.; Hedges, S.B. Phylogenetics, classification, and biogeography of the treefrogs (Amphibia: Anura: Arboranae). Zootaxa 2016, 4104, 1–109. [Google Scholar] [CrossRef]
- Chun, S.; Chung, E.; Voloshina, I.; Chong, J.R.; Lee, H.; Min, M.S. Genetic Diversity of Korean Tree Frog (Hyla suweonensis and Hyla japonica): Assessed by Mitochondrial Cytochrome b Gene and Cytochrome Oxidase Subunit I Gene. Korean J. Herpetol. 2012, 4, 31–41. [Google Scholar]
- Hua, H.; Zheng, R.-Q.; Zhang, J.-Y.; Chen, W.; Yu, X.-Y.; Chen, P. A review of cryptic species in amphibians. Chin. Bull. Life Sci. 2012, 24, 483–491. [Google Scholar]
- Yu, S.; Lee, H. Comparative karyological analysis of the Korean tree frogs, Hyla japonica and Hyla suweonensis (Anura, Hylidae). Korean J. Zool. 1989, 33, 1–5. [Google Scholar]
- Yang, S.Y.; Park, B.S. Speciation of the two species of the genus Hyla (anura) in Korea. Korean J. Zool. 1988, 31, 11–20. [Google Scholar]
- Yang, S.Y.; Min, M.S.; Kim, J.B.; Suh, J.H. Intra and inter specific diversity and speciation of two tree frogs in the genus Hyla. Korean J. Genet. 1997, 19, 71–87. [Google Scholar]
- Jang, Y.; Hahm, E.H.; Lee, H.J.; Park, S.; Won, Y.J.; Choe, J.C. Geographic variation in advertisement calls in a tree frog species: Gene flow and selection hypotheses. PLoS ONE 2011, 6, e23297. [Google Scholar] [CrossRef]
- Park, S.; Jeong, G.; Jang, Y. No reproductive character displacement in male advertisement signals of Hyla japonica in relation to the sympatric H. suweonensis. Behav. Ecol. Sociobiol. 2013, 67, 1345–1355. [Google Scholar] [CrossRef]
- Borzée, A.; Park, S.; Kim, A.; Kim, H.-T.; Jang, Y. Morphometrics of two sympatric species of tree frogs in Korea: A morphological key for the critically endangered Hyla suweonensis in relation to H. japonica. Anim. Cells Syst. 2013, 17, 348–356. [Google Scholar] [CrossRef]
- Borzee, A.; Kim, J.Y.; Da Cunha, M.A.; Lee, D.; Sin, E.; Oh, S.; Yi, Y.; Jang, Y. Temporal and spatial differentiation in microhabitat use: Implications for reproductive isolation and ecological niche specification. Integr. Zool. 2016, 11, 375–387. [Google Scholar] [CrossRef]
- Borzée, A.; Kim, J.Y.; Jang, Y. Asymmetric competition over calling sites in two closely related treefrog species. Sci. Rep. 2016, 6, 32569. [Google Scholar] [CrossRef]
- Li, J.T.; Wang, J.S.; Nian, H.H.; Litvinchuk, S.N.; Wang, J.; Li, Y.; Rao, D.Q.; Klaus, S. Amphibians crossing the bering land bridge: Evidence from holarctic treefrogs (Hyla, Hylidae, Anura). Mol. Phylogenetics Evol. 2015, 87, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Dufresnes, C.; Litvinchuk, S.N.; Borzée, A.; Jang, Y.; Li, J.T.; Miura, I.; Perrin, N.; Stöck, M. Phylogeography reveals an ancient cryptic radiation in East-Asian tree frogs (Hyla japonica group) and complex relationships between continental and island lineages. BMC Evol. Biol. 2016, 16, 253. [Google Scholar] [CrossRef] [PubMed]
- Borzée, A.; Kong, S.; Didinger, C.; Nguyen, H.; Jang, Y. A ring-species or a ring of species? Phylogenetic relationship between two treefrog species, Dryophytes suweonensis and D. immaculatus, around the Yellow Sea. Herpetol. J. 2018, 28, 160–170. [Google Scholar]
- Ministry of Environment. Hyla suweonensis; Ministry of Environment: Seoul, Korea, 2012. Available online: http://www.me.go.kr/web/4245/ysg/common/board (accessed on 30 January 2013).
