
Gene Networks and Pathways Involved in Escherichia coli Response to Multiple Stressors

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
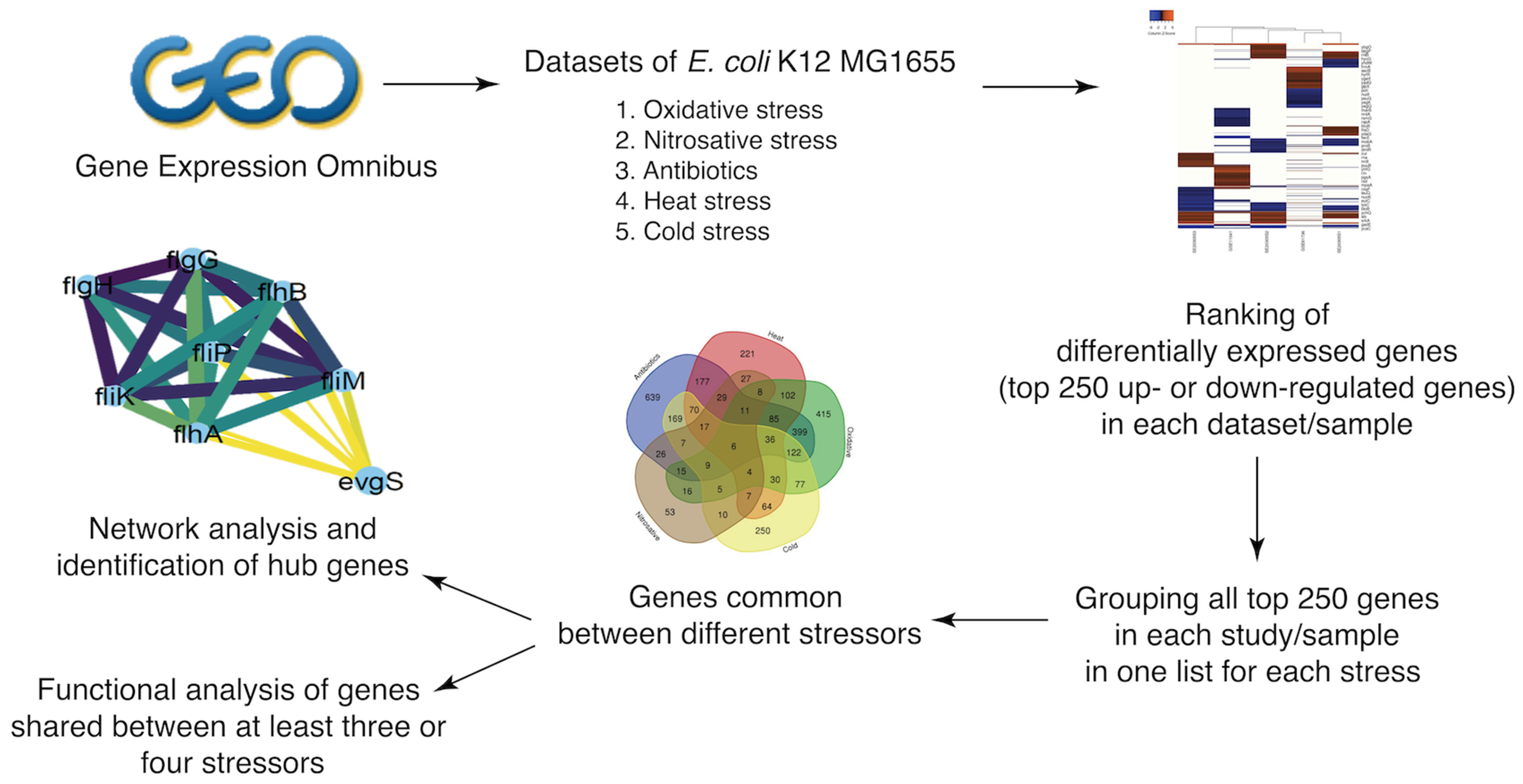
2.1. Selection of Stressors, Growth Conditions, and Data Sets
2.2. Identification of Differentially Expressed Genes under Selected Stressors
2.3. Identification of Genes That Are Differentially Expressed under Multiple Stressors
2.4. Generation of Protein–Protein Interaction Networks
2.5. Determination of Functional Categories from Gene Lists
3. Results
3.1. Details of Data Sets Used in Our Analysis
3.2. Differentially Expressed Genes (DEGs) under Selected Stressors
3.3. Identification of Genes Affected by Multiple Stressors
3.4. Functional Categories Involved in E. coli Response to Multiple Stressors
3.5. Network Analysis of DEGs and Identification of Hub Genes
4. Discussion
- osmB encodes an outer membrane lipoprotein, known to be upregulated by osmotic shock and stationary phase [16,17,18]. Although the function of osmB is not fully characterized [19], its transcriptional regulation was thoroughly studied and found to depend on the response regulator, RcsB, and its transcription was found to increase following the activation of the RcsCDB phosphorelay [20]. Our analysis indicated that osmY, osmF, and osmC were downregulated under three, two, and one stressor, respectively.
- nrdH and nrdI: NrdH, a gluatredoxin-like protein, serves as the electron donor for NrdEF and is reduced by thioredoxin reductase [21]. nrdH is part of the nrdHIEF operon, while nrdEF encodes class Ib ribonucleotide reductase (RNR), catalyzing the reduction of ribonucleotides (NTP) to deoxyribonucleotides (dNTP), a key reaction for DNA synthesis, replication, and repair. nrdE encodes the alpha 2 subunit, harboring the site for nucleotide reduction, and nrfF encodes the beta 2 subunit, which contains the metallocofactor required for catalysis at alpha 1 subunit [22]. Our analysis indicated nrdE, nrdF to be upregulated under at least four stressors. NrdI is a flavodoxin that mediates the generation of the tyrosyl radical cofactor of NrdF. Oxidative stress was reported to induce nrdHIEF expression (up to 23.4-fold), suggesting that E. coli overexpresses various reductases and electron donors to increase the cell’s free radical scavenging capacity to cope with oxidative stress [23]. Our analysis indicated that this operon was upregulated under oxidative stress, antibiotic treatment, cold, heat, and nitrosative stress.
- yqjI (nfeR) is part of the yqjH-yqjI operon. yqjI encodes a DNA-binding transcription repressor that regulates the expression of the NADPH-dependent ferric siderophore reductase yqjH [24]. YqjH is required for iron homeostasis in E. coli and it is also part of the Fur regulon [25]. Our analysis indicated that yqjH was upregulated under antibiotic and heat stress. In aerobic conditions, iron is present as insoluble iron hydroxides; consequently, bacteria produce siderophores, which are extracellular ferric chelators, to mobilize iron [26]. Once ferri-siderophores complexes are transferred through membranes to the cytoplasm, they are either degraded by esterase or reduced by ferric siderophore reductases to release ferrous ions [27].
