Whole Transcriptome Analyses Reveal Differential mRNA and microRNA Expression Profiles in Primary Human Dermal Fibroblasts Infected with Clinical or Vaccine Strains of Varicella Zoster Virus

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. RNA-Sequencing
2.3. Differentially Expressed Genes (DEGs) and Gene Ontology Analysis
2.4. Three Dimensional (3D)-Multidimensional Scaling (MDS)
2.5. Small RNA-seq Library Construction and Sequencing
2.6. Bioinformatic Prediction of miRNA Targets and Gene Ontology
2.7. Real-time - Quantitative Polymerase Chain Reaction (RT-qPCR)
2.8. Transfection of miRNA Inhibitors
2.9. Statistical Analysis
3. Results
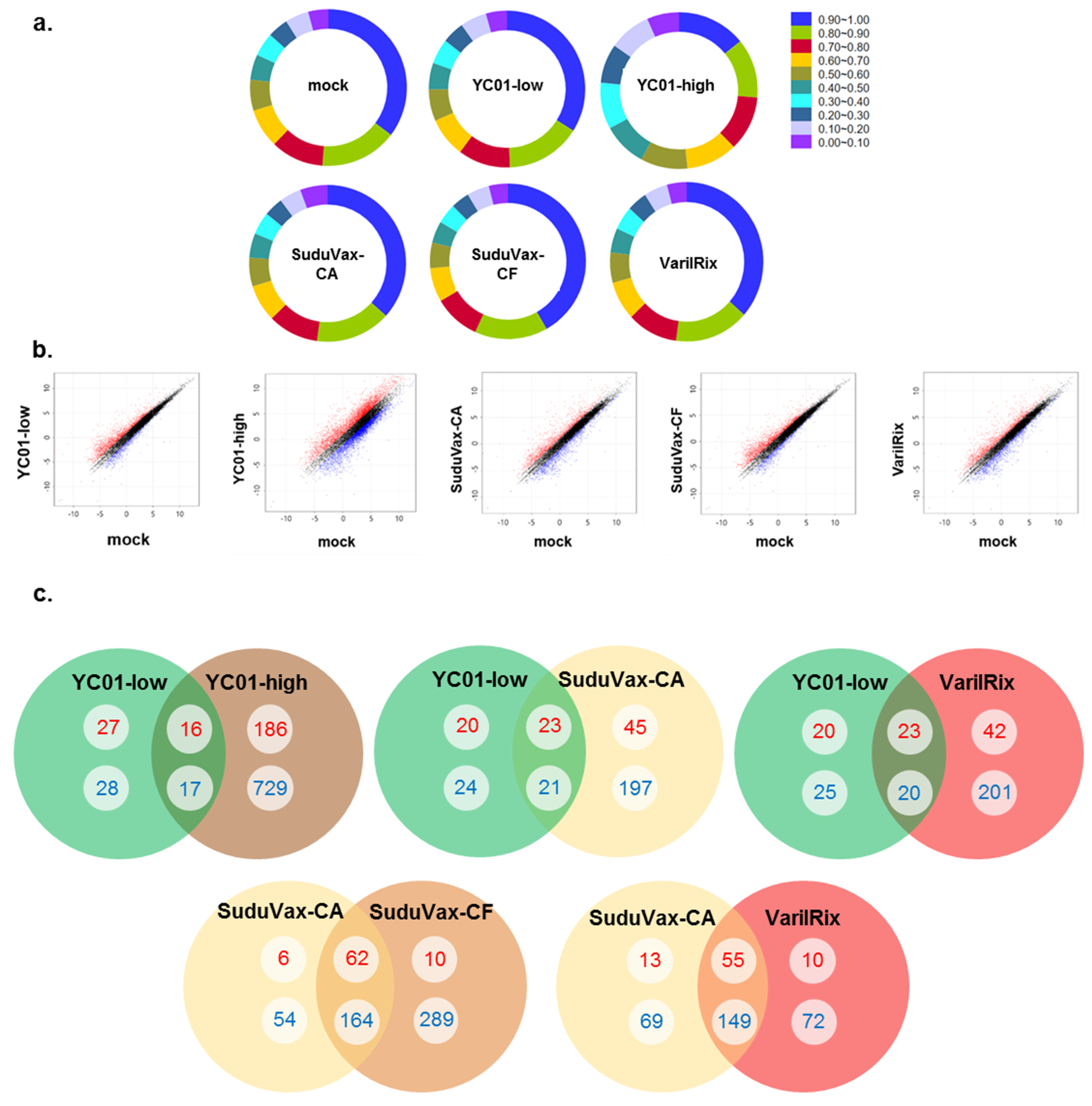
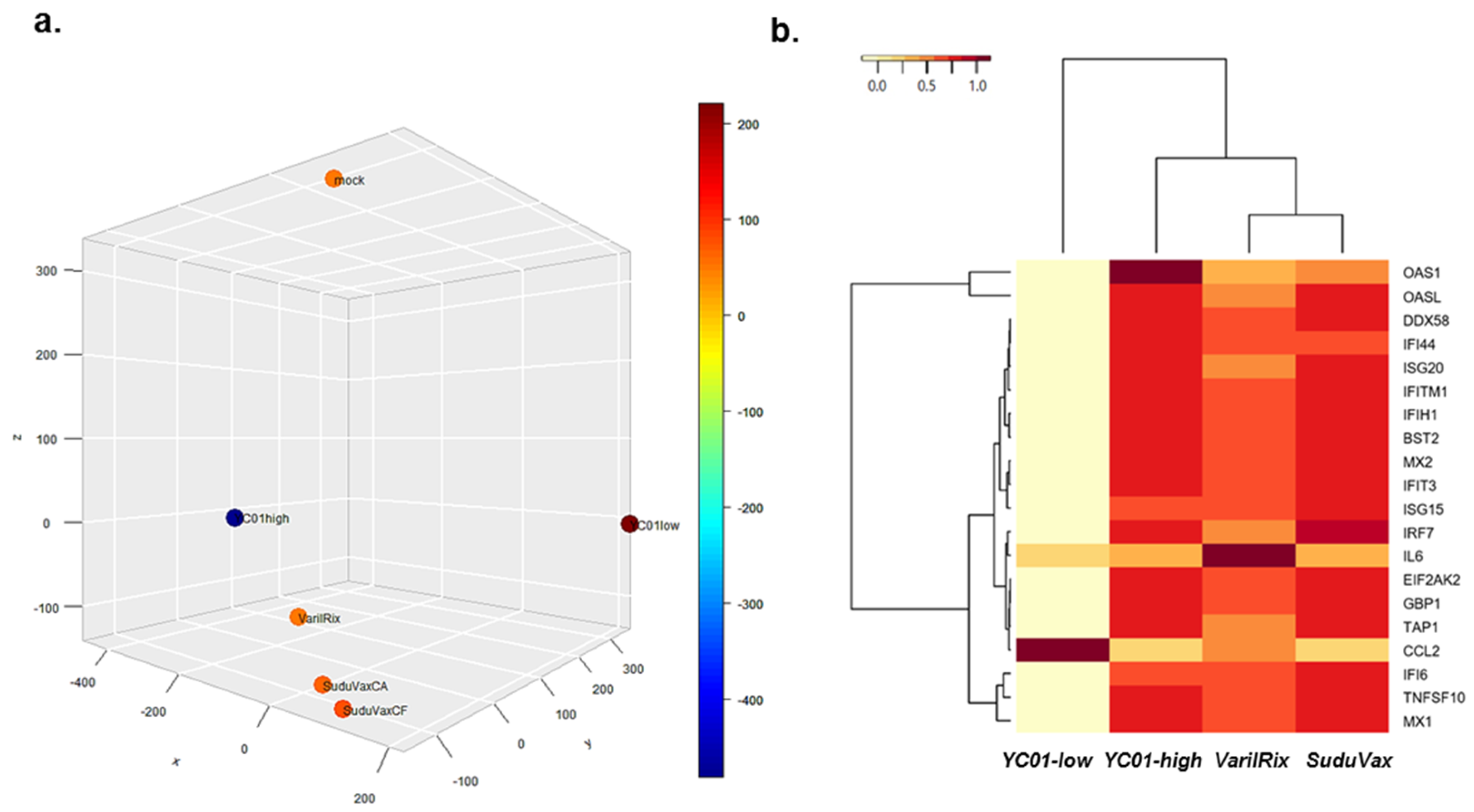
3.1. Analysis of High-Throughput RNA-seq Transcriptome
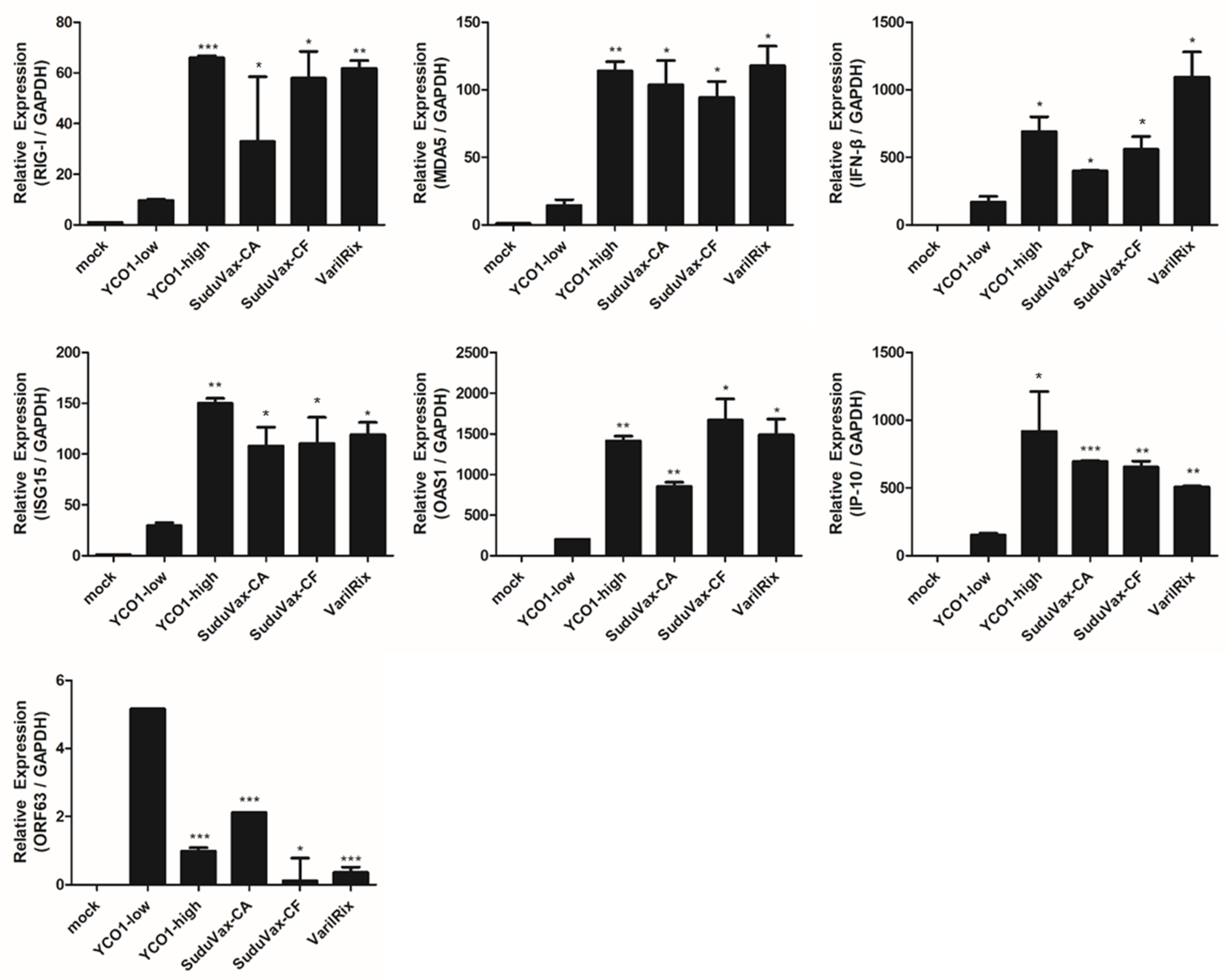
3.2. Distinct Changes in Host DEGs in Wildtype and Attenuated VZV-Infected HDFs
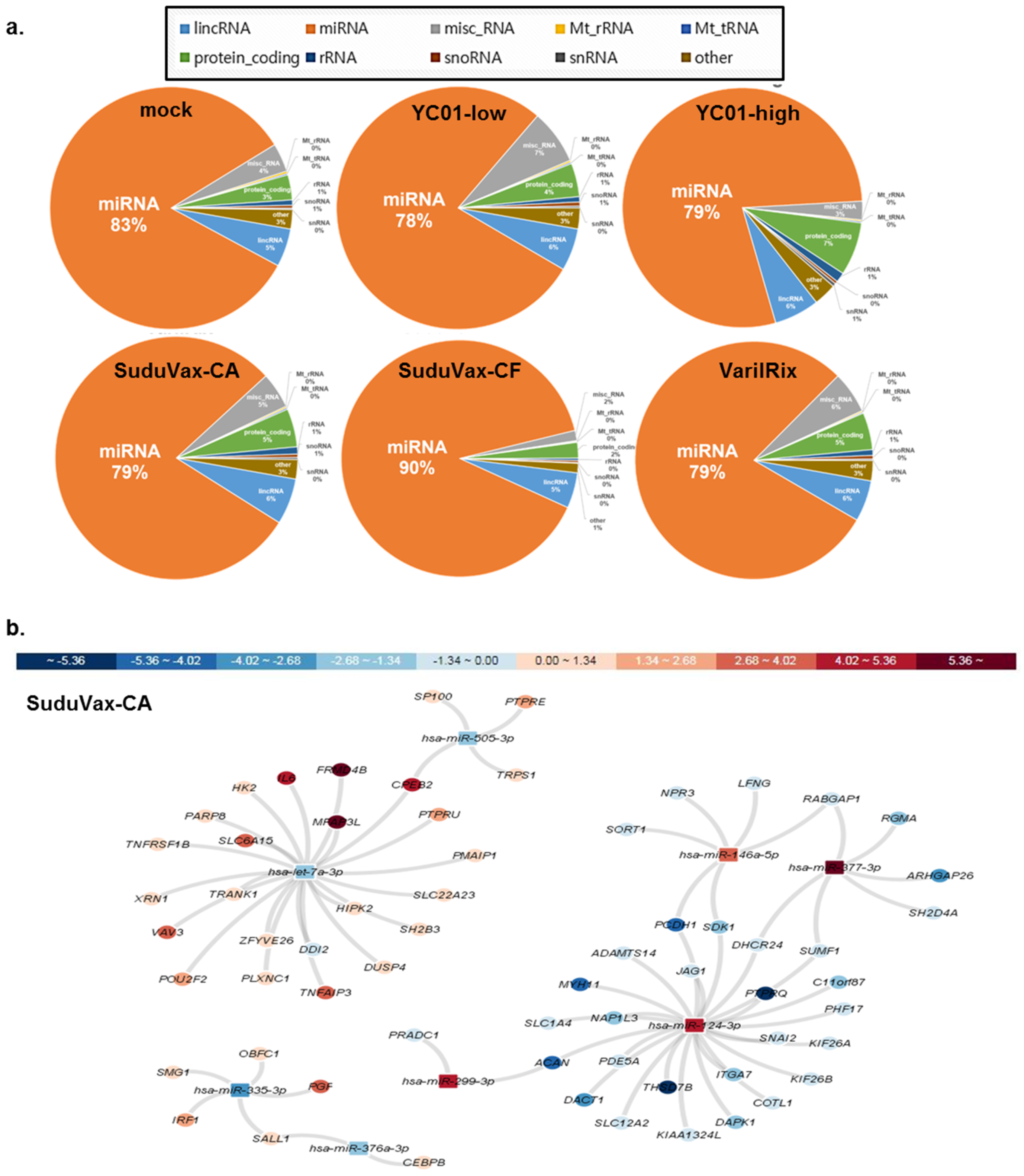
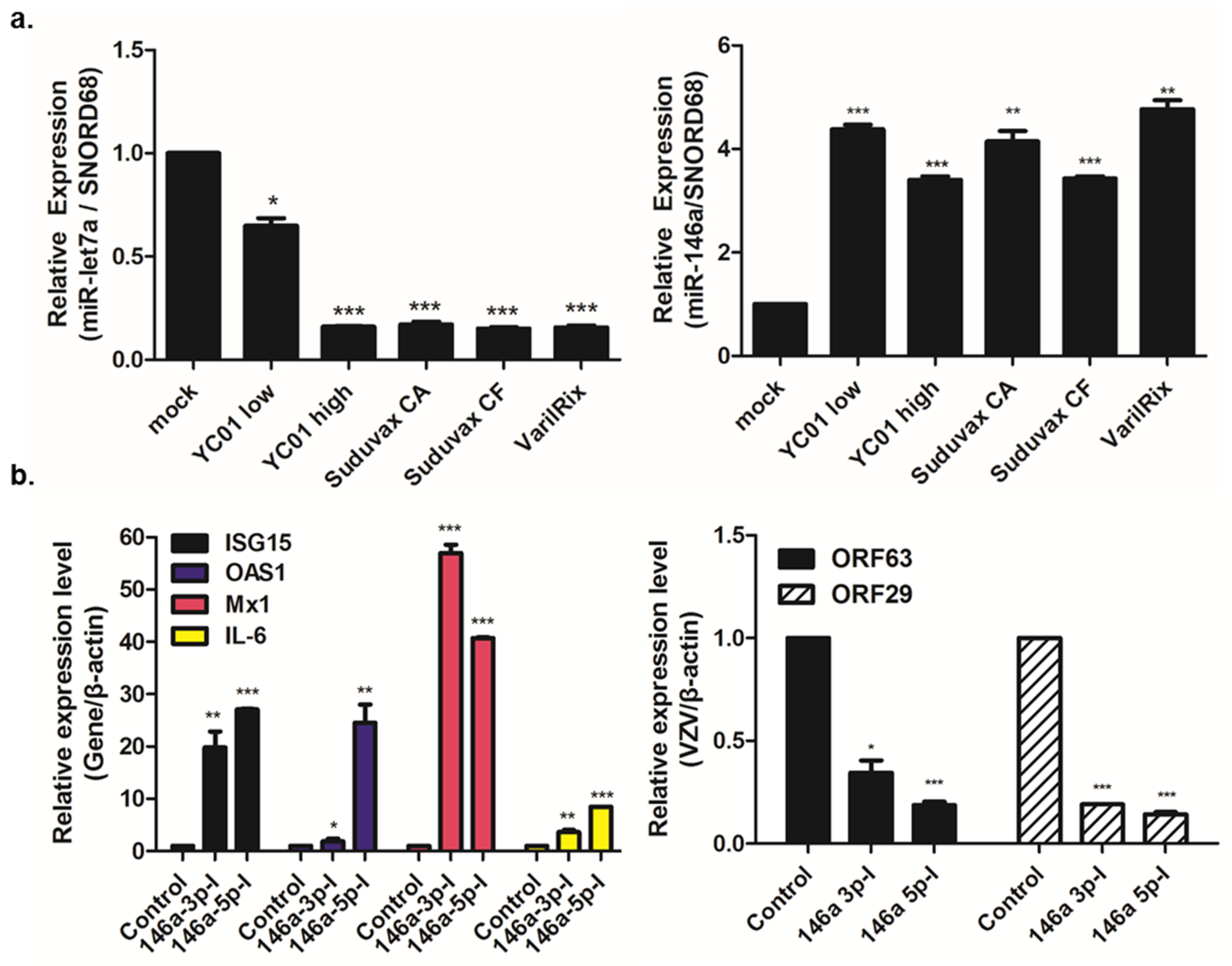
3.3. Distinct microRNA Expression Patterns in Response to Wildtype and Attenuated