- IUCN SSC Amphibian Specialist Group. Dryophytes suweonensis (amended version of 2014 assessment). IUCN Red List Threat. Species 2017. [Google Scholar] [CrossRef]
- Borzée, A.; Seliger, B. Dryophytes suweonensis (Suweon Treefrog). Herpetol. Rev. Geogr. Distrib. 2018, 49, 707. [Google Scholar]
- Borzée, A.; Yu, S.H.; Jang, Y. Dryophytes suweonensis (Suweon Treefrog). Herpetol. Rev. 2016, 47, 418. [Google Scholar]
- Borzée, A.; Kim, K.; Heo, K.; Jablonski, P.G.; Jang, Y. Impact of land reclamation and agricultural water regime on the distribution and conservation status of the endangered Dryophytes suweonensis. PeerJ 2017, 5, e3872. [Google Scholar] [CrossRef]
- Roh, G.; Borzée, A.; Jang, Y. Spatiotemporal distributions and habitat characteristics of the endangered treefrog, Hyla suweonensis, in relation to sympatric H. japonica. Ecol. Inform. 2014, 24, 78–84. [Google Scholar] [CrossRef]
- Kuzmin, S.; Maslova, I.; Matsui, M.; Liang, F.; Kaneko, Y. Dryophytes Japonicus (Amended Version of 2014 Assessment); Amphibian Specialist Group, Ed.; The IUCN Red List of Threatened Species (IUCN): Gland, Switzerland, 2017. [Google Scholar]
- Kuramoto, M. Systematic implications of hybridization experiments with some eurasian treefrogs (genus Hyla). Copeia 1984, 3, 609–616. [Google Scholar] [CrossRef]
- Dufresnes, C.; Borzée, A.; Horn, A.; Stöck, M.; Ostini, M.; Sermier, R.; Wassef, J.; Litvinchuck, S.N.; Kosch, T.A.; Waldman, B.; et al. Sex-chromosome homomorphy in Palearctic tree frogs proceeds from both turnovers and X-Y recombination. Mol. Biol. Evol. 2015, 32, 2328–2337. [Google Scholar] [CrossRef]
- Kim, I.-H.; Ham, C.-H.; Jang, S.-W.; Kim, E.-Y.; Kim, J.-B. Determination of Breeding Season, and Daily Pattern of Calling Behavior of the Endangered Suweon-Tree Frog (Hyla suweonensis). Korean J. Herpetol. 2012, 4, 23–29. [Google Scholar]
- Park, S.-R.; Cheon, S.-M.; Yang, S.Y. The classification of call types in genus Hyla in habitats around South Korea. Korean J. Herpetol. 1996, 39, 207–214. [Google Scholar]
- Borzée, A.; Andersen, D.; Jang, Y. Population trend inferred from aural surveys for calling anurans in Korea. PeerJ 2018, 6, e5568. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.H.; Son, S.H.; Kang, S.W.; Kim, J.B. Distribution and habitat characteristics of the endangered Suweon-Tree Frog (Hyla suweonensis). Korean Soc. Herpetol. 2012, 4, 15–22. [Google Scholar]
- Smith, M.A.; Green, D.M. Dispersal and the metapopulation paradigm in amphibian ecology and conservation: Are all amphibian populations metapopulations? Ecography 2005, 28, 110–128. [Google Scholar] [CrossRef]
- Angelone, S.; Holderegger, R. Population genetics suggests effectiveness of habitat connectivity measures for the European tree frog in Switzerland. J. Appl. Ecol. 2009, 46, 879–887. [Google Scholar] [CrossRef]
- Broquet, T.; Berset-Braendli, L.; Emaresi, G.; Fumagalli, L. Buccal swabs allow efficient and reliable microsatellite genotyping in amphibians. Conserv. Genet. 2007, 8, 509–511. [Google Scholar] [CrossRef]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Arens, P.; Westende, W.V.; Bugter, R.; Smulders, M.J.; Vosman, B. Microsatellite markers for the European tree frog Hyla arborea. Mol. Ecol. 2000, 9, 1944–1946. [Google Scholar] [CrossRef]