- yhcN encodes a putative periplasmic protein involved in response to hydrogen peroxide and acid stress. A knockout strain lacking yhcN was found to be more sensitive to hydrogen peroxide and was more able to form biofilm than its parental strain [28]. YhcN was first reported to be upregulated in response to cytoplasmic acid stress by Kannan, et al. [29].
- prs encodes ribose-phosphate diphosphokinase, which transfers a pyrophosphoryl group from ATP to ribose 5-phosphate, synthesizing PRPP that is utilized in the biosynthesis of purine and pyrimidine nucleotides [41].
- purF encodes an amidophosphoribosyl transferase, which catalyzes the pathway flux-controlling step in de novo purine biosynthesis. In this reaction, 5-phospho-β-D-ribosyl-amine (PRA) is formed from PRPP and glutamine. It is feedback-regulated by GMP and AMP [42].
- purD encodes a phosphoribosylamine glycine ligase, the second enzyme in the de novo purine biosynthesis pathway. It catalyzes the ligation of glycine PRA to produce 5-phospho-ribosyl-glycineamide (GAR) [43].
- purN and purT encode two different GAR transformylases, with no significant homology; each of them can catalyze the third step in de novo purine biosynthesis, producing 5-phospho-ribosyl-N-formylglycinamide (FGAR). PurN transfers a formyl group from 10-formyl-tetrahydrofolate, while PurT utilizes formate, after the hydrolysis of 10-formyl-tetrahydrofolate by purU [44].
- purL encodes a phosphoribosylformylglycinamide synthetase, which catalyzes the fourth step in the E. coli de novo purine biosynthesis pathway. In this reaction, 5-phosphoribosyl-N-formylglycineamidine (FGAM) is formed from FGAR, glutamine, and ATP [45].
- purC encodes a phosphoribosylaminoimidazole-succinocarboxamide synthase, which catalyzes the formation of 4-(N-succinylcarboxamide)-5-aminoimidazole ribonucleotide (SAICAR) from CAIR [48]. This is similar to another reaction catalyzed by PurA (adenylosuccinate synthetase) that also utilizes aspartate and a ribonucleoside triphosphate [49]. Our analysis indicated that purA was downregulated under antibiotic treatment and heat stress.
- purB encodes adenylosuccinate lyase, which catalyzes two reactions in de novo purine nucleotide biosynthesis. It converts 5-aminoimidazole-4-carboxamide SAICAR to ribonucleotide (AICAR) and catalyzes the breakdown of adenylosuccinate to AMP [38].
- purH encodes the bifunctional AICAR transformylase/IMP cyclohydrolase, which catalyzes the last two steps of the de novo purine biosynthetic pathway converting AICAR to IMP [50].
- glyA encodes a serine hydroxymethyltransferase, which catalyzes the conversion of serine to glycine through forming 5,10-mTHF. It is activated by MetR, and repressed by MetR and PurR [51].
- gcvT is part of gcvTHP operon. It encodes the T-protein in the glycine cleavage system, which catalyzes glycine degradation and the formation of 5,10- methylenetetrahydrofolate 5,10-mTHF. The other two genes encode GcvH, or H-protein, and GcvP, or P protein [52,53,54]. Expression of the glycine cleavage enzyme system is induced by glycine, activated by GcvA, and repressed by PurR and GcvA [55,56]. Both gcvH and gcvP in our analysis were downregulated under three and four stressors, respectively, indicating the many-sided roles of GcvTHP beyond amino acid metabolism. GcvA is also the activator of gcvB. Both were reported to be related to stress response. The hdeAB operon, which encodes chaperone-like functions, was reported to be activated by GcvB and repressed by GcvA. Both HdeA and HdeB protect periplasmic proteins from aggregation by acid stress [57]. gcvB encodes a small regulatory RNA. gcvB knockout mutant was found to be more sensitive to oxidative stress and accumulate more endogenous reactive oxygen species than wild type. The role of gcvB in oxidative stress was also found to be conferred by increasing OxyR expression [58]. These findings suggest that gcvB and gcvA products allow E. coli to survive in presence of both oxidative stress and low pH. Our findings expand their regulatory functions to cold, heat, and antibiotic treatment.
- pyrC encodes a dihydroorotase, which catalyzes the third reaction in the de novo pyrimidine biosynthesis pathway. By looking for other genes involved in pyrimidine metabolism in our data, we found seven more genes to be downregulated: pyrB, pyrD, and pyrI were downregulated under four stressors while pyrE, pyrF, pyrG, and pyrL were downregulated under two stressors.
- carA is part of the carAB operon. It encodes the amidotransferase component, CarA, while the CarB subunit is the synthetase component of the carbamoylphosphate synthetase, involved in L- arginine synthetic pathway, along with the de novo uridine monophosphate (UMP) biosynthetic pathway (part of pyrimidine biosynthesis). We found carB to be downregulated under four stressors.
- codB and codA form the codBA operon. CodB is a cytosine permease, which brings cytosine into the cell [59], while CodA is a cytosine deaminase (CDA), which catalyzes cytosine deamination into uracil. It is one of the enzymes in the pyrimidine salvage pathway, permitting the cell to utilize cytosine for pyrimidine nucleotide synthesis [60,61].
- cyoB encodes subunit I of the cytochrome bo3 complex. It is a part of the cyoABCDE operon. Our analysis indicated that cyoA, cyoC, cyoD, and cyoE were downregulated under two out of five stressors.
- nuoC is part of the nuoABCEFGHIJKLMN operon, representing the 13 subunits of NADH ubiquinone oxidoreductase. Our analysis detected the downregulation of nuoE, nuoF, nuoH, nuoI, and nuoJ under four stressors, and of nuoB, nuoG, nuoK, nuoL, nuoM, and nuoN under three stressors, while nuoA was detectably downregulated under two stressors.
- fadL encodes the component of a channel involved in the import of long-chain (C12-C18) fatty acids (LCFA) across the bacterial outer membrane [68]. It is part of the fatty acid-degrading (fad), regulon which includes eight more genes involved in fatty acids catabolism [69]. Our analysis indicated that fadD, fadE, fadI, and fadR were downregulated under at least two stressors, suggesting that, during stress, E. coli downregulates the uptake of exogenous fatty acids along with its metabolism and degradation which generates various reduced cofactors. As previously reported, LCFA degradation generates oxidative stress with high levels of reactive oxygen species [70,71].