VZV Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gershon, A.A.; Breuer, J.; Cohen, J.I.; Cohrs, R.J.; Gershon, M.D.; Gilden, D.; Seward, J.F.; Grose, C.; Hambleton, S.; Kennedy, P.G.E.; et al. Varicella zoster virus infection. Nat. Rev. Dis. Primers 2015, 1, 15016. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.G.E.; Gershon, A.A. Clinical features of varicella-zoster virus infection. Viruses 2018, 10, 609. [Google Scholar] [CrossRef] [PubMed]
- Gershon, A.A.; Katz, S.L. Perspective on live varicella vaccine. J. Infect. Dis. 2008, 197, S242–S245. [Google Scholar] [CrossRef] [PubMed]
- Gershon, A.A. Varicella-zoster vaccine. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Takahashi, M.; Otsuka, T.; Okuno, Y.; Asano, Y.; Yazaki, T. Live vaccine used to prevent the spread of varicella in children in hospital. Lancet 1974, 2, 1288–1290. [Google Scholar] [CrossRef]
- Depledge, D.P.; Yamanishi, K.; Gomi, Y.; Gershon, A.A.; Breuer, J. Deep sequencing of distinct preparations of the live attenuated varicella-zoster virus vaccine reveals a conserved core of attenuating single-nucleotide polymorphisms. J. Virol. 2016, 90, 8698–8704. [Google Scholar] [CrossRef]
- Cohrs, R.J.; Gilden, D.H.; Gomi, Y.; Yamanishi, K.; Cohen, J.I. Comparison of virus transcription during lytic infection of the Oka parental and vaccine strains of Varicella-Zoster virus. J. Virol. 2006, 80, 2076–2082. [Google Scholar] [CrossRef]
- Gomi, Y.; Sunamachi, H.; Mori, Y.; Nagaike, K.; Takahashi, M.; Yamanishi, K. Comparison of the complete DNA sequences of the Oka varicella vaccine and its parental virus. J. Virol. 2002, 76, 11447–11459. [Google Scholar] [CrossRef]
- Quinlivan