- Arens, P.; Bugter, R.; Van’t Westende, W.; Zollinger, R.; Stronks, J.; Vos, C.C.; Smulders, M.J. Microsatellite variation and population structure of a recovering Tree frog (Hyla arborea L.) metapopulation. Conserv. Genet. 2006, 7, 825–835. [Google Scholar] [CrossRef]
- Stöck, M.; Horn, A.; Grossen, C.; Lindtke, D.; Sermier, R.; Betto-Colliard, C.; Dufresnes, C.; Bonjour, E.; Dumas, Z.; Luquet, E.; et al. Ever-young sex chromosomes in European tree frogs. PLoS Biol. 2011, 9, e1001062. [Google Scholar] [CrossRef] [PubMed]
- Moreira, C.N. Population Analysis of Hyla arborea and Hyla meridionalis (Amphibia, Anura) in Portugal: A molecular genetic and bioacoustic approach. Ph.D. Thesis, Universidade de Lisboa, Lisbon, Portugal, 2012. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.J.; Spurgin, L.G.; Collar, N.J.; Komdeur, J.; Burke, T.; Richardson, D.S. The impact of translocations on neutral and functional genetic diversity within and among populations of the Seychelles warbler. Mol. Ecol. 2014, 23, 2165–2177. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Fu, Y.-X. Statistical Tests of Neutrality of Mutations Against Population Growth, Hitchhiking and Background Selection. Genetics 1997, 147, 915–925. [Google Scholar]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef]
- Sundberg, K.; Carroll, H.; Snell, Q.; Clement, M.J. Incomparability of results between phylogenetic search programs. In Proceedings of the International Conference on Bioinformatics & Computational Biology, Las Vegas, NV, USA, 14–17 July 2008. [Google Scholar]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, C.; Weetman, D.; Hutchinson, W. Estimation and adjustment of microsatellite null alleles in nonequilibrium populations. Mol. Ecol. Resour. 2006, 6, 255–256. [Google Scholar] [CrossRef]
- Rousset, F. genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J. FSTAT (version 1.2): A computer program to calculate F-statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Leberg, P.L. Estimating allelic richness: Effects of sample size and bottlenecks. Mol. Ecol. 2002, 11, 2445–2449. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: Columbia, SC, USA, 1987. [Google Scholar]
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar]
- Broquet, T.; Angelone, S.; Jaquiéry, J.; Joly, P.; Léna, J.P.; Lengagne, T.; Plénet, S.; Luquet, E.; Perrin, N. Disconnection can drive genetic signatures of bottleneck: A case study in european tree frogs. Conserv. Biol. 2010, 24, 1596–1605. [Google Scholar] [CrossRef]
- Fan, L.; Zheng, H.; Milne, R.I.; Zhang, L.; Mao, K. Strong population bottleneck and repeated demographic expansions of Populus adenopoda (Salicaceae) in subtropical China. Ann. Bot. 2018, 121, 665–679. [Google Scholar] [CrossRef]
- Hey, J. Isolation with migration models for more than two populations. Mol. Biol. Evol. 2010, 27, 905–920. [Google Scholar] [CrossRef]
- Hey, J. The divergence of chimpanzee species and subspecies as revealed in multipopulation isolation-with-migration analyses. Mol. Biol. Evol. 2010, 27, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, B.M. Power and sample size for nested analysis of molecular variance. Mol. Ecol. 2009, 18, 3961–3966. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Pritchard, J.K.; Wen, X.; Falush, D. Documentation for Structure Software: Version 2.2; University of Chicago: Chicago, IL, USA, 2007; Available online: http://pritch.bsd.uchicago.edu/software (accessed on 17 April 2020).