- cvpA encodes colicin V production protein [72] and is located directly upstream of purF and repressed by PurR, the main repressor of purine synthetic pathway [73]. Its deletion mutant in EHEC was highly sensitive to bile and was deemed important in cell envelope homeostasis in response to stressors, such as deoxycholate bile salt [74].
- gtrB (yfdH) and gtrS (yfdI) are part of the yfdGHI operon. Our analysis indicated that yfdG was downregulated under two stressors. yfdG, yfdH, and yfdI are homologous to the type IV O antigen modification genes (gtrAIV, gtrBIV, and gtrIV) in the genome of Shigella flexneri NCTC 8296 [77].
- rnb encodes the ribonuclease II enzyme (RNase II), involved in the specific degradation of mRNA in the 3′ to 5′ direction [78].
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guo, M.S.; Gross, C.A. Stress-induced remodeling of the bacterial proteome. Curr. Biol. CB 2014, 24, R424–R434. [Google Scholar] [CrossRef]
- Gadgil, M.; Kapur, V.; Hu, W.S. Transcriptional response of Escherichia coli to temperature shift. Biotechnol. Prog. 2005, 21, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Dragosits, M.; Mozhayskiy, V.; Quinones-Soto, S.; Park, J.; Tagkopoulos, I. Evolutionary potential, cross-stress behavior and the genetic basis of acquired stress resistance in Escherichia coli. Mol. Syst. Biol. 2013, 9, 643. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, T.S.; Csonka, L.N.; Paliy, O. Genome-wide transcriptional responses of Escherichia coli K-12 to continuous osmotic and heat stresses. J. Bacteriol. 2008, 190, 3712–3720. [Google Scholar] [CrossRef] [PubMed]
- Giuliodori, A.M.; Gualerzi, C.O.; Soto, S.; Vila, J.; Tavío, M.M. Review on bacterial stress topics. Ann. N. Y. Acad. Sci. 2007, 1113, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Raivio, T.L.; Leblanc, S.K.; Price, N.L. The Escherichia coli Cpx envelope stress response regulates genes of diverse function that impact antibiotic resistance and membrane integrity. J. Bacteriol. 2013, 195, 2755–2767. [Google Scholar] [CrossRef] [PubMed]
- Poole, K. Bacterial stress responses as determinants of antimicrobial resistance. J. Antimicrob. Chemother. 2012, 67, 2069–2089. [Google Scholar] [CrossRef]
- Bhatia, R.P.; Kirit, H.A.; Predeus, A.V.; Bollback, J.P. Transcriptomic profiling of Escherichia coli K-12 in response to a compendium of stressors. Sci. Rep. 2022, 12, 8788. [Google Scholar] [CrossRef]
- Pavlopoulos, G.A.; Malliarakis, D.; Papanikolaou, N.; Theodosiou, T.; Enright, A.J.; Iliopoulos, I. Visualizing genome and systems biology: Technologies, tools, implementation techniques and trends, past, present and future. GigaScience 2015, 4, 38. [Google Scholar] [CrossRef]
- Yoon, S.H.; Han, M.J.; Jeong, H.; Lee, C.H.; Xia, X.X.; Lee, D.H.; Shim, J.H.; Lee, S.Y.; Oh, T.K.; Kim, J.F. Comparative multi-omics systems analysis of Escherichia coli strains B and K-12. Genome Biol. 2012, 13, R37. [Google Scholar] [CrossRef]
- Barrett, T.; Edgar, R. Mining microarray data at NCBI’s Gene Expression Omnibus (GEO). Methods Mol. Biol. 2006, 338, 175–190. [Google Scholar] [CrossRef]
- Von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Gutierrez, C.; Barondess, J.; Manoil, C.; Beckwith, J. The use of transposon TnphoA to detect genes for cell envelope proteins subject to a common regulatory stimulus. Analysis of osmotically regulated genes in Escherichia coli. J. Mol. Biol. 1987, 195, 289–297. [Google Scholar] [CrossRef]
- Hengge-Aronis, R.; Klein, W.; Lange, R.; Rimmele, M.; Boos, W. Trehalose synthesis genes are controlled by the putative sigma factor encoded by rpoS and are involved in stationary-phase thermotolerance in Escherichia coli. J. Bacteriol. 1991, 173, 7918–7924. [Google Scholar] [CrossRef]
- Jung, J.U.; Gutierrez, C.; Martin, F.; Ardourel, M.; Villarejo, M. Transcription of osmB, a gene encoding an Escherichia coli lipoprotein, is regulated by dual signals. Osmotic stress and stationary phase. J. Biol. Chem. 1990, 265, 10574–10581. [Google Scholar] [CrossRef]
- Jung, J.U.; Gutierrez, C.; Villarejo, M.R. Sequence of an osmotically inducible lipoprotein gene. J. Bacteriol. 1989, 171, 511–520. [Google Scholar] [CrossRef]
- Boulanger, A.; Francez-Charlot, A.; Conter, A.; Castanié-Cornet, M.P.; Cam, K.; Gutierrez, C. Multistress regulation in Escherichia coli: Expression of osmB involves two independent promoters responding either to sigmaS or to the RcsCDB His-Asp phosphorelay. J. Bacteriol. 2005, 187, 3282–3286. [Google Scholar] [CrossRef]
