High-Resolution Composition Analysis of an Inactivated Polyvalent Foot-and-Mouth Disease Vaccine



Abstract
1. Introduction
2. Results
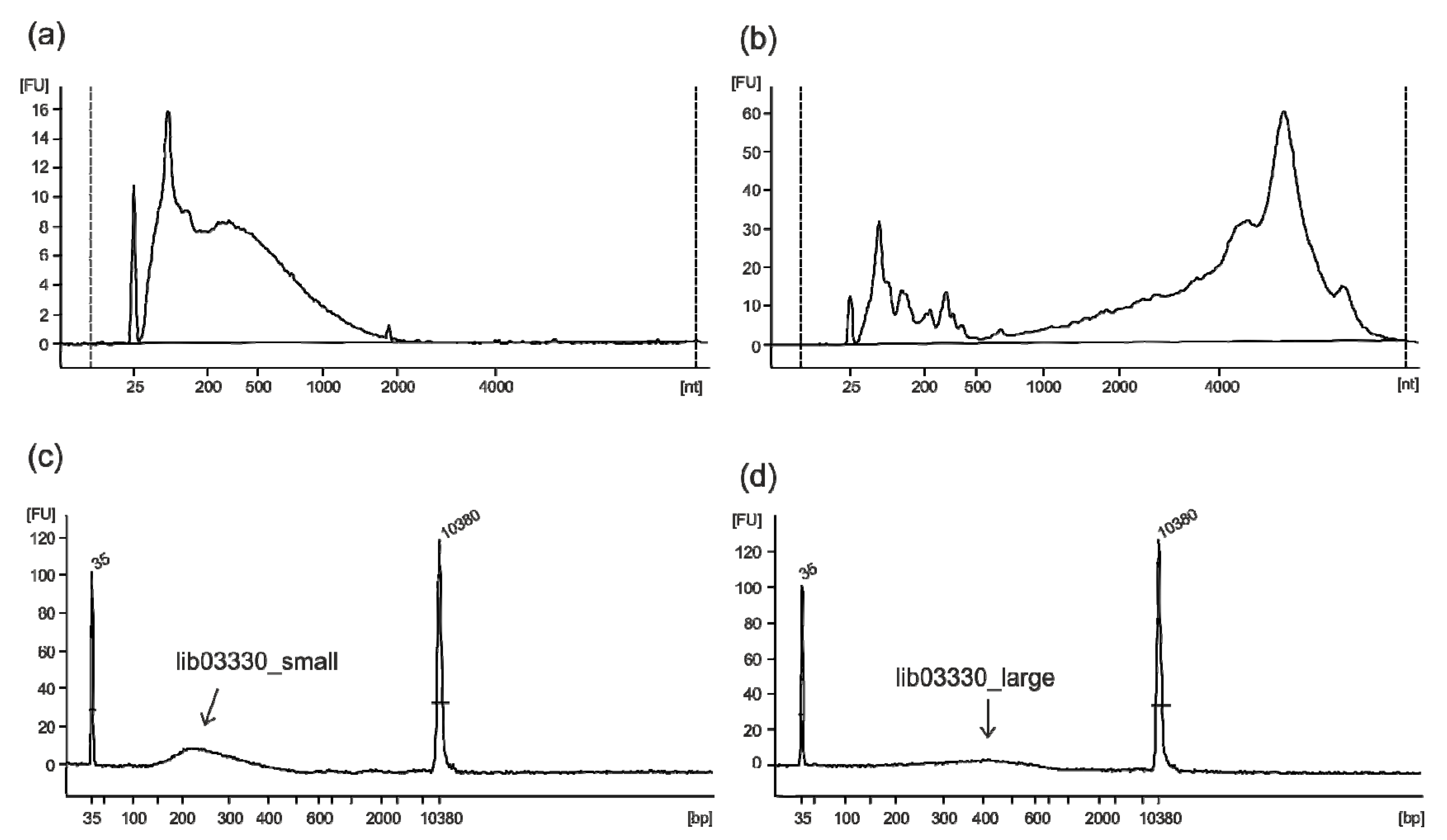
2.1. FMD Vaccines Contain Highly Fragmented Nucleic Acids due to BEI Inactivation
2.2. Vaccine Purity
2.3. Attempted Identification of Strain Composition Using Megablast
2.4. Reference-Guided Assembly Approach on Full Genomes
2.5. De Novo Assembly of Full-Genomes
2.6. Distribution of FMDV RNA
3. Discussion
4. Materials and Methods
4.1. Nucleic Acid Extraction, Library Preparation, and Sequencing
4.2. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klein, J. Understanding the molecular epidemiology of foot-and-mouth-disease virus. Infect. Genet. Evol. 2009, 9, 153–161. [Google Scholar] [CrossRef]
- Alexandersen, S.; Mowat, N. Foot-and-mouth disease: Host range and pathogenesis. Curr. Top. Microbiol. Immunol. 2005, 288, 9–42. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.M.Z.H.; Rahman, M.F.; Rahman, M.B.; Rahman, M.M. Foot and mouth disease and its effects on morbidity, mortality, milk yield and draft power in Bangladesh. Asian-Aust. J. Anim. Sci. 1993, 6, 423–426. [Google Scholar] [CrossRef]
- FAO. Foot-and-Mouth Disease Situation—Monthly Report September 2016; FAO: Roma, Italy, 2016. [Google Scholar]
- Stenfeldt, C.; Eschbaumer, M.; Rekant, S.I.; Pacheco, J.M.; Smoliga, G.R.; Hartwig, E.J.; Rodriguez, L.L.; Arzt, J. The Foot-and-Mouth Disease Carrier State Divergence in Cattle. J. Virol. 2016, 90, 6344–6364. [Google Scholar] [CrossRef] [PubMed]
- USDA. Factsheet: Foot and Mouth Disease and Vaccine Use; Aphis, Ed.; USDA: Washington, DC, USA, 2018.
- Cox, S.J.; Voyce, C.; Parida, S.; Reid, S.M.; Hamblin, P.A.; Hutchings, G.; Paton, D.J.; Barnett, P.V. Effect of emergency FMD vaccine antigen payload on protection, sub-clinical infection and persistence following direct contact challenge of cattle. Vaccine 2006, 24, 3184–3190. [Google Scholar] [CrossRef] [PubMed]
- Parida, S. Vaccination against foot-and-mouth disease virus: Strategies and effectiveness. Exp. Rev. Vaccines 2009, 8, 347–365. [Google Scholar] [CrossRef]
- Gao, Y.; Sun, S.Q.; Guo, H.C. Biological function of Foot-and-mouth disease virus non-structural proteins and non-coding elements. Virol. J. 2016, 13, 107. [Google Scholar] [CrossRef]
- Domingo, E. Chapter 2—Molecular basis of genetic variation of viruses: Error-prone replication. In Virus as Populations; Domingo, E., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 35–71. [Google Scholar] [CrossRef]
- Domingo, E.; Baranowski, E.; Escarmís, C.; Sobrino, F. Foot-and-mouth disease virus. Comp. Immunol Microbiol. Infect. Dis. 2002, 25, 297–308. [Google Scholar] [CrossRef]
- Fernandez-Sainz, I.; Gavitt, T.D.; Koster, M.; Ramirez-Medina, E.; Rodriguez, Y.Y.; Wu, P.; Silbart, L.K.; de Los Santos, T.; Szczepanek, S.M. The VP1 G-H loop hypervariable epitope contributes to protective immunity against Foot and Mouth Disease Virus in swine. Vaccine 2019, 37, 3435–3442. [Google Scholar] [CrossRef]
- Kitching, R.P.; Knowles, N.J.; Samuel, A.R.; Donaldson, A.I. Development of foot-and-mouth disease virus strain characterisation—A review. Trop. Anim. Health Prod. 1989, 21, 153–166. [Google Scholar] [CrossRef]
- Singh, R.K.; Sharma, G.K.; Mahajan, S.; Dhama, K.; Basagoudanavar, S.H.; Hosamani, M.; Sreenivasa, B.P.; Chaicumpa, W.; Gupta, V.K.; Sanyal, A. Foot-and-Mouth Disease Virus: Immunobiology, Advances in Vaccines and Vaccination Strategies Addressing Vaccine Failures-An Indian Perspective. Vaccines 2019, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Ullah, H.; Siddique, M.A.; Al Amin, M.; Das, B.C.; Sultana, M.; Hossain, M.A. Re-emergence of circulatory foot-and-mouth disease virus serotypes Asia1 in Bangladesh and VP1 protein heterogeneity with vaccine strain IND 63/72. Lett. Appl. Microbiol. 2015, 60, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Paton, D.; Duffy, S.; Bartels, C.; Knight-Jones, T. Foot and Mouth Disease Vaccination and Post-Vaccination Monitoring; The Food and Agriculture Organization of the United Nations & World Organisation for Animal Health: Roma, Italy; Paris, France, 2016. [Google Scholar]
- Paton, D.J.; Valarcher, J.F.; Bergmann, I.; Matlho, O.G.; Zakharov, V.M.; Palma, E.L.; Thomson, G.R. Selection of foot and mouth disease vaccine strains—A review. Rev. Sci. Tech. 2005, 24, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Je, S.H.; Kwon, T.; Yoo, S.J.; Lee, D.U.; Seo, S.W.; Byun, J.J.; Shin, J.Y.; Lyoo, Y.S. Genetic identification and serological evaluation of commercial inactivated foot-and-mouth disease virus vaccine in pigs. Clin. Exp. Vaccine Res. 2018, 7, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Amaral-Doel, C.M.; Owen, N.E.; Ferris, N.P.; Kitching, R.P.; Doel, T.R. Detection of foot-and-mouth disease viral sequences in clinical specimens and ethyleneimine-inactivated preparations by the polymerase chain reaction. Vaccine 1993, 11, 415–421. [Google Scholar] [CrossRef]
- Ludi, A.B.; Horton, D.L.; Li, Y.; Mahapatra, M.; King, D.P.; Knowles, N.J.; Russell, C.A.; Paton, D.J.; Wood, J.L.; Smith, D.J.; et al. Antigenic variation of foot-and-mouth disease virus serotype A. J. Gen. Virol. 2014, 95, 384–392. [Google Scholar] [CrossRef]
- Scheuch, M.; Höper, D.; Beer, M. RIEMS: A software pipeline for sensitive and comprehensive taxonomic classification of reads from metagenomics datasets. BMC Bioinform. 2015, 16, 69. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Park, J.H.; Tark, D.; Lee, K.N.; Chun, J.E.; Lee, H.S.; Ko, Y.J.; Kye, S.J.; Kim, Y.J.; Oem, J.K.; Ryoo, S.; et al. Control of type O foot-and-mouth disease by vaccination in Korea, 2014-2015. J. Vet. Sci. 2018, 19, 271–279. [Google Scholar] [CrossRef]
- Mahapatra, M.; Parida, S. Foot and mouth disease vaccine strain selection: Current approaches and future perspectives. Exp. Rev. Vaccines 2018, 17, 577–591. [Google Scholar] [CrossRef]
- Park, J.H. Requirements for improved vaccines against foot-and-mouth disease epidemics. Clin. Exp. Vaccine Res. 2013, 2, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Mee, E.T.; Minor, P.D.; Martin, J. High resolution identity testing of inactivated poliovirus vaccines. Vaccine 2015, 33, 3533–3541. [Google Scholar] [CrossRef] [PubMed]
- Sarcey, E.; Serres, A.; Tindy, F.; Chareyre, A.; Ng, S.; Nicolas, M.; Vetter, E.; Bonnevay, T.; Abachin, E.; Mallet, L. Quantifying low-frequency revertants in oral poliovirus vaccine using next generation sequencing. J. Virol. Methods 2017, 246, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.; Dill, V.; Shalaby, M.A.; Eschbaumer, M.; Bohlken-Fascher, S.; Hoffmann, B.; Czerny, C.P.; Abd El Wahed, A. Serotyping of foot-and-mouth disease virus using oxford nanopore sequencing. J. Virol. Methods 2019, 263, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Logan, G.; Freimanis, G.L.; King, D.J.; Valdazo-Gonzalez, B.; Bachanek-Bankowska, K.; Sanderson, N.D.; Knowles, N.J.; King, D.P.; Cottam, E.M. A universal protocol to generate consensus level genome sequences for foot-and-mouth disease virus and other positive-sense polyadenylated RNA viruses using the Illumina MiSeq. BMC Genom. 2014, 15, 828. [Google Scholar] [CrossRef] [PubMed]
- Moser, L.A.; Ramirez-Carvajal, L.; Puri, V.; Pauszek, S.J.; Matthews, K.; Dilley, K.A.; Mullan, C.; McGraw, J.; Khayat, M.; Beeri, K.; et al. A Universal Next-Generation Sequencing Protocol To Generate Noninfectious Barcoded cDNA Libraries from High-Containment RNA Viruses. mSystems 2016, 1, e00039-15. [Google Scholar] [CrossRef]
- Wylezich, C.; Papa, A.; Beer, M.; Höper, D. A Versatile Sample Processing Workflow for Metagenomic Pathogen Detection. Sci. Rep. 2018, 8, 13108. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Vaccine Component | Closest Relative in the Nucleotide Database | |||
|---|---|---|---|---|
| Serotype | Strain | Accession No. | Nucleotide Identity [%] | |
| Component 1 | O | o1manisa iso87 | AY593823.1 | 99 |
| Component 2 | O | Akesu/58 | AF511039.1 | 91 |
| Component 3 | A | a22iraq-95 iso95 | AY593762.1 | 99 |
| Component 4 | A | Malaysia 97 | KJ933864.1 | 99 |
| Component 5 | Asia1 | As1/Shamir/89 | JF739177.1 | 100 |
| Component 1 | Component 2 | Component 3 | Component 4 | Component 5 | |
|---|---|---|---|---|---|
| Component 1 | 100.0 | 89.4 | 86.0 | 85.8 | 85.8 |
| Component 2 | 89.4 | 100.0 | 85.0 | 84.8 | 84.8 |
| Component 3 | 86.0 | 85.0 | 100.0 | 89.3 | 85.7 |
| Component 4 | 85.8 | 84.8 | 89.3 | 100.0 | 85.7 |
| Component 5 | 85.8 | 84.8 | 85.7 | 85.7 | 100.0 |
| Reference | Highly Stringent Mapping 1 | Less Stringent Mapping 2 | ||
|---|---|---|---|---|
| Unique Matching Reads * | % of All Reads | Unique Matching Reads * | % of All Reads | |
| Component 1 | 29,823 | 49.9 | 32,452 | 54.3 |
| Component 5 | 6635 | 11.1 | 7920 | 13.2 |
| Component 4 | 6439 | 10.8 | 7917 | 13.2 |
| Component 3 | 5320 | 8.9 | 6489 | 10.9 |
| Component 2 | 3242 | 5.4 | 3934 | 6.6 |
| Total | 51,459 | 86.1 | 58,712 | 98.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forth, L.F.; Höper, D.; Beer, M.; Eschbaumer, M. High-Resolution Composition Analysis of an Inactivated Polyvalent Foot-and-Mouth Disease Vaccine. Pathogens 2020, 9, 63. https://doi.org/10.3390/pathogens9010063
Forth LF, Höper D, Beer M, Eschbaumer M. High-Resolution Composition Analysis of an Inactivated Polyvalent Foot-and-Mouth Disease Vaccine. Pathogens. 2020; 9(1):63. https://doi.org/10.3390/pathogens9010063
Chicago/Turabian StyleForth, Leonie F., Dirk Höper, Martin Beer, and Michael Eschbaumer. 2020. "High-Resolution Composition Analysis of an Inactivated Polyvalent Foot-and-Mouth Disease Vaccine" Pathogens 9, no. 1: 63. https://doi.org/10.3390/pathogens9010063
APA StyleForth, L. F., Höper, D., Beer, M., & Eschbaumer, M. (2020). High-Resolution Composition Analysis of an Inactivated Polyvalent Foot-and-Mouth Disease Vaccine. Pathogens, 9(1), 63. https://doi.org/10.3390/pathogens9010063