- Porras-Hurtado, L.; Ruiz, Y.; Santos, C.; Phillips, C.; Carracedo, Á.; Lareu, M. An overview of STRUCTURE: Applications, parameter settings and supporting software. Front. Genet. 2013, 1, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pisa, G.; Orioli, V.; Spilotros, G.; Fabbri, E.; Randi, E.; Bani, L. Detecting a hierarchical genetic population structure: The case study of the Fire Salamander (Salamandra salamandra) in Northern Italy. Ecol. Evol. 2015, 5, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Putman, A.I.; Carbone, I. Challenges in analysis and interpretation of microsatellite data for population genetic studies. Ecol. Evol. 2014, 4, 4399–4428. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Lee, C.; Abdool, A.; Huang, C.H. PCA-based population structure inference with generic clustering algorithms. BMC Bioinform. 2009, 10, S73. [Google Scholar] [CrossRef]
- van Heerwaarden, J.; Odong, T.L.; van Eeuwijk, F.A. Maximizing genetic differentiation in core collections by PCA-based clustering of molecular marker data. Appl. Genet. 2013, 126, 763–772. [Google Scholar] [CrossRef]
- Odong, T.L.; Van Heerwaarden, J.; van Hintum, T.J.; van Eeuwijk, F.A.; Jansen, J. Improving hierarchical clustering of genotypic data via principal component analysis. Crop Sci. 2013, 53, 1546–1554. [Google Scholar] [CrossRef]
- Box, G.E.; Tidwell, P.W. Transformation of the independent variables. Technometrics 1962, 4, 531–550. [Google Scholar] [CrossRef]
- Tabachnick, B.; Fidell, L. Using Multivariate Statistics, 6th ed.; Pearson Education Limited: Harlow, UK, 2014. [Google Scholar]
- Bryson, R.W., Jr.; Smith, B.T.; Nieto-Montes de Oca, A.; García-Vázquez, U.O.; Riddle, B.R. The role of mitochondrial introgression in illuminating the evolutionary history of Nearctic treefrogs. Zool. J. Linn. Soc. 2014, 172, 103–116. [Google Scholar] [CrossRef]
- Joly, S.; McLenachan, P.A.; Lockhart, P.J. A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am. Nat. 2009, 174, E54–E70. [Google Scholar] [CrossRef] [PubMed]
- De Hert, K.; Jacquemyn, H.; Van Glabeke, S.; Roldán-Ruiz, I.; Vandepitte, K.; Leus, L.; Honnay, O. Reproductive isolation and hybridization in sympatric populations of three Dactylorhiza species (Orchidaceae) with different ploidy levels. Ann. Bot. 2012, 109, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Vähä, J.P.; Primmer, C.R. Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol. Ecol. 2006, 15, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J.; Burgess, K.S.; Milne, R.; Fu, C.N.; Li, D.Z.; Gao, L.M. Asymmetrical natural hybridization varies among hybrid swarms between two diploid Rhododendron species. Ann. Bot. 2017, 120, 51–61. [Google Scholar] [CrossRef]
- Burgarella, C.; Lorenzo, Z.; Jabbour-Zahab, R.; Lumaret, R.; Guichoux, E.; Petit, R.J.; Soto, A.; Gil, L. Detection of hybrids in nature: Application to oaks (Quercus suber and Q. ilex). Heredity 2009, 102, 442–452. [Google Scholar] [CrossRef]