- Jordan, A.; Aslund, F.; Pontis, E.; Reichard, P.; Holmgren, A. Characterization of Escherichia coli NrdH. A glutaredoxin-like protein with a thioredoxin-like activity profile. J. Biol. Chem. 1997, 272, 18044–18050. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.; Aragall, E.; Gibert, I.; Barbe, J. Promoter identification and expression analysis of Salmonella typhimurium and Escherichia coli nrdEF operons encoding one of two class I ribonucleotide reductases present in both bacteria. Mol. Microbiol. 1996, 19, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Monje-Casas, F.; Jurado, J.; Prieto-Alamo, M.J.; Holmgren, A.; Pueyo, C. Expression analysis of the nrdHIEF operon from Escherichia coli. Conditions that trigger the transcript level in vivo. J. Biol. Chem. 2001, 276, 18031–18037. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, Y.; Outten, F.W. Fur and the novel regulator YqjI control transcription of the ferric reductase gene yqjH in Escherichia coli. J. Bacteriol. 2011, 193, 563–574. [Google Scholar] [CrossRef]
- McHugh, J.P.; Rodríguez-Quinoñes, F.; Abdul-Tehrani, H.; Svistunenko, D.A.; Poole, R.K.; Cooper, C.E.; Andrews, S.C. Global iron-dependent gene regulation in Escherichia coli. A new mechanism for iron homeostasis. J. Biol. Chem. 2003, 278, 29478–29486. [Google Scholar] [CrossRef]
- Köster, W. ABC transporter-mediated uptake of iron, siderophores, heme and vitamin B12. Res. Microbiol. 2001, 152, 291–301. [Google Scholar] [CrossRef]
- Andrews, S.C.; Robinson, A.K.; Rodríguez-Quiñones, F. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003, 27, 215–237. [Google Scholar] [CrossRef]
- Lee, J.; Hiibel, S.R.; Reardon, K.F.; Wood, T.K. Identification of stress-related proteins in Escherichia coli using the pollutant cis-dichloroethylene. J. Appl. Microbiol. 2010, 108, 2088–2102. [Google Scholar] [CrossRef]
- Kannan, G.; Wilks, J.C.; Fitzgerald, D.M.; Jones, B.D.; Bondurant, S.S.; Slonczewski, J.L. Rapid acid treatment of Escherichia coli: Transcriptomic response and recovery. BMC Microbiol. 2008, 8, 37. [Google Scholar] [CrossRef]
- Dufour, A.; Furness, R.B.; Hughes, C. Novel genes that upregulate the Proteus mirabilis flhDC master operon controlling flagellar biogenesis and swarming. Mol. Microbiol. 1998, 29, 741–751. [Google Scholar] [CrossRef]
- Beloin, C.; Valle, J.; Latour-Lambert, P.; Faure, P.; Kzreminski, M.; Balestrino, D.; Haagensen, J.A.; Molin, S.; Prensier, G.; Arbeille, B.; et al. Global impact of mature biofilm lifestyle on Escherichia coli K-12 gene expression. Mol. Microbiol. 2004, 51, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Makaroff, C.A.; Zalkin, H. Regulation of Escherichia coli purF. Analysis of the control region of a pur regulon gene. J. Biol. Chem. 1985, 260, 10378–10387. [Google Scholar] [CrossRef]
- Rolfes, R.J.; Zalkin, H. Escherichia coli gene purR encoding a repressor protein for purine nucleotide synthesis. Cloning, nucleotide sequence, and interaction with the purF operator. J. Biol. Chem. 1988, 263, 19653–19661. [Google Scholar] [CrossRef]
- Aiba, A.; Mizobuchi, K. Nucleotide sequence analysis of genes purH and purD involved in the de novo purine nucleotide biosynthesis of Escherichia coli. J. Biol. Chem. 1989, 264, 21239–21246. [Google Scholar] [CrossRef]
- Flannigan, K.A.; Hennigan, S.H.; Vogelbacker, H.H.; Gots, J.S.; Smith, J.M. Purine biosynthesis in Escherichia coli K12: Structure and DNA sequence studies of the purHD locus. Mol. Microbiol. 1990, 4, 381–392. [Google Scholar] [CrossRef]
- Meng, L.M.; Kilstrup, M.; Nygaard, P. Autoregulation of PurR repressor synthesis and involvement of purR in the regulation of purB, purC, purL, purMN and guaBA expression in Escherichia coli. Eur. J. Biochem. 1990, 187, 373–379. [Google Scholar] [CrossRef]
- Tiedeman, A.A.; DeMarini, D.J.; Parker, J.; Smith, J.M. DNA sequence of the purC gene encoding 5′-phosphoribosyl-5-aminoimidazole-4-N-succinocarboxamide synthetase and organization of the dapA-purC region of Escherichia coli K-12. J. Bacteriol. 1990, 172, 6035–6041. [Google Scholar] [CrossRef]
- He, B.; Smith, J.M.; Zalkin, H. Escherichia coli purB gene: Cloning, nucleotide sequence, and regulation by purR. J. Bacteriol. 1992, 174, 130–136. [Google Scholar] [CrossRef]
- Watanabe, W.; Sampei, G.; Aiba, A.; Mizobuchi, K. Identification and sequence analysis of Escherichia coli purE and purK genes encoding 5′-phosphoribosyl-5-amino-4-imidazole carboxylase for de novo purine biosynthesis. J. Bacteriol. 1989, 171, 198–204. [Google Scholar] [CrossRef]
- Smith, J.M.; Daum, H.A., 3rd. Nucleotide sequence of the purM gene encoding 5′-phosphoribosyl-5-aminoimidazole synthetase of Escherichia coli K12. J. Biol. Chem. 1986, 261, 10632–10636. [Google Scholar] [CrossRef]
- Khorana, H.G.; Fernandes, J.F.; Kornberg, A. Pyrophosphorylation of ribose 5-phosphate in the enzymatic synthesis of 5-phosphorylribose 1-pyrophosphate. J. Biol. Chem. 1958, 230, 941–948. [Google Scholar] [CrossRef]
- Messenger, L.J.; Zalkin, H. Glutamine phosphoribosylpyrophosphate amidotransferase from Escherichia coli. Purification and properties. J. Biol. Chem. 1979, 254, 3382–3392. [Google Scholar] [CrossRef]
