Human Herpesvirus Sequencing in the Genomic Era: The Growing Ranks of the Herpetic Legion


Abstract
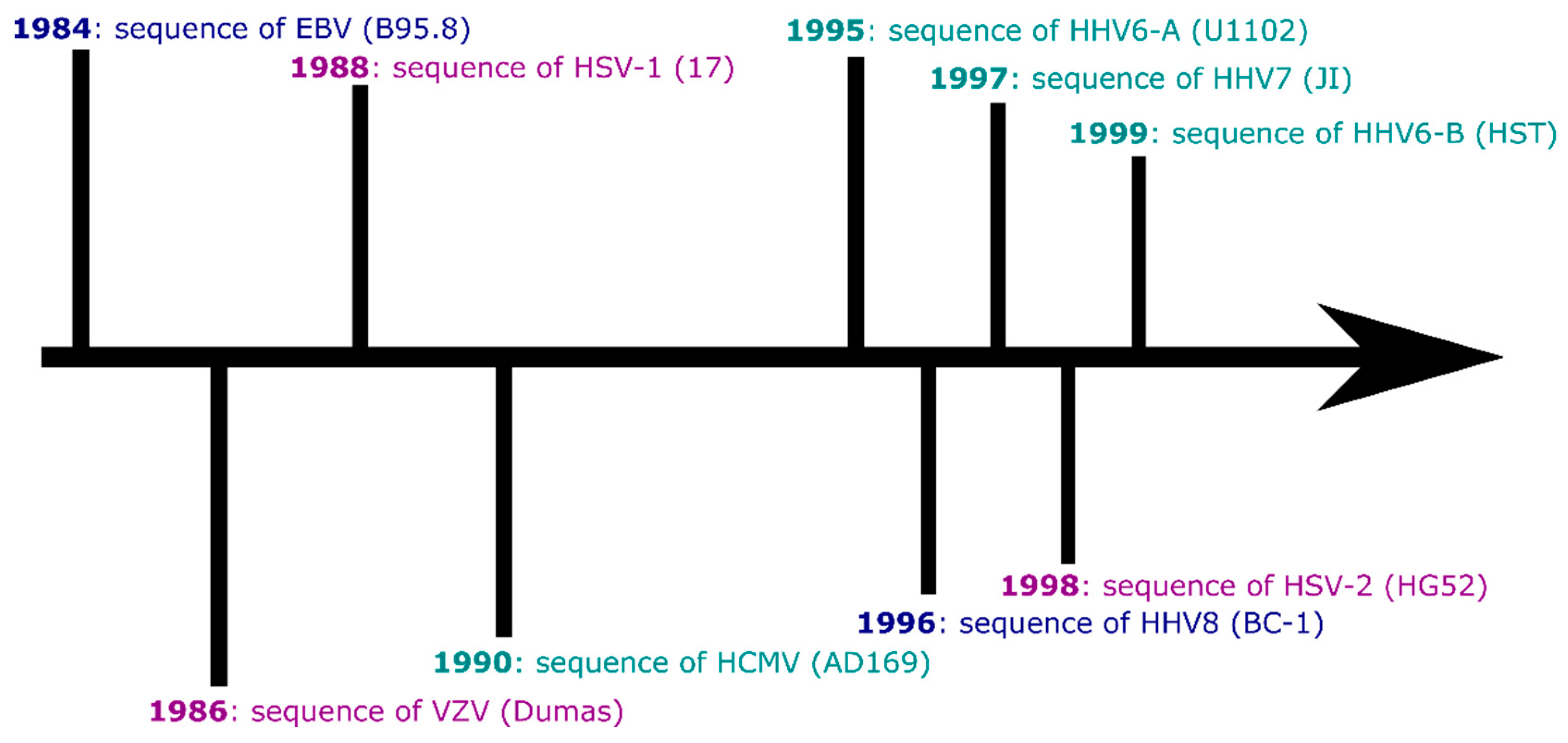
1. Introduction
2. Sequencing Herpesviruses: From PCR to High-Throughput and Target Enriched Sequencing
2.1. Alphaherpesviruses
2.1.1. Herpes Simplex Virus 1
2.1.2. Herpes Simplex Virus 2
2.1.3. Varicella-Zoster Virus
2.2. Betaherpesviruses
2.2.1. Cytomegalovirus
2.2.2. Human Herpesvirus 6A and 6B
HHV6A
HHV6B
2.2.3. Human Herpesvirus 7
2.3. Gammaherpesviruses
2.3.1. Epstein-Barr Virus
2.3.2. KSHV
2.4. Long-Read Sequencing of Herpesvirus Genomes and Getting to Finished Genomes
3. Conclusions and Future Directions for Human Herpesvirus Sequencing Studies
Funding
Acknowledgments
Conflicts of Interest
References
- Schowalter, R.M.; Pastrana, D.V.; Pumphrey, K.A.; Moyer, A.L.; Buck, C.B. Merkel Cell Polyomavirus and Two Previously Unknown Polyomaviruses Are Chronically Shed from Human Skin. Cell Host Microbe 2010, 7, 509–515. [Google Scholar] [CrossRef]
- Ismail, A.M.; Cui, T.; Dommaraju, K.; Singh, G.; Dehghan, S.; Seto, J.; Shrivastava, S.; Fedorova, N.B.; Gupta, N.; Stockwell, T.B.; et al. Genomic analysis of a large set of currently—And historically—Important human adenovirus pathogens. Emerg. Microbes Infect. 2018, 7, 1–22. [Google Scholar] [CrossRef]
- Frisque, R.J.; Bream, G.L.; Cannella, M.T. Human polyomavirus JC virus genome. J. Virol. 1984, 51, 458–469. [Google Scholar]
- Bravo, I.G.; Félez-Sánchez, M. Papillomaviruses: Viral evolution, cancer and evolutionary medicine. Evol. Med. Public Health 2015, 2015, 32–51. [Google Scholar] [CrossRef]
- Renzette, N.; Bhattacharjee, B.; Jensen, J.D.; Gibson, L.; Kowalik, T.F. Extensive Genome-Wide Variability of Human Cytomegalovirus in Congenitally Infected Infants. PLoS Pathog. 2011, 7, e1001344. [Google Scholar] [CrossRef]
- Depledge, D.P.; Palser, A.L.; Watson, S.J.; Lai, I.Y.-C.; Gray, E.R.; Grant, P.; Kanda, R.K.; Leproust, E.; Kellam, P.; Breuer, J. Specific Capture and Whole-Genome Sequencing of Viruses from Clinical Samples. PLoS ONE 2011, 6, e27805. [Google Scholar] [CrossRef]
- Tillieux, S.L.; Halsey, W.S.; Thomas, E.S.; Voycik, J.J.; Sathe, G.M.; Vassilev, V. Complete DNA Sequences of Two Oka Strain Varicella-Zoster Virus Genomes. J. Virol. 2008, 82, 11023–11044. [Google Scholar] [CrossRef]
- Cunningham, C.; Gatherer, D.; Hilfrich, B.; Baluchova, K.; Dargan, D.J.; Thomson, M.; Griffiths, P.D.; Wilkinson, G.W.G.; Schulz, T.F.; Davison, A.J. Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens. J. Gen. Virol. 2010, 91, 605–615. [Google Scholar] [CrossRef]
- Green, E.D.; Rubin, E.M.; Olson, M.V. The future of DNA sequencing. Nature 2017, 550, 179–181. [Google Scholar] [CrossRef]
- Cha, T.A.; Tom, E.; Kemble, G.W.; Duke, G.M.; Mocarski, E.S.; Spaete, R.R. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 1996, 70, 78–83. [Google Scholar]
- Tweedy, J.G.; Escriva, E.; Topf, M.; Gompels, U.A. Analyses of Tissue Culture Adaptation of Human Herpesvirus-6A by Whole Genome Deep Sequencing Redefines the Reference Sequence and Identifies Virus Entry Complex Changes. Viruses 2018, 10, 16. [Google Scholar] [CrossRef]
- Depledge, D.P.; Kundu, S.; Jensen, N.J.; Gray, E.R.; Jones, M.; Steinberg, S.; Gershon, A.; Kinchington, P.R.; Schmid, D.S.; Balloux, F.; et al. Deep Sequencing of Viral Genomes Provides Insight into the Evolution and Pathogenesis of Varicella Zoster Virus and Its Vaccine in Humans. Mol. Biol. Evol. 2014, 31, 397–409. [Google Scholar] [CrossRef]
- Tamburro, K.M.; Yang, D.; Poisson, J.; Fedoriw, Y.; Roy, D.; Lucas, A.; Sin, S.-H.; Malouf, N.; Moylan, V.; Damania, B.; et al. Vironome of Kaposi sarcoma associated herpesvirus-inflammatory cytokine syndrome in an AIDS patient reveals co-infection of human herpesvirus 8 and human herpesvirus 6A. Virology 2012, 433, 220–225. [Google Scholar] [CrossRef]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. [Google Scholar] [CrossRef]