- Araya-Anchetta, A.; Scoles, G.A.; Giles, J.; Busch, J.D.; Wagner, D.M. Hybridization in natural sympatric populations of Dermacentor ticks in northwestern North America. Ecol. Evol. 2013, 3, 714–724. [Google Scholar] [CrossRef]
- Starr, T.N.; Gadek, K.E.; Yoder, J.B.; Flatz, R.; Smith, C.I. Asymmetric hybridization and gene flow between Joshua trees (Agavaceae: Yucca) reflect differences in pollinator host specificity. Mol. Ecol. 2013, 22, 437–449. [Google Scholar] [CrossRef]
- Kartavtsev, Y. Phylogenetics & Evolutionary Biology Sequence Diversity at Cyt-b and Co-1 mtDNA Genes in Animal Taxa. Phylogenetic Evol. Biol. 2013, 1, 1–5. [Google Scholar]
- Hillis, D.M.; Bull, J.J. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst. Biol. 1993, 42, 182–192. [Google Scholar] [CrossRef]
- Borzée, A. Why are anurans threatened? The case of Dryophytes suweonensis. Ph.D. Thesis, Seoul National University, Seoul, Korea, 2018. [Google Scholar]
- Frankham, R. Inbreeding and extinction: Island populations. Conserv. Biol. 1998, 12, 665–675. [Google Scholar] [CrossRef]
- Spielman, D.; Brook, B.W.; Frankham, R. Most species are not driven to extinction before genetic factors impact them. Proc. Natl. Acad. Sci. USA 2004, 101, 15261–15264. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R. Genetics and extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Jiggins, C.D.; Mallet, J. Bimodal hybrid zones and speciation. Trends Ecol. Evol. 2000, 15, 250–255. [Google Scholar] [CrossRef]
- Borzée, A.; Ahn, J.; Kim, S.; Heo, K.; Jang, Y. Seoul, keep your paddies! Implications for the conservation of hylid species. Anim. Syst. Evol. Divers. 2015, 31, 176–181. [Google Scholar] [CrossRef]
- Yang, Z.; Rannala, B. Molecular phylogenetics: Principles and practice. Nat. Rev. Genet. 2012, 13, 303–314. [Google Scholar] [CrossRef]
- Grobler, J.; Mafumo, H.; Minter, L. Genetic differentiation among five populations of the South African ghost frog, Heleophryne natalensis. Biochem. Syst. Ecol. 2003, 31, 1023–1032. [Google Scholar] [CrossRef]
- Andersen, L.W.; Fog, K.; Damgaard, C. Habitat fragmentation causes bottlenecks and inbreeding in the European tree frog (Hyla arborea). Proc. R. Soc. B Biol. Sci. 2004, 271, 1293–1302. [Google Scholar] [CrossRef]
- Borzée, A.; Jang, Y. The International Society for Ecological Modelling; Elsevier: Jeju, Korea, 2017. [Google Scholar]
- Bilton, D.T.; Freeland, J.R.; Okamura, B. Dispersal in freshwater invertebrates. Annu. Rev. Ecol. Syst. 2001, 32, 159–181. [Google Scholar] [CrossRef]
- Lamb, T.; Avise, J.C. Directional introgression of mitochondrial DNA in a hybrid population of tree frogs: The influence of mating behavior. Proc. Natl. Acad. Sci. USA 1986, 83, 2526–2530. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.A. A case of interbreeding between Rana aurora and Bufo boreas (Amphibia, Anura). J. Herpetol. 1977, 11, 92–94. [Google Scholar] [CrossRef]