- Cheng, Y.S.; Shen, Y.; Rudolph, J.; Stern, M.; Stubbe, J.; Flannigan, K.A.; Smith, J.M. Glycinamide ribonucleotide synthetase from Escherichia coli: Cloning, overproduction, sequencing, isolation, and characterization. Biochemistry 1990, 29, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.L.; McCorkle, G.M.; Zalkin, H. purU, a source of formate for purT-dependent phosphoribosyl-N-formylglycinamide synthesis. J. Bacteriol. 1993, 175, 7066–7073. [Google Scholar] [CrossRef][Green Version]
- Sampei, G.; Mizobuchi, K. The organization of the purL gene encoding 5′-phosphoribosylformylglycinamide amidotransferase of Escherichia coli. J. Biol. Chem. 1989, 264, 21230–21238. [Google Scholar] [CrossRef]
- Mueller, E.J.; Oh, S.; Kavalerchik, E.; Kappock, T.J.; Meyer, E.; Li, C.; Ealick, S.E.; Stubbe, J. Investigation of the ATP binding site of Escherichia coli aminoimidazole ribonucleotide synthetase using affinity labeling and site-directed mutagenesis. Biochemistry 1999, 38, 9831–9839. [Google Scholar] [CrossRef]
- Schrimsher, J.L.; Schendel, F.J.; Stubbe, J.; Smith, J.M. Purification and characterization of aminoimidazole ribonucleotide synthetase from Escherichia coli. Biochemistry 1986, 25, 4366–4371. [Google Scholar] [CrossRef]
- Zhang, Y.; Morar, M.; Ealick, S.E. Structural biology of the purine biosynthetic pathway. Cell. Mol. Life Sci. CMLS 2008, 65, 3699–3724. [Google Scholar] [CrossRef]
- Nelson, S.W.; Binkowski, D.J.; Honzatko, R.B.; Fromm, H.J. Mechanism of action of Escherichia coli phosphoribosylaminoimidazolesuccinocarboxamide synthetase. Biochemistry 2005, 44, 766–774. [Google Scholar] [CrossRef]
- Wolan, D.W.; Cheong, C.G.; Greasley, S.E.; Wilson, I.A. Structural insights into the human and avian IMP cyclohydrolase mechanism via crystal structures with the bound XMP inhibitor. Biochemistry 2004, 43, 1171–1183. [Google Scholar] [CrossRef]
- Schirch, V.; Hopkins, S.; Villar, E.; Angelaccio, S. Serine hydroxymethyltransferase from Escherichia coli: Purification and properties. J. Bacteriol. 1985, 163, 1. [Google Scholar] [CrossRef]
- Okamura-Ikeda, K.; Ohmura, Y.; Fujiwara, K.; Motokawa, Y. Cloning and nucleotide sequence of the gcv operon encoding the Escherichia coli glycine-cleavage system. Eur. J. Biochem. 1993, 216, 539–548. [Google Scholar] [CrossRef]
- Stauffer, L.T.; Plamann, M.D.; Stauffer, G.V. Cloning and characterization of the glycine-cleavage enzyme system of Escherichia coli. Gene 1986, 44, 219–226. [Google Scholar] [CrossRef]
- Kikuchi, G. The glycine cleavage system: Composition, reaction mechanism, and physiological significance. Mol. Cell. Biochem. 1973, 1, 169–187. [Google Scholar] [CrossRef]
- Meedel, T.H.; Pizer, L.I. Regulation of one-carbon biosynthesis and utilization in Escherichia coli. J. Bacteriol. 1974, 118, 905–910. [Google Scholar] [CrossRef]
- Wilson, R.L.; Stauffer, L.T.; Stauffer, G.V. Roles of the GcvA and PurR proteins in negative regulation of the Escherichia coli glycine cleavage enzyme system. J. Bacteriol. 1993, 175, 5129–5134. [Google Scholar] [CrossRef]
- Stauffer, L.T.; Stauffer, G.V. Antagonistic Roles for GcvA and GcvB in hdeAB Expression in Escherichia coli. ISRN Microbiol. 2012, 2012, 697308. [Google Scholar] [CrossRef]
- Ju, X.; Fang, X.; Xiao, Y.; Li, B.; Shi, R.; Wei, C.; You, C. Small RNA GcvB Regulates Oxidative Stress Response of Escherichia coli. Antioxidants 2021, 10, 1774. [Google Scholar] [CrossRef]
- Danielsen, S.; Kilstrup, M.; Barilla, K.; Jochimsen, B.; Neuhard, J. Characterization of the Escherichia coli codBA operon encoding cytosine permease and cytosine deaminase. Mol. Microbiol. 1992, 6, 1335–1344. [Google Scholar] [CrossRef]
- De Haan, P.G.; Felix, H.S.; Peters, R. Mapping of the gene for cytosine deaminase on the Escherichia coli chromosome. Antonie Van Leeuwenhoek 1972, 38, 257–263. [Google Scholar] [CrossRef]
- Ahmad, S.I.; Pritchard, R.H. Location of gene specifying cytosine deaminase in Escherichia coli. Mol. Gen. Genet. MGG 1972, 118, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Shayanfar, S.; Broumand, A.; Pillai, S.D. Acid stress induces differential accumulation of metabolites in Escherichia coli O26:H11. J. Appl. Microbiol. 2018, 125, 1911–1919. [Google Scholar] [CrossRef] [PubMed]
- Drazic, A.; Kutzner, E.; Winter, J.; Eisenreich, W. Metabolic Response of Escherichia coli upon Treatment with Hypochlorite at Sub-Lethal Concentrations. PLoS ONE 2015, 10, e0125823. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhao, X.; Wu, J.; Lou, X.; Yang, H. Comparison of metabolic response between the planktonic and air-dried Escherichia coli to electrolysed water combined with ultrasound by 1H NMR spectroscopy. Food Res. Int. 2019, 125, 108607. [Google Scholar] [CrossRef]
- Mao, Q.; Liu, J.; Wiertzema, J.R.; Chen, D.; Chen, P.; Baumler, D.J.; Ruan, R.; Chen, C. Identification of Quinone Degradation as a Triggering Event for Intense Pulsed Light-Elicited Metabolic Changes in Escherichia coli by Metabolomic Fingerprinting. Metabolites 2021, 11, 102. [Google Scholar] [CrossRef]
- Guest, R.L.; Wang, J.; Wong, J.L.; Raivio, T.L. A Bacterial Stress Response Regulates Respiratory Protein Complexes to Control Envelope Stress Adaptation. J. Bacteriol. 2017, 199, 20. [Google Scholar] [CrossRef]
- Maurer, L.M.; Yohannes, E.; Bondurant, S.S.; Radmacher, M.; Slonczewski, J.L. pH regulates genes for flagellar motility, catabolism, and oxidative stress in Escherichia coli K-12. J. Bacteriol. 2005, 187, 304–319. [Google Scholar] [CrossRef]
- Nunn, W.D.; Simons, R.W. Transport of long-chain fatty acids by Escherichia coli: Mapping and characterization of mutants in the fadL gene. Proc. Natl. Acad. Sci. USA 1978, 75, 3377–3381. [Google Scholar] [CrossRef]
- Feng, Y.; Cronan, J.E. A new member of the Escherichia coli fad regulon: Transcriptional regulation of fadM (ybaW). J. Bacteriol. 2009, 191, 6320–6328. [Google Scholar] [CrossRef]
- Agrawal, S.; Jaswal, K.; Shiver, A.L.; Balecha, H.; Patra, T.; Chaba, R. A genome-wide screen in Escherichia coli reveals that ubiquinone is a key antioxidant for metabolism of long-chain fatty acids. J. Biol. Chem. 2017, 292, 20086–20099. [Google Scholar] [CrossRef]
- Jaswal, K.; Shrivastava, M.; Roy, D.; Agrawal, S.; Chaba, R. Metabolism of long-chain fatty acids affects disulfide bond formation in Escherichia coli and activates envelope stress response pathways as a combat strategy. PLoS Genet. 2020, 16, e1009081. [Google Scholar] [CrossRef]
- Fath, M.J.; Mahanty, H.K.; Kolter, R. Characterization of a purF operon mutation which affects colicin V production. J. Bacteriol. 1989, 171, 3158–3161. [Google Scholar] [CrossRef]
- Cho, B.K.; Federowicz, S.A.; Embree, M.; Park, Y.S.; Kim, D.; Palsson, B. The PurR regulon in Escherichia coli K-12 MG1655. Nucleic Acids Res. 2011, 39, 6456–6464. [Google Scholar] [CrossRef]
- Warr, A.R.; Giorgio, R.T.; Waldor, M.K. Genetic analysis of the role of the conserved inner membrane protein CvpA in EHEC resistance to deoxycholate. J. Bacteriol. 2020, 203, 6. [Google Scholar] [CrossRef]
- Molloy, M.P.; Herbert, B.R.; Slade, M.B.; Rabilloud, T.; Nouwens, A.S.; Williams, K.L.; Gooley, A.A. Proteomic analysis of the Escherichia coli outer membrane. Eur. J. Biochem. 2000, 267, 2871–2881. [Google Scholar] [CrossRef]
- Chen, H.; Wilson, J.; Ercanbrack, C.; Smith, H.; Gan, Q.; Fan, C. Genome-Wide Screening of Oxidizing Agent Resistance Genes in Escherichia coli. Antioxidants 2021, 10, 861. [Google Scholar] [CrossRef]
- Adams, M.M.; Allison, G.E.; Verma, N.K. Type IV O antigen modification genes in the genome of Shigella flexneri NCTC 8296. Microbiology 2001, 147, 851–860. [Google Scholar] [CrossRef]
- Cheng, Z.F.; Deutscher, M.P. Purification and characterization of the Escherichia coli exoribonuclease RNase R. Comparison with RNase II. J. Biol. Chem. 2002, 277, 21624–21629. [Google Scholar] [CrossRef]
- Seshasayee, A.S.; Fraser, G.M.; Babu, M.M.; Luscombe, N.M. Principles of transcriptional regulation and evolution of the metabolic system in E. coli. Genome Res. 2009, 19, 79–91. [Google Scholar] [CrossRef]
- Grainger, D.C.; Hurd, D.; Harrison, M.; Holdstock, J.; Busby, S.J. Studies of the distribution of Escherichia coli cAMP-receptor protein and RNA polymerase along the E. coli chromosome. Proc. Natl. Acad. Sci. USA 2005, 102, 17693–17698. [Google Scholar] [CrossRef]
- Grainger, D.C.; Aiba, H.; Hurd, D.; Browning, D.F.; Busby, S.J. Transcription factor distribution in Escherichia coli: Studies with FNR protein. Nucleic Acids Res. 2007, 35, 269–278. [Google Scholar] [CrossRef]
- Cho, B.K.; Barrett, C.L.; Knight, E.M.; Park, Y.S.; Palsson, B. Genome-scale reconstruction of the Lrp regulatory network in Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 19462–19467. [Google Scholar] [CrossRef]
- Anderson, B.W.; Schumacher, M.A.; Yang, J.; Turdiev, A.; Turdiev, H.; Schroeder, J.W.; He, Q.; Lee, V.T.; Brennan, R.G.; Wang, J.D. The nucleotide messenger (p)ppGpp is an anti-inducer of the purine synthesis transcription regulator PurR in Bacillus. Nucleic Acids Res. 2022, 50, 847–866. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stressor | Subtype | Media |
|---|---|---|
| Heat | GSE15534 (45 °C 10 min) | M9 complete medium |
| GSE40557 (58 °C f = 2, 58 °C f = 3, 60 °C f = 3, 71 °C) | Brain Heart Infusion (BHI) | |
| GSE42675 (42 °C 10 min) | M9 minimal medium | |
| GSE11041 (46 °C till 1 × 107 CFU/mL) | Tryptone soya broth (TSB) | |
| GSE20305 (45 °C 10 min) | Modified morpholinopropane sulfonate (MOPS) minimal medium | |
| Oxidative stress | GSE20305 (H2O2 10 min and 90 min) | Modified MOPS minimal medium |
| GSE61736 (H2O2 till OD600 0.3–0.4) | MOPS medium | |
| GSE56133 (10 uM H2O2 60 min) | Luria-Bertani (LB) broth | |
| GSE58176 (Tellurite 0.5 µg/mL 15 min) | LB broth | |
| GSE19370 (300 μM peroxynitrite 5 min, 300 μM H2O2 5 min) | Defined media containing 2 mM glycerol | |
| Cold | GSE 11041 (15 °C till 1 × 107 CFU/mL) | TSB |
| GSE61736 (15 °C 4 h) | LB broth | |
| GSE20305 (16 °C 10 min, 30 min and 90 min) | Modified MOPS minimal medium | |
| Nitrosative | GSE60522 (Dipropylenetriamine (DPTA) 10 min) | MOPS minimal medium |
| Antibiotics | GSE56133 (Ampicillin 1 h, Gentamycin 1 h, Kanamycin 1 h, Norfloxacin 1 h) | LB broth |
| GSE57084 (Enorfloxacin, Tetracycline) | Fresh Evans medium | |
| GSE47221 (Amoxicillin) | Fresh Evans medium | |
| GSE37026 (Colicin 30 min and 60 min) | LB broth | |
| GSE10160 (Cefsulodin 10 μg/mL 5 min, 20 min and 40 min, Cefsulodin 60 μg/mL 10 min, Mecillinam 0.03 μg/mL 5 min, 20 min and 40 min, Mecillinam 0.3 μg/mL 60 min) | LB broth |
| Functional Category | Function Type | N Genes | Names of Genes | Stress | ||||
|---|---|---|---|---|---|---|---|---|
| Antibiotic | Oxidative | Cold | Heat | Nitrosative | ||||
| Metabolic processes | Transferses | 2 | tusb, rlmB | × | × | × | × | |
| Hydrolases | 2 | ycaC, yliE | × | × | × | × | ||
| Amino acids biosynthesis and metabolism | 2 | ilvM, fbaB | × | × | × | × | × | |
| Oxidoreductase | 1 | Ndh | × | × | × | × | ||
| Lipid metabolism | 1 | pgpC | × | × | × | × | ||
| Starch and sucrose metabolism | 1 | treF | × | × | × | × | ||
| Secondary metabolites biosynthesis | 1 | cadA | × | × | × | × | ||
| Cellular response | DNA damage | 8 | ycgB, blc, gadX, gadW, yqjI, iraD, sulA, ybaV | × | × | × | × | × |
| Stress | 4 | rmf, uspG, mqsA, bolA | × | × | × | × | × | |
| Oxidative stress | 1 | grxA | × | × | × | × | ||
| Acid stress | 5 | slp, ydeP, ygaC, ycgZ, mgrB | × | × | × | × | ||
| Osmotic stress | 1 | osmB | × | × | × | × | × | |
| Phosphate starvation | 1 | psiE | × | × | × | × | ||
| Heat | 1 | ldhA | × | × | × | × | ||
| Nitrogen starvation | 1 | yeaG | × | × | × | × | ||
| Trasnport | Amino acid | 3 | leuE, gltP, alaE | × | × | × | × | × |
| Sugar | 1 | bglF | × | × | × | × | ||
| Iron | 2 | fecI, fepD | × | × | × | × | × | |
| Metal ion | 2 | corA, zntA | × | × | × | × | × | |
| Bacterial secretion | 1 | gspO | × | × | × | × | ||
| Multidug efflux | 1 | emrD | × | × | × | × | ||
| Others | 3 | tehA, ybhS, ytfL | × | × | × | × | ||
| Cell adhesion and biofilm formation | 10 | ychH, yhcN, yodD, bssS, ycfJ, ymgA, dgcZ, cnu, tomB, ybfG | × | × | × | × | × | |
| Transcription regulation | 4 | eutR, yddm, zntR, dsdC | × | × | × | × | × | |
| DNA repair | 5 | recF, nrdE, nrdF, nrdH, nrdI | × | × | × | × | ||
| Motility | Formation and regulation | 1 | flgL | × | × | × | × | |
| Others | 3 | bluf, sra, essQ | × | × | × | × | ||
| Uncharacterized proteins | 15 | yqfA, yhfG, yhhA, ybgS, arpA, yqaE, yfdY, yaiY, yebE, ydiE, yjcB, yiiX, ycjF, yihF, yidX | × | × | × | × | × | |
| Functional Category | Function Type | N Genes | Names of Genes | Stress | ||||
|---|---|---|---|---|---|---|---|---|
| Antibiotic | Oxidative | Cold | Heat | Nitrosative | ||||
| Transport | B lactam resistance | 3 | oppB, oppC, ampG | × | × | × | × | |
| Electron | 1 | rsxC | × | × | × | × | ||
| Iron | 2 | fecC, efeo | × | × | × | × | × | |
| Nucleoside | 1 | tsx | × | × | × | × | ||
| Fatty acid | 1 | fadL | × | × | × | × | × | |
| Sugar | 4 | gatA, fruB, ptsG, mglA | × | × | × | × | × | |
| Amino acid | 10 | livM, livG, livF, artQ, artp, artJ, lysP, plaP, pheP, codB | × | × | × | × | ||
| Others | 7 | xanP, uraA, yeiB, thiQ, thiP, potB, potD | × | a × | × | × | × | |
| Metabolic pathways | Purine biosynthesis | 12 | purD, purL, purB, purH, purM, purN, prs, purE, purT, purK, purF, purC | × | × | × | × | × |
| Antibiotics biosynthesis | 7 | accC, gph, aceE, icd, ilvC, gcd, dapB | × | × | × | × | ||
| Pyrimidine metabolism | 8 | pyrB, pyrD, pyrI, carB, carA, upp, pyrC, coda | × | × | × | × | × | |
| Argnine biosynthesis | 3 | argD, gdhA, alaA | × | × | × | × | ||
| Glycine, serine and threonine metabolism | 6 | thrA, thrB, gcvT, gcvP, lysC, glyA | × | × | × | × | ||
| Cysteine methionine metabolism | 3 | ynjE, metE, metC | × | × | × | × | ||
| Amino acid biosynthesis | 2 | trpE, cysJ | × | × | × | × | × | |
| Lipopolysaccharide biosynthesis | 4 | eptC, waaL, waaC, lpxH | × | × | × | × | ||
| Pyruvate metabolism | 2 | pfo, aldA | × | × | × | × | × | |
| Transferase | 4 | hsdM, gtrB, lipB, opgH | × | × | × | × | × | |
| Hydrolase | 8 | mgtA, frmB, ydcP, hypB, ybhC, yliE, ravA, rnb | × | × | × | × | × | |
| Oxidative phosphorylation | 8 | atpF, nuoJ, nuoC, nuoE, nuoF, nuoI, nuoH, cyoB | × | × | × | × | ||
| Others | 6 | speA, bioD, pntA, gatD, ycaO, hypD | × | × | × | × | ||
| Cellular Response | Stimulus | 2 | tsgA, borD | × | × | × | × | × |
| Acidic pH | 3 | evgS, yagU, yqgB | × | × | × | × | ||
| Motility | Flagellum biogenesis and protien export | 3 | fliP, flhA, flhB | × | × | × | × | |
| Flagellar assembly | 4 | fliM, flgH, flgG, fliK | × | × | × | × | ||
| Peptidoglycan | Biosynthetic process | 4 | dacA, mipA, lpoA, murI | × | × | × | × | × |
| Transcription | Regulation | 3 | fis, mprA, suhB | × | × | × | × | × |
| Others | 3 | cvpA, gtrS, yeiP | × | × | × | × | × | |
| Uncharacterized | Proteins | 3 | ymfI, ydiJ, yedE | × | × | × | × | |
| Function | Function Type | N Genes | Name of Genes |
|---|---|---|---|
| Metabolic processes | Transferses | 21 | tusb, rlmB, yjgX, ydiU, elaA, yafK, tusE, ldtc, alaC, trmN, opgE, tdcD, wecH, opgC, yjaB, fic, lnt, maa, yafE, rlmE, rlmG |
| Hydrolases | 15 | ycaC, yliE, yfcI, yadD, sixA, casE, ygbF, glpG, rnd, yhjJ, yahA, cdd, dbpA, phoA, fes | |
| Amino acids biosynthesis and metabolism | 16 | ilvG, metA, gltA, asnA, ilvC, tdcG, argI, argF, argH, ilvM, acnA, fbaB, puuB, puuA, eutQ, yhfx | |
| Oxidoreductase | 11 | nrdG, nrdD, dadA, mhpB, dmsC, torZ, dusC, nirB, qorA, ndh, hcr | |
| Lipopolysaccharide biosynthesis | 7 | wcaA, waaZ, wcaF, wzxC, wzzB, wcaD, wcaE | |
| Lipid metabolism | 6 | yihG, pgpC, clsC, yegS, fadD, yiiD | |
| Ascorbate and aldarate metabolism | 5 | garD, gudD, lgoD, sgbE, lyxK | |
| Carbohydrate metabolism | 5 | fsaB, araC, fucR, mlc, glmS | |
| Starch and sucrose metabolism | 5 | glgA, amyA, bglB, treF, pgm | |
| Secondary metabolites biosynthesis | 4 | cadA, entF, cysN, ubiX | |
| Carbon metabolism | 3 | acs, gntK, mqo | |
| Galactose metabolism | 2 | dgoK, ebgC | |
| Other pathways | 6 | ybdZ, thiC, pyrF, atpC, torY, hofM, | |
| Cellular response | DNA damage | 19 | ycgB, blc, gadX, gadW, yqjI, iraD, sulA, ydjM, sbmC, yidQ, yqiJ, yedV, betT, elaB, yhcF, rcnB, dinD, yedk, dinF |
| Stress | 15 | rmf, uspG, mqsA, bolA, cbpA, cbpM, yjbJ, uspB, nemR, rclR, rclB, cpxP, pspG, rpoS, srkA | |
| Oxidative stress | 12 | grxA, msrA, wrbA, degP, sufA, clpA, yhbO, rseC, soxR, sufE, yfcG, ytfK | |
| Antibiotic | 7 | bcr, entS, mdtO, ydaC, yibA, yojI, ymdB | |
| Acid stress | 7 | slp, ydeP, ygaC, iraM, frc, ycgZ, mgrB | |
| Heat | 7 | ybeD, rpoH, htpG, hspQ, eutD, hslJ, ldhA | |
| Cold | 5 | ydfK, ynaE, cspF, cspD, cspG | |
| Osmotic stress | 4 | otsB, otsA, osmB, osmY | |
| Phosphate starvation | 4 | psiE, waaH, appY, appA | |
| Nitrogen starvation | 3 | ycjX, yeaH, yeaG | |
| pH elevation | 3 | gadB, adiC, gadA | |
| Starvation | 2 | gpp, dps | |
| Trasnport | Amino acid | 13 | leuE, gltP, alaE, ydgI, yehX, yifK, potF, proY, proX, artJ, ydjN, proP, yhdW |
| Sugar | 15 | bglF, lgoT, nanT, kdgT, srlA, ascF, fruA, araF, xylH, frvB, gntP, alsA, yicJ, ytfT, glvC | |
| Iron | 7 | fecI, fecR, fiu, fepC, fepB, fepG, fepD | |
| Metal ion | 4 | corA, zntA, mntH, mgtA | |
| Bacterial secretion | 3 | yidC, gspK, gspO | |
| Multidug efflux | 3 | emrD, mdtJ, mdtI | |
| Others | 21 | tehA, ybhS, mlaE, yhbE, yhjD, yjhF, yfdV, ygaY, ycgH, araJ, yphD, hsrA, garP, nepI, cusB, adeP, yidK, yhhQ, mscM, ytfL, phoE | |
| Membrane Component | 25 | ypjA, ppdD, nfrB, yaiO, wzyE, yedR, yfjD, ygbE, yhdU, fliR, yfbV, fxsA, yqiK, yqjE, alx, ycdZ, ydhI, ybhL, yeiH, yohJ, yhfL, yjiJ, creD, ychE, ydgK | |
| Cell adhesion and biofilm formation | 31 | dosC, ychH, yhcN, yodD, bssS, ycfJ, ymgA, dgcZ, cnu, tomB, ybfG, tqsA, csgF, ymgC, ariR, mqsR, csgB, bhsA, sdiA, yjaA, yghO, fimG, elfA, sfmD, ydeR, glgS, yjcZ, yfcU, ydeT, fimD, ybgQ | |
| Transcription regulation | 24 | csiE, dctR, nsrR, hcaR, stpA, arsR, soxS, sxy, hyfR, yidL, norR, rof, yhfY, yieP, greA, chaB, ydjF, yiaG, eutR, yddm, zntR, dsdC, Crl, ykgA | |
| Signal peptide | 19 | yibG, ycbK, rzpQ, yncD, ybbC, sslE, ydbL, yjfY, ymgD, ybaY, ygdI, ypfG, eco, ypeC, yqjC, ytfJ, yjfN, ykfB, ybcW | |
| DNA repair | 18 | recF, yegP, umuD, mutM, uvrA, phr, lexA, recQ, recN, polB, alkA, yebG, nrdE, nrdF, nrdI, nrdH, yhcG, prlC | |
| Motility | Formation and regulation | 2 | flgL, ydiV |
| Toxin-Antitoxin | 2 | hokD, higA | |
| Others | 16 | infA, rdlD, rybB, ryhB, rttR, insZ, msyB, TfaS, ftsA, tfaD, bluf, sra, rsxA, ybaV, essQ, essD | |
| Uncharacterized proteins | 64 | aroM, yqfA, yhfG, yhhA, ybgS, arpA, yqaE, yfdY, yaiY, yebE, ydiE, yjcB, yiiX, ycjF, yihF, yidX, yccJ, yifE, YheO, yffB, ydbA, ygeN, yfaH, yrhA, yacL, ygeQ, YifN, YjhE, YkiA, yehH, yehQ, ycgX, yjfK, yhiJ, yidB, ymfD, yddH, ydeJ, ynbE, yrbL, tpr, ycbJ, yhcO, sgcQ, yfbP, yaeH, ydiH, yecT, yfbN, ygbA, yodC, tfaP, ytfI, rem, yagN, yfdT, yffL, yggI, yfeS, ymgG, ydcy, ymjA, yqeB, yccM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelwahed, E.K.; Hussein, N.A.; Moustafa, A.; Moneib, N.A.; Aziz, R.K. Gene Networks and Pathways Involved in Escherichia coli Response to Multiple Stressors. Microorganisms 2022, 10, 1793. https://doi.org/10.3390/microorganisms10091793
Abdelwahed EK, Hussein NA, Moustafa A, Moneib NA, Aziz RK. Gene Networks and Pathways Involved in Escherichia coli Response to Multiple Stressors. Microorganisms. 2022; 10(9):1793. https://doi.org/10.3390/microorganisms10091793
Chicago/Turabian StyleAbdelwahed, Eman K., Nahla A. Hussein, Ahmed Moustafa, Nayera A. Moneib, and Ramy K. Aziz. 2022. "Gene Networks and Pathways Involved in Escherichia coli Response to Multiple Stressors" Microorganisms 10, no. 9: 1793. https://doi.org/10.3390/microorganisms10091793
APA StyleAbdelwahed, E. K., Hussein, N. A., Moustafa, A., Moneib, N. A., & Aziz, R. K. (2022). Gene Networks and Pathways Involved in Escherichia coli Response to Multiple Stressors. Microorganisms, 10(9), 1793. https://doi.org/10.3390/microorganisms10091793