- Greninger, A.L.; Roychoudhury, P.; Xie, H.; Casto, A.; Cent, A.; Pepper, G.; Koelle, D.M.; Huang, M.-L.; Wald, A.; Johnston, C.; et al. Ultrasensitive Capture of Human Herpes Simplex Virus Genomes Directly from Clinical Samples Reveals Extraordinarily Limited Evolution in Cell Culture. mSphere 2018, 3, e00283-18. [Google Scholar] [CrossRef]
- Casto, A.M.; Roychoudhury, P.; Xie, H.; Selke, S.; Perchetti, G.A.; Wofford, H.; Huang, M.-L.; Verjans, G.M.G.M.; Gottlieb, G.S.; Wald, A.; et al. Large, Stable, Contemporary Interspecies Recombination Events in Circulating Human Herpes Simplex Viruses. J. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Suárez, N.M.; Wilkie, G.S.; Hage, E.; Camiolo, S.; Holton, M.; Hughes, J.; Maabar, M.; Vattipally, S.B.; Dhingra, A.; Gompels, U.A.; et al. Human Cytomegalovirus Genomes Sequenced Directly From Clinical Material: Variation, Multiple-Strain Infection, Recombination, and Gene Loss. J. Infect. Dis. 2019, 220, 781–791. [Google Scholar] [CrossRef]
- Correia, S.; Bridges, R.; Wegner, F.; Venturini, C.; Palser, A.; Middeldorp, J.M.; Cohen, J.I.; Lorenzetti, M.A.; Bassano, I.; White, R.E.; et al. Sequence Variation of Epstein-Barr Virus: Viral Types, Geography, Codon Usage, and Diseases. J. Virol. 2018, 92, e01132-18. [Google Scholar] [CrossRef]
- Lassalle, F.; Depledge, D.P.; Reeves, M.B.; Brown, A.C.; Christiansen, M.T.; Tutill, H.J.; Williams, R.J.; Einer-Jensen, K.; Holdstock, J.; Atkinson, C.; et al. Islands of linkage in an ocean of pervasive recombination reveals two-speed evolution of human cytomegalovirus genomes. Virus Evol. 2016, 2. [Google Scholar] [CrossRef]
- Wegner, F.; Lassalle, F.; Depledge, D.P.; Balloux, F.; Breuer, J. Co-evolution of sites under immune selection shapes Epstein-Barr Virus population structure. Mol. Biol. Evol. 2019. [Google Scholar] [CrossRef]
- Pontremoli, C.; Forni, D.; Clerici, M.; Cagliani, R.; Sironi, M. Possible European Origin of Circulating Varicella Zoster Virus Strains. J. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Szpara, M.L.; Gatherer, D.; Ochoa, A.; Greenbaum, B.; Dolan, A.; Bowden, R.J.; Enquist, L.W.; Legendre, M.; Davison, A.J. Evolution and Diversity in Human Herpes Simplex Virus Genomes. J. Virol. 2014, 88, 1209–1227. [Google Scholar] [CrossRef]
- Balloux, F.; Brønstad Brynildsrud, O.; van Dorp, L.; Shaw, L.P.; Chen, H.; Harris, K.A.; Wang, H.; Eldholm, V. From Theory to Practice: Translating Whole-Genome Sequencing (WGS) into the Clinic. Trends Microbiol. 2018, 26, 1035–1048. [Google Scholar] [CrossRef]
- Wertheim, J.O.; Smith, M.D.; Smith, D.M.; Scheffler, K.; Kosakovsky Pond, S.L. Evolutionary origins of human herpes simplex viruses 1 and 2. Mol. Biol. Evol. 2014, 31, 2356–2364. [Google Scholar] [CrossRef]
- Tang, J.W.; Coward, L.J.; Davies, N.W.S.; Geretti, A.M.; Howard, R.S.; Hirsch, N.P.; Ward, K.N. Brain stem encephalitis caused by primary herpes simplex 2 infection in a young woman. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1323–1325. [Google Scholar] [CrossRef]
- Akhtar, L.N.; Bowen, C.D.; Renner, D.W.; Pandey, U.; Della Fera, A.N.; Kimberlin, D.W.; Prichard, M.N.; Whitley, R.J.; Weitzman, M.D.; Szpara, M.L. Genotypic and Phenotypic Diversity of Herpes Simplex Virus 2 within the Infected Neonatal Population. mSphere 2019, 4, e00590-18. [Google Scholar] [CrossRef]
- Wald, A.; Corey, L. Persistence in the Population: Epidemiology, Transmission. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007; ISBN 9780521827140. [Google Scholar]
- Looker, K.J.; Magaret, A.S.; Turner, K.M.E.; Vickerman, P.; Gottlieb, S.L.; Newman, L.M. Global estimates of prevalent and incident herpes simplex virus type 2 infections in 2012. PLoS ONE 2015, 10, e114989. [Google Scholar] [CrossRef]
- Underdown, S.J.; Kumar, K.; Houldcroft, C. Network analysis of the hominin origin of Herpes Simplex virus 2 from fossil data. Virus Evol. 2017, 3, vex026. [Google Scholar] [CrossRef]
- Ayoub, H.H.; Chemaitelly, H.; Abu-Raddad, L.J. Characterizing the transitioning epidemiology of herpes simplex virus type 1 in the USA: Model-based predictions. BMC Med. 2019, 17, 57. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Dalrymple, M.A.; Davison, A.J.; Dolan, A.; Frame, M.C.; McNab, D.; Perry, L.J.; Scott, J.E.; Taylor, P. The Complete DNA Sequence of the Long Unique Region in the Genome of Herpes Simplex Virus Type 1. J. Gen. Virol. 1988, 69, 1531–1574. [Google Scholar] [CrossRef]
- Szpara, M.L.; Parsons, L.; Enquist, L.W. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J. Virol. 2010, 84, 5303–5313. [Google Scholar] [CrossRef]
- Kolb, A.W.; Larsen, I.V.; Cuellar, J.A.; Brandt, C.R. Genomic, Phylogenetic, and Recombinational Characterization of Herpes Simplex Virus 2 Strains. J. Virol. 2015, 89, 6427–6434. [Google Scholar] [CrossRef]
- Koelle, D.M.; Norberg, P.; Fitzgibbon, M.P.; Russell, R.M.; Greninger, A.L.; Huang, M.-L.; Stensland, L.; Jing, L.; Magaret, A.S.; Diem, K.; et al. Worldwide circulation of HSV-2 × HSV-1 recombinant strains. Sci. Rep. 2017, 7, 44084. [Google Scholar] [CrossRef]
- Bowen, C.D.; Paavilainen, H.; Renner, D.W.; Palomäki, J.; Lehtinen, J.; Vuorinen, T.; Norberg, P.; Hukkanen, V.; Szpara, M.L. Comparison of Herpes Simplex Virus 1 Strains Circulating in Finland Demonstrates the Uncoupling of Whole-Genome Relatedness and Phenotypic Outcomes of Viral Infection. J. Virol. 2019, 93, e01824-18. [Google Scholar] [CrossRef]
- Bowen, C.D.; Renner, D.W.; Shreve, J.T.; Tafuri, Y.; Payne, K.M.; Dix, R.D.; Kinchington, P.R.; Gatherer, D.; Szpara, M.L. Viral forensic genomics reveals the relatedness of classic herpes simplex virus strains KOS, KOS63, and KOS79. Virology 2016, 492, 179–186. [Google Scholar] [CrossRef]
- Pandey, U.; Renner, D.W.; Thompson, R.L.; Szpara, M.L.; Sawtell, N.M. Inferred father-to-son transmission of herpes simplex virus results in near-perfect preservation of viral genome identity and in vivo phenotypes. Sci. Rep. 2017, 7, 13666. [Google Scholar] [CrossRef]
- Shipley, M.M.; Renner, D.W.; Ott, M.; Bloom, D.C.; Koelle, D.M.; Johnston, C.; Szpara, M.L. Genome-Wide Surveillance of Genital Herpes Simplex Virus Type 1 From Multiple Anatomic Sites Over Time. J. Infect. Dis. 2018, 218, 595–605. [Google Scholar] [CrossRef]
- Dolan, A.; Jamieson, F.E.; Cunningham, C.; Barnett, B.C.; McGeoch, D.J. The genome sequence of herpes simplex virus type 2. J. Virol. 1998, 72, 2010–2021. [Google Scholar]
- Newman, R.M.; Lamers, S.L.; Weiner, B.; Ray, S.C.; Colgrove, R.C.; Diaz, F.; Jing, L.; Wang, K.; Saif, S.; Young, S.; et al. Genome Sequencing and Analysis of Geographically Diverse Clinical Isolates of Herpes Simplex Virus 2. J. Virol. 2015, 89, 8219–8232. [Google Scholar] [CrossRef]
- Burrel, S.; Boutolleau, D.; Ryu, D.; Agut, H.; Merkel, K.; Leendertz, F.H.; Calvignac-Spencer, S. Ancient Recombination Events Between Human Herpes Simplex Viruses. Mol. Biol. Evol. 2017, 25, 1910–1920. [Google Scholar] [CrossRef]
- Burrel, S.; Desire, N.; Marlet, J.; Dacheux, L.; Seang, S.; Caumes, E.; Bourhy, H.; Agut, H.; Boutolleau, D. Genetic Diversity within Alphaherpesviruses: Characterization of a Novel Variant of Herpes Simplex Virus 2. J. Virol. 2015, 89, 12273–12283. [Google Scholar] [CrossRef] [PubMed]
- Seward, J.; Jumaan, A. VZV: Persistence in the Population. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007; ISBN 9780521827140. [Google Scholar]
- Cohen, J.I. Herpes Zoster. N. Engl. J. Med. 2013, 369, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Breuer, J.; Pacou, M.; Gauthier, A.; Brown, M.M. Herpes zoster as a risk factor for stroke and TIA: A retrospective cohort study in the UK. Neurology 2014, 82, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Muir, W.B.; Nichols, R.; Breuer, J. Phylogenetic analysis of varicella-zoster virus: Evidence of intercontinental spread of genotypes and recombination. J. Virol. 2002, 76, 1971–1979. [Google Scholar] [CrossRef]
- McGeoch, D.J. Lineages of varicella-zoster virus. J. Gen. Virol. 2009, 90, 963–969. [Google Scholar] [CrossRef]
- Breuer, J. Molecular Genetic Insights Into Varicella Zoster Virus (VZV), the vOka Vaccine Strain, and the Pathogenesis of Latency and Reactivation. J. Infect. Dis. 2018, 218, S75–S80. [Google Scholar] [CrossRef]
- Weinert, L.A.; Depledge, D.P.; Kundu, S.; Gershon, A.A.; Nichols, R.A.; Balloux, F.; Welch, J.J.; Breuer, J. Rates of vaccine evolution show strong effects of latency: Implications for varicella zoster virus epidemiology. Mol. Biol. Evol. 2015, 32, 1020–1028. [Google Scholar] [CrossRef]
- Jensen, N.J.; Rivailler, P.; Tseng, H.F.; Quinlivan, M.L.; Radford, K.; Folster, J.; Harpaz, R.; LaRussa, P.; Jacobsen, S.; Scott Schmid, D. Revisiting the genotyping scheme for varicella-zoster viruses based on whole-genome comparisons. J. Gen. Virol. 2017, 98, 1434–1438. [Google Scholar] [CrossRef]
- Masse, M.J.; Karlin, S.; Schachtel, G.A.; Mocarski, E.S.; Dickson, M.; Jarvis, M.A.; Hahn, G.; Nelson, J.A.; Myers, R.M.; Shenk, T.E. Human cytomegalovirus origin of DNA replication (oriLyt) resides within a highly complex repetitive region. Proc. Natl. Acad. Sci. USA 1992, 89, 5246–5250. [Google Scholar] [CrossRef]
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C.; et al. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312. [Google Scholar] [CrossRef]
- Murrell, I.; Wilkie, G.S.; Davison, A.J.; Statkute, E.; Fielding, C.A.; Tomasec, P.; Wilkinson, G.W.G.; Stanton, R.J. Genetic Stability of Bacterial Artificial Chromosome-Derived Human Cytomegalovirus during Culture In Vitro. J. Virol. 2016, 90, 3929–3943. [Google Scholar] [CrossRef] [PubMed]
- Sijmons, S.; Thys, K.; Mbong Ngwese, M.; Van Damme, E.; Dvorak, J.; Van Loock, M.; Li, G.; Tachezy, R.; Busson, L.; Aerssens, J.; et al. High-Throughput Analysis of Human Cytomegalovirus Genome Diversity Highlights the Widespread Occurrence of Gene-Disrupting Mutations and Pervasive Recombination. J. Virol. 2015, 89, 7673–7695. [Google Scholar] [CrossRef] [PubMed]
- Hage, E.; Wilkie, G.S.; Linnenweber-Held, S.; Dhingra, A.; Suárez, N.M.; Schmidt, J.J.J.; Kay-Fedorov, P.C.; Mischak-Weissinger, E.; Heim, A.; Schwarz, A.; et al. Characterization of Human Cytomegalovirus Genome Diversity in Immunocompromised Hosts by Whole-Genome Sequencing Directly From Clinical Specimens. J. Infect. Dis. 2017, 215, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Martí-Carreras, J.; Maes, P. Human cytomegalovirus genomics and transcriptomics through the lens of next-generation sequencing: Revision and future challenges. Virus Genes 2019, 55, 138–164. [Google Scholar] [CrossRef]
- Suárez, N.M.; Musonda, K.G.; Escriva, E.; Njenga, M.; Agbueze, A.; Camiolo, S.; Davison, A.J.; Gompels, U.A. Multiple-Strain Infections of Human Cytomegalovirus with High Genomic Diversity are Common In Breast Milk from HIV-Positive Women in Zambia. J. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Baraniak, I.; Kropff, B.; Ambrose, L.; McIntosh, M.; McLean, G.R.; Pichon, S.; Atkinson, C.; Milne, R.S.B.; Mach, M.; Griffiths, P.D.; et al. Protection from cytomegalovirus viremia following glycoprotein B vaccination is not dependent on neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2018, 115, 6273–6278. [Google Scholar] [CrossRef]
- Houldcroft, C.J.; Bryant, J.M.; Depledge, D.P.; Margetts, B.K.; Simmonds, J.; Nicolaou, S.; Tutill, H.J.; Williams, R.; Worth, A.J.J.; Marks, S.D.; et al. Detection of Low Frequency Multi-Drug Resistance and Novel Putative Maribavir Resistance in Immunocompromised Pediatric Patients with Cytomegalovirus. Front. Microbiol. 2016, 7, 1317. [Google Scholar] [CrossRef]
- Piret, J.; Boivin, G. Herpesvirus Resistance to Antiviral Drugs. In Antimicrobial Drug Resistance; Springer International Publishing: Cham, Switzerland, 2017; pp. 1185–1211. [Google Scholar]
- Cudini, J.; Roy, S.; Houldcroft, C.J.; Bryant, J.M.; Depledge, D.P.; Tutill, H.; Veys, P.; Williams, R.; Worth, A.J.J.; Tamuri, A.U.; et al. Human cytomegalovirus haplotype reconstruction reveals high diversity due to superinfection and evidence of within-host recombination. Proc. Natl. Acad. Sci. USA 2019, 116, 5693–5698. [Google Scholar] [CrossRef]
- Ablashi, D.; Agut, H.; Alvarez-Lafuente, R.; Clark, D.A.; Dewhurst, S.; DiLuca, D.; Flamand, L.; Frenkel, N.; Gallo, R.; Gompels, U.A.; et al. Classification of HHV-6A and HHV-6B as distinct viruses. Arch. Virol. 2014, 159, 863–870. [Google Scholar] [CrossRef]
- Zhang, E.; Bell, A.J.; Wilkie, G.S.; Suárez, N.M.; Batini, C.; Veal, C.D.; Armendáriz-Castillo, I.; Neumann, R.; Cotton, V.E.; Huang, Y.; et al. Inherited Chromosomally Integrated Human Herpesvirus 6 Genomes Are Ancient, Intact, and Potentially Able To Reactivate from Telomeres. J. Virol. 2017, 91, e01137-17. [Google Scholar] [CrossRef]
- Clark, D.A. Clinical and laboratory features of human herpesvirus 6 chromosomal integration. Clin. Microbiol. Infect. 2016, 22, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Gravel, A.; Dubuc, I.; Morissette, G.; Sedlak, R.H.; Jerome, K.R.; Flamand, L. Inherited chromosomally integrated human herpesvirus 6 as a predisposing risk factor for the development of angina pectoris. Proc. Natl. Acad. Sci. USA 2015, 112, 8058–8063. [Google Scholar] [CrossRef] [PubMed]
- Tweedy, J.; Spyrou, M.A.; Hubacek, P.; Kuhl, U.; Lassner, D.; Gompels, U.A. Analyses of germline, chromosomally integrated human herpesvirus 6A and B genomes indicate emergent infection and new inflammatory mediators. J. Gen. Virol. 2015, 96, 370–389. [Google Scholar] [CrossRef] [PubMed]
- Telford, M.; Navarro, A.; Santpere, G. Whole genome diversity of inherited chromosomally integrated HHV-6 derived from healthy individuals of diverse geographic origin. Sci. Rep. 2018, 8, 3472. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Gray, E.R.; Kundu, S.; Cooray, S.; Poulsen, A.; Aaby, P.; Breuer, J. Evolution of cocirculating varicella-zoster virus genotypes during a chickenpox outbreak in Guinea-Bissau. J. Virol. 2014, 88, 13936–13946. [Google Scholar] [CrossRef] [PubMed]
- Gompels, U.A.; Nicholas, J.; Lawrence, G.; Jones, M.; Thomson, B.J.; Martin, M.E.D.; Efstathiou, S.; Craxton, M.; Macaulay, H.A. The DNA Sequence of Human Herpesvirus-6: Structure, Coding Content, and Genome Evolution. Virology 1995, 209, 29–51. [Google Scholar] [CrossRef] [PubMed]
- Gravel, A.; Ablashi, D.; Flamand, L. Complete Genome Sequence of Early Passaged Human Herpesvirus 6A (GS Strain) Isolated from North America. Genome Announc. 2013, 1, e00012-13. [Google Scholar] [CrossRef]
- Tweedy, J.; Spyrou, M.A.; Donaldson, C.D.; Depledge, D.; Breuer, J.; Gompels, U.A. Complete Genome Sequence of the Human Herpesvirus 6A Strain AJ from Africa Resembles Strain GS from North America. Genome Announc. 2015, 3, e01498-14. [Google Scholar] [CrossRef]
- Tweedy, J.; Spyrou, M.; Pearson, M.; Lassner, D.; Kuhl, U.; Gompels, U. Complete Genome Sequence of Germline Chromosomally Integrated Human Herpesvirus 6A and Analyses Integration Sites Define a New Human Endogenous Virus with Potential to Reactivate as an Emerging Infection. Viruses 2016, 8, 19. [Google Scholar] [CrossRef]
- Greninger, A.L.; Roychoudhury, P.; Makhsous, N.; Hanson, D.; Chase, J.; Krueger, G.; Xie, H.; Huang, M.-L.; Saunders, L.; Ablashi, D.; et al. Copy Number Heterogeneity, Large Origin Tandem Repeats, and Interspecies Recombination in Human Herpesvirus 6A (HHV-6A) and HHV-6B Reference Strains. J. Virol. 2018, 92, e00135-18. [Google Scholar] [CrossRef]
- Engdahl, E.; Gustafsson, R.; Huang, J.; Biström, M.; Bomfim, I.L.; Stridh, P.; Khademi, M.; Brenner, N.; Butt, J.; Michel, A.; et al. Increased serological response against human herpesvirus 6A is associated with risk for multiple sclerosis. bioRxiv 2019, 737932. [Google Scholar] [CrossRef]
- Mullins, T.B.; Krishnamurthy, K. Roseola Infantum (Exanthema Subitum, Sixth Disease); StatPearls Publishing: Bethesda, MD, USA, 2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK448190/ (accessed on 12 October 2019).
- Greninger, A.L.; Knudsen, G.M.; Roychoudhury, P.; Hanson, D.J.; Sedlak, R.H.; Xie, H.; Guan, J.; Nguyen, T.; Peddu, V.; Boeckh, M.; et al. Comparative genomic, transcriptomic, and proteomic reannotation of human herpesvirus 6. BMC Genom. 2018, 19, 204. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Ikoma, M.; Zerr, D.M.; Basom, R.S.; Peddu, V.; Huang, M.-L.; Hall Sedlak, R.; Jerome, K.R.; Boeckh, M.; Barcy, S. RNA Sequencing of the In Vivo Human Herpesvirus 6B Transcriptome To Identify Targets for Clinical Assays Distinguishing between Latent and Active Infections. J. Virol. 2019, 93, e01419-18. [Google Scholar] [CrossRef] [PubMed]
- Perlejewski, K.; Popiel, M.; Laskus, T.; Nakamura, S.; Motooka, D.; Stokowy, T.; Lipowski, D.; Pollak, A.; Lechowicz, U.; Caraballo Cortés, K.; et al. Next-generation sequencing (NGS) in the identification of encephalitis-causing viruses: Unexpected detection of human herpesvirus 1 while searching for RNA pathogens. J. Virol. Methods 2015, 226, 1–6. [Google Scholar] [CrossRef]
- Sharp, C.; Golubchik, T.; Gregory, W.F.; McNaughton, A.L.; Gow, N.; Selvaratnam, M.; Mirea, A.; Foster, D.; Andersson, M.; Klenerman, P.; et al. Human Herpes Virus 6 (HHV-6)—Pathogen or Passenger? A pilot study of clinical laboratory data and next generation sequencing. bioRxiv 2018, 236083. [Google Scholar] [CrossRef]
- Mori, Y.; Yamanishi, K. HHV-6A, 6B, and 7: Pathogenesis, Host Response, and Clinical Disease. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007; ISBN 9780521827140. [Google Scholar]
- Yoshikawa, T.; Yoshida, J.; Hamaguchi, M.; Kubota, T.; Akimoto, S.; Ihira, M.; Nishiyama, Y.; Asano, Y. Human herpesvirus 7-associated meningitis and optic neuritis in a patient after allogeneic stem cell transplantation. J. Med. Virol. 2003, 70, 440–443. [Google Scholar] [CrossRef]
- Corral, Í.; Sainz de la Maza, S.; Rodríguez, M.; Kawiorski, M.-M.; López-Martínez, M.-J.; Galán, J.-C. Molecular detection of human herpesvirus 7 DNA in cerebrospinal fluid from adult patients with neurological disorders. J. Neurovirol. 2018, 24, 333–338. [Google Scholar] [CrossRef]
- Readhead, B.; Haure-Mirande, J.-V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 2018, 99, 64–82.e7. [Google Scholar] [CrossRef]
- Frenkel, N.; Schirmer, E.C.; Wyatt, L.S.; Katsafanas, G.; Roffman, E.; Danovich, R.M.; June, C.H. Isolation of a new herpesvirus from human CD4+ T cells. Proc. Natl. Acad. Sci. USA 1990, 87, 748–752. [Google Scholar] [CrossRef]
- Nicholas, J. Determination and analysis of the complete nucleotide sequence of human herpesvirus. J. Virol. 1996, 70, 5975–5989. [Google Scholar]
- Donaldson, C.D.; Clark, D.A.; Kidd, I.M.; Breuer, J.; Depledge, D.D. Genome Sequence of Human Herpesvirus 7 Strain UCL-1. Genome Announc. 2013, 1, e00830-13. [Google Scholar] [CrossRef] [PubMed]
- Worth, A.J.J.; Houldcroft, C.J.; Booth, C. Severe Epstein–Barr virus infection in primary immunodeficiency and the normal host. Br. J. Haematol. 2016, 175, 559–576. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.H.; Macsween, K.F.; Higgins, C.D.; Thomas, R.; McAulay, K.; Williams, H.; Harrison, N.; Reid, S.; Conacher, M.; Douglas, J.; et al. A Cohort Study among University Students: Identification of Risk Factors for Epstein-Barr Virus Seroconversion and Infectious Mononucleosis. Clin. Infect. Dis. 2006, 43, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Li, T.; Hung, G.-C.; Li, B.; Tsai, S.; Lo, S.-C. Identification and characterization of EBV genomes in spontaneously immortalized human peripheral blood B lymphocytes by NGS technology. BMC Genom. 2013, 14, 804. [Google Scholar] [CrossRef]
- Kwok, H.; Wu, C.W.; Palser, A.L.; Kellam, P.; Sham, P.C.; Kwong, D.L.W.; Chiang, A.K.S. Genomic diversity of Epstein-Barr virus genomes isolated from primary nasopharyngeal carcinoma biopsy samples. J. Virol. 2014, 88, 10662–10672. [Google Scholar] [CrossRef]
- Chiara, M.; Manzari, C.; Lionetti, C.; Mechelli, R.; Anastasiadou, E.; Buscarinu, M.C.; Ristori, G.; Salvetti, M.; Picardi, E.; D’Erchia, A.M.; et al. Geographic Population Structure in Epstein-Barr Virus Revealed by Comparative Genomics. Genome Biol. Evol. 2016, 8, 3284. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef]
- Okuno, Y.; Murata, T.; Sato, Y.; Muramatsu, H.; Ito, Y.; Watanabe, T.; Okuno, T.; Murakami, N.; Yoshida, K.; Sawada, A.; et al. Defective Epstein–Barr virus in chronic active infection and haematological malignancy. Nat. Microbiol. 2019, 4, 404. [Google Scholar] [CrossRef]
- Xu, M.; Yao, Y.; Chen, H.; Zhang, S.; Cao, S.-M.; Zhang, Z.; Luo, B.; Liu, Z.; Li, Z.; Xiang, T.; et al. Genome sequencing analysis identifies Epstein–Barr virus subtypes associated with high risk of nasopharyngeal carcinoma. Nat. Genet. 2019, 51, 1131–1136. [Google Scholar] [CrossRef]
- Grande, B.M.; Gerhard, D.S.; Jiang, A.; Griner, N.B.; Abramson, J.S.; Alexander, T.B.; Allen, H.; Ayers, L.W.; Bethony, J.M.; Bhatia, K.; et al. Genome-wide discovery of somatic coding and non-coding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 2019, 133, 131–1324. [Google Scholar] [CrossRef]
- Panea, R.I.; Love, C.L.; Shingleton, J.R.; Reddy, A.; Bailey, J.A.; Moormann, A.M.; Otieno, J.A.; Ong’echa, J.M.; Oduor, C.I.; Schroeder, K.M.S.; et al. The whole genome landscape of Burkitt lymphoma subtypes. Blood 2019. [Google Scholar] [CrossRef] [PubMed]
- Beral, V.; Peterman, T.; Berkelman, R.; Jaffe, H.W. Kaposi’s sarcoma among persons with AIDS: A sexually transmitted infection? Lancet 1990, 335, 123–128. [Google Scholar] [CrossRef]
- Chang, Y.; Cesarman, E.; Pessin, M.; Lee, F.; Culpepper, J.; Knowles, D.; Moore, P. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Olp, L.N.; Jeanniard, A.; Marimo, C.; West, J.T.; Wood, C. Whole-Genome Sequencing of Kaposi’s Sarcoma-Associated Herpesvirus from Zambian Kaposi’s Sarcoma Biopsy Specimens Reveals Unique Viral Diversity. J. Virol. 2015, 89, 12299–12308. [Google Scholar] [CrossRef] [PubMed]
- Sallah, N.; Palser, A.L.; Watson, S.J.; Labo, N.; Asiki, G.; Marshall, V.; Newton, R.; Whitby, D.; Kellam, P.; Barroso, I. Genome-Wide Sequence Analysis of Kaposi Sarcoma-Associated Herpesvirus Shows Diversification Driven by Recombination. J. Infect. Dis. 2018, 218, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Osawa, M.; Mine, S.; Ota, S.; Kato, K.; Sekizuka, T.; Kuroda, M.; Kataoka, M.; Fukumoto, H.; Sato, Y.; Kanno, T.; et al. Establishing and characterizing a new primary effusion lymphoma cell line harboring Kaposi’s sarcoma–associated herpesvirus. Infect. Agent. Cancer 2016, 11, 37. [Google Scholar] [CrossRef][Green Version]
- De Leo, A.; Deng, Z.; Vladimirova, O.; Chen, H.-S.; Dheekollu, J.; Calderon, A.; Myers, K.A.; Hayden, J.; Keeney, F.; Kaufer, B.B.; et al. LANA oligomeric architecture is essential for KSHV nuclear body formation and viral genome maintenance during latency. PLoS Pathog. 2019, 15, e1007489. [Google Scholar] [CrossRef]
- Awazawa, R.; Utsumi, D.; Katano, H.; Awazawa, T.; Miyagi, T.; Hayashi, K.; Matori, S.; Uezato, H.; Takahashi, K. High Prevalence of Distinct Human Herpesvirus 8 Contributes to the High Incidence of Non-acquired Immune Deficiency Syndrome-Associated Kaposi’s Sarcoma in Isolated Japanese Islands. J. Infect. Dis. 2017, 216, 850–858. [Google Scholar] [CrossRef]
- Depledge, D.P.; Mohr, I.; Wilson, A.C. Going the Distance: Optimizing RNA-Seq Strategies for Transcriptomic Analysis of Complex Viral Genomes. J. Virol. 2018, 93, e01342-18. [Google Scholar] [CrossRef]
- López-Muñoz, A.D.; Rastrojo, A.; Alcamí, A. Complete Genome Sequence of Herpes Simplex Virus 2 Strain 333. Microbiol. Resour. Announc. 2018, 7, e00870-18. [Google Scholar] [CrossRef] [PubMed]
- Karamitros, T.; Harrison, I.; Piorkowska, R.; Katzourakis, A.; Magiorkinis, G.; Mbisa, J.L. De Novo Assembly of Human Herpes Virus Type 1 (HHV-1) Genome, Mining of Non-Canonical Structures and Detection of Novel Drug-Resistance Mutations Using Short- and Long-Read Next Generation Sequencing Technologies. PLoS ONE 2016, 11, e0157600. [Google Scholar] [CrossRef] [PubMed]
- Karamitros, T.; van Wilgenburg, B.; Wills, M.; Klenerman, P.; Magiorkinis, G. Nanopore sequencing and full genome de novo assembly of human cytomegalovirus TB40/E reveals clonal diversity and structural variations. BMC Genom. 2018, 19, 577. [Google Scholar] [CrossRef] [PubMed]
- Yajima, M.; Ikuta, K.; Kanda, T. Rapid CRISPR/Cas9-Mediated Cloning of Full-Length Epstein-Barr Virus Genomes from Latently Infected Cells. Viruses 2018, 10, 171. [Google Scholar] [CrossRef]
- Depledge, D.P.; Srinivas, K.P.; Sadaoka, T.; Bready, D.; Mori, Y.; Placantonakis, D.G.; Mohr, I.; Wilson, A.C. Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat. Commun. 2019, 10, 754. [Google Scholar] [CrossRef]
- Boldogkői, Z.; Szűcs, A.; Balázs, Z.; Sharon, D.; Snyder, M.; Tombácz, D. Transcriptomic study of Herpes simplex virus type-1 using full-length sequencing techniques. Sci. Data 2018, 5, 180266. [Google Scholar] [CrossRef]
- Prazsák, I.; Moldován, N.; Balázs, Z.; Tombácz, D.; Megyeri, K.; Szűcs, A.; Csabai, Z.; Boldogkői, Z. Long-read sequencing uncovers a complex transcriptome topology in varicella zoster virus. BMC Genom. 2018, 19, 873. [Google Scholar] [CrossRef]
- Tombácz, D.; Prazsák, I.; Moldován, N.; Szűcs, A.; Boldogkői, Z. Lytic Transcriptome Dataset of Varicella Zoster Virus Generated by Long-Read Sequencing. Front. Genet. 2018, 9, 460. [Google Scholar] [CrossRef]
- Boldogkői, Z.; Moldován, N.; Balázs, Z.; Snyder, M.; Tombácz, D. Long-Read Sequencing—A Powerful Tool in Viral Transcriptome Research. Trends Microbiol. 2019, 27, 578–592. [Google Scholar] [CrossRef]
- Eckert, S.E.; Chan, J.Z.-M.; Houniet, D.; The Pathseek Consortium, J.; Breuer, J.; Speight, G. Enrichment by hybridisation of long DNA fragments for Nanopore sequencing. Microb. Genom. 2016, 2, e000087. [Google Scholar] [CrossRef]
- Ba Abdullah, M.M.; Palermo, R.D.; Palser, A.L.; Grayson, N.E.; Kellam, P.; Correia, S.; Szymula, A.; White, R.E. Heterogeneity of the Epstein-Barr Virus (EBV) Major Internal Repeat Reveals Evolutionary Mechanisms of EBV and a Functional Defect in the Prototype EBV Strain B95-8. J. Virol. 2017, 91, e00920-17. [Google Scholar] [CrossRef] [PubMed]
- Palser, A.L.; Grayson, N.E.; White, R.E.; Corton, C.; Correia, S.; Ba Abdullah, M.M.; Watson, S.J.; Cotten, M.; Arrand, J.R.; Murray, P.G.; et al. Genome diversity of Epstein-Barr virus from multiple tumor types and normal infection. J. Virol. 2015, 89, 5222–5237. [Google Scholar] [CrossRef] [PubMed]
- Sijmons, S.; Thys, K.; Corthout, M.; Van Damme, E.; Van Loock, M.; Bollen, S.; Baguet, S.; Aerssens, J.; Van Ranst, M.; Maes, P. A Method Enabling High-Throughput Sequencing of Human Cytomegalovirus Complete Genomes from Clinical Isolates. PLoS ONE 2014, 9, e95501. [Google Scholar] [CrossRef] [PubMed]
- Colgrove, R.; Diaz, F.; Newman, R.; Saif, S.; Shea, T.; Young, S.; Henn, M.; Knipe, D.M. Genomic sequences of a low passage herpes simplex virus 2 clinical isolate and its plaque-purified derivative strain. Virology 2014, 450, 140–145. [Google Scholar] [CrossRef]
- Pandey, U.; Szpara, M.L. Herpes Simplex Virus Disease Management and Diagnostics in the Era of High-Throughput Sequencing. Clin. Microbiol. Newsl. 2019, 41, 41–48. [Google Scholar] [CrossRef]
- Nakamura, K.; Oshima, T.; Morimoto, T.; Ikeda, S.; Yoshikawa, H.; Shiwa, Y.; Ishikawa, S.; Linak, M.C.; Hirai, A.; Takahashi, H.; et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 2011, 39, e90. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Sinha, A.; Bird, S.W.; Jayamohan, H.; Edwards, H.S.; Schoeniger, J.S.; Patel, K.D.; Branda, S.S.; Bartsch, M.S. Systematic and stochastic influences on the performance of the MinION nanopore sequencer across a range of nucleotide bias. Sci. Rep. 2018, 8, 3159. [Google Scholar] [CrossRef]
- Miller, S.; Naccache, S.N.; Samayoa, E.; Messacar, K.; Arevalo, S.; Federman, S.; Stryke, D.; Pham, E.; Fung, B.; Bolosky, W.J.; et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 2019, 29, 831–842. [Google Scholar] [CrossRef]
- Depledge, D.P.; Cudini, J.; Kundu, S.; Atkinson, C.; Brown, J.R.; Haque, T.; Houldcroft, C.J.; Koay, E.S.; McGill, F.; Milne, R.; et al. High Viral Diversity and Mixed Infections in Cerebral Spinal Fluid From Cases of Varicella Zoster Virus Encephalitis. J. Infect. Dis. 2018, 218, 1592–1601. [Google Scholar] [CrossRef]
- Bartha, I.; Carlson, J.M.; Brumme, C.J.; McLaren, P.J.; Brumme, Z.L.; John, M.; Haas, D.W.; Martinez-Picado, J.; Dalmau, J.; López-Galíndez, C.; et al. A genome-to-genome analysis of associations between human genetic variation, HIV-1 sequence diversity, and viral control. Elife 2013, 2, e01123. [Google Scholar] [CrossRef]
- Lees, J.A.; Ferwerda, B.; Kremer, P.H.C.; Wheeler, N.E.; Serón, M.V.; Croucher, N.J.; Gladstone, R.A.; Bootsma, H.J.; Rots, N.Y.; Wijmega-Monsuur, A.J.; et al. Joint sequencing of human and pathogen genomes reveals the genetics of pneumococcal meningitis. Nat. Commun. 2019, 10, 2176. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, N.; Svarovskaia, E.S.; Mo, H.; Osinusi, A.O.; Brainard, D.M.; Subramanian, G.M.; McHutchison, J.G.; Zeuzem, S.; Fellay, J. Adaptation of hepatitis C virus to interferon lambda polymorphism across multiple viral genotypes. Elife 2019, 8, e42542. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.Z.; Niemann, J.; Højholt Iversen, K.; Fotakis, A.; Gopalakrishnan, S.; Sinding, M.S.; Ellegaard, M.R.; Allentoft, M.E.; Lanigan, L.T.; Taurozzi, A.J.; et al. Stone Age “Chewing Gum” Yields 5700 Year-Old Human Genome and Oral Microbiome; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2018. [Google Scholar]
- Gurdasani, D.; Barroso, I.; Zeggini, E.; Sandhu, M.S. Genomics of disease risk in globally diverse populations. Nat. Rev. Genet. 2019, 20, 520–535. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Virus | Whole Genomes in Genbank * (as of 28/08/2019) |
|---|---|
| Human herpesvirus 1—herpes simplex virus 1 | 288 |
| Human herpesvirus 2—herpes simplex virus 2 | 378 |
| Human herpesvirus 3—varicella-zoster virus | 247 |
| Human herpesvirus 4—Epstein-Barr virus | 1043 |
| Human herpesvirus 5—cytomegalovirus | 315 |
| Human herpesvirus 6 (unclassified) | 28 |
| Human herpesvirus 6A | 91 |
| Human herpesvirus 6B | 102 |
| Human herpesvirus 7 | 3 |
| Human herpesvirus 8—Kaposi’s sarcoma-associated herpesvirus | 33 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Houldcroft, C.J. Human Herpesvirus Sequencing in the Genomic Era: The Growing Ranks of the Herpetic Legion. Pathogens 2019, 8, 186. https://doi.org/10.3390/pathogens8040186
Houldcroft CJ. Human Herpesvirus Sequencing in the Genomic Era: The Growing Ranks of the Herpetic Legion. Pathogens. 2019; 8(4):186. https://doi.org/10.3390/pathogens8040186
Chicago/Turabian StyleHouldcroft, Charlotte J. 2019. "Human Herpesvirus Sequencing in the Genomic Era: The Growing Ranks of the Herpetic Legion" Pathogens 8, no. 4: 186. https://doi.org/10.3390/pathogens8040186
APA StyleHouldcroft, C. J. (2019). Human Herpesvirus Sequencing in the Genomic Era: The Growing Ranks of the Herpetic Legion. Pathogens, 8(4), 186. https://doi.org/10.3390/pathogens8040186