- Reading, C. Interspecific spawning between common frogs (Rana temporaria) and common toads (Bufo bufo). J. Zool. 1984, 203, 95–101. [Google Scholar] [CrossRef]
- Kim, Y.E. Differential Antipredator Behavior between Hyla japonica and H. suweonensis Suggests Separate Evolution. Master’s Thesis, Ewha Womans University, Seoul, Korea, 2016. [Google Scholar]
- Borzée, A.; Heo, K.; Jang, Y. Relationship between agro-environmental variables and breeding Hylids in rice paddies. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fuller, D.Q.; Qin, L.; Harvey, E. Evidence for a late onset of agriculture in the Lower Yangtze region and challenges for an archaeobotany of rice. In Human Migrations in Continental East Asia and Taiwan: Genetic, Linguistic and Archaeological Evidence; Sanchez-Mazas, A., Blench, R., Ross, M., Lin, M., Pejros, I., Eds.; Taylor & Francis: London, UK, 2008; pp. 40–83. [Google Scholar]
- Borzée, A.; Yu, A.-Y.; Jang, Y. Variation in the persistence of two Hylid species in relation to behavioural and physiological traits. Ethol. Ecol. Evol. 2018, 30, 515–533. [Google Scholar] [CrossRef]
- Anderson, E. Hybridization of the habitat. Evol. Int. J. Org. Evol. 1948, 2, 1–9. [Google Scholar] [CrossRef]
- Whitmore, D.H. Introgressive hybridization of smallmouth bass (Micropterus dolomieui) and Guadalupe bass (M. treculi). Copeia 1983, 3, 672–679. [Google Scholar] [CrossRef]
- Schlefer, E.K.; Romano, M.A.; Guttman, S.I.; Ruth, S.B. Effects of twenty years of hybridization in a disturbed habitat on Hyla cinerea and Hyla gratiosa. J. Herpetol. 1986, 20, 210–221. [Google Scholar] [CrossRef]
- Fuller, D.Q.; Harvey, E.; Qin, L. Presumed domestication? Evidence for wild rice cultivation and domestication in the fifth millennium BC of the Lower Yangtze region. Antiquity 2007, 81, 316–331. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence | Primer Developed/Used by | Annealing Temperature |
|---|---|---|---|
| WHA1-9-F | 5′-CGTTTGGACGTGATGCTG-3′ | Arens et al. [51] 2000, Arens et al. [52] 2006, Moreira [54] 2012 | 47 |
| WHA1-9-R | 5′-GAGGAGTTTCTTCACAAGGGG-3′ | ||
| WHA5-201-F | 5′-TCATGGACTGTCGTCATGGT-3′ | Arens et al. [51] 2000, Arens et al. [52] 2006, Moreira [54] 2012 | 47 |
| WHA5-201-R | 5′-AGGTAAATGGAATCTGGGTGTG-3′ | ||
| WHA5-22A-F | 5′-TTACAGCAACAGCAAATGG-3′ | Arens et al. [51] 2000, Arens et al. [52] 2006, Moreira [54] 2012, Stöck et al. 2011, Dusfresnes et al. 2015 | 50 |
| WHA5-22A-R | 5′-ATCAGGGACTGGGTCTGT-3′ | ||
| WHA1-104-F | 5′-ACTTGGGACAGCCAGTATGTTTT-3′ | Arens et al. [51] 2000, Arens et al. [52] 2006, Moreira [54] 2012 | 47 |
| WHA1-104-R | 5′-TGAGCTGGTGGGTATAACCTAAC-3′ | ||
| WHA1-25-F | 5′-AAGAATCTGCCGCAAAGAAG-3′ | Arens et al. [51] 2000, Arens et al. [52] 2006, Moreira [54] 2012 | 47 |
| WHA1-25-R | 5′-TAGGAAGGGACAGGAGGTCA-3′ | ||
| WHA5-57-F | 5′-TTGTCCTGACATGCACACCT-3′ | Arens et al. [51] 2000, Moreira [54] 2012 | |
| WHA5-57-R | 5′-CGTGTCTAACCCCAGCTCAT-3′ | ||
| WHA1-140-F | 5′-ATGTGCCATAGAAATGAAGG-3′ | Arens et al. [51] 2000, Arens et al. [52] 2006, Moreira [54] 2012 | 47 |
| WHA1-140-R | 5′-AGGCTTGCTGCTATTATGTC-3′ | ||
| WHA1-60-F | 5′-TAGGTCATGTATAGCCTGTT-3′ | Arens et al. [51] 2000, Arens et al. [52] 2006 | 47 |
| WHA1-60-R | 5′-TCTGTTTACTTCAGGGGT-3′ | ||
| WHA1-20-F | 5′-GTCCCTTCCTGAATAAGTGTCG-3′ | Arens et al. [51] 2000, Arens et al. [52] 2006 | 47 |
| WHA1-20-R | 5′-CCATTCCCTCCTGGCTTT-3′ |
| Variables | PC1 | PC2 | PC3 | PC4 | PC5 |
|---|---|---|---|---|---|
| WHA1-9-F | 0.87 | 0.07 | −0.08 | 0.00 | 0.04 |
| WHA1-9-R | 0.86 | 0.04 | −0.03 | 0.04 | 0.09 |
| WHA5-201-F | −0.09 | 0.01 | 0.99 | 0.00 | 0.03 |
| WHA5-201-R | −0.09 | 0.01 | 0.99 | −0.01 | 0.03 |
| WHA5-22A-F | 0.04 | −0.01 | 0.02 | 0.04 | 0.87 |
| WHA5-22A-R | −0.07 | 0.03 | 0.00 | −0.22 | 0.82 |
| WHA1-104-F | 0.71 | 0.27 | −0.08 | 0.10 | −0.35 |
| WHA1-104-R | 0.36 | 0.24 | −0.10 | 0.08 | −0.32 |
| WHA1-25-F | 0.07 | 0.11 | 0.00 | 0.96 | −0.09 |
| WHA1-25-R | 0.04 | 0.13 | 0.00 | 0.96 | −0.10 |
| WHA1-140-F | 0.15 | 0.97 | 0.02 | 0.12 | −0.01 |
| WHA1-140-R | 0.15 | 0.97 | 0.02 | 0.12 | −0.01 |
| Eigenvalues | 3.28 | 2.09 | 1.78 | 1.45 | 1.26 |
| % of variance | 27.32 | 17.42 | 14.86 | 12.12 | 10.47 |
| F-Statistics Analyses | All Populations | Dj Populations | Ds Populations | p-Value |
|---|---|---|---|---|
| Gene diversity (He) | 0.28 | 0.246 | 0.29 | 0.746 |
| Number of alleles (Ae) | 2.564 | 2.12 | 2.167 | 0.928 |
| Allelic richness (AR) | 1.267 | 1.231 | 1.281 | 0.700 |
| Inbreeding (FIS) | 0.576 | 0.62 | 0.369 | 0.251 |
| Binary Logistic Regression | B | SE | df | p-Value |
|---|---|---|---|---|
| PC1 | −6.87 | 1.34 | 1 | <0.001 |
| PC2 | 15.94 | 4.26 | 1 | 0.046 |
| PC3 | 4.61 | 1.42 | 1 | 0.211 |
| PC4 | −5.76 | 1.81 | 1 | <0.001 |
| PC5 | 1.82 | 0.74 | 1 | 0.294 |
| Sampling locality | −0.05 | 0.03 | 1 | <0.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borzée, A.; Fong, J.J.; Nguyen, H.Q.; Jang, Y. Large-Scale Hybridisation as an Extinction Threat to the Suweon Treefrog (Hylidae: Dryophytes suweonensis). Animals 2020, 10, 764. https://doi.org/10.3390/ani10050764
Borzée A, Fong JJ, Nguyen HQ, Jang Y. Large-Scale Hybridisation as an Extinction Threat to the Suweon Treefrog (Hylidae: Dryophytes suweonensis). Animals. 2020; 10(5):764. https://doi.org/10.3390/ani10050764
Chicago/Turabian StyleBorzée, Amaël, Jonathan J. Fong, Hoa Quynh Nguyen, and Yikweon Jang. 2020. "Large-Scale Hybridisation as an Extinction Threat to the Suweon Treefrog (Hylidae: Dryophytes suweonensis)" Animals 10, no. 5: 764. https://doi.org/10.3390/ani10050764
APA StyleBorzée, A., Fong, J. J., Nguyen, H. Q., & Jang, Y. (2020). Large-Scale Hybridisation as an Extinction Threat to the Suweon Treefrog (Hylidae: Dryophytes suweonensis). Animals, 10(5), 764. https://doi.org/10.3390/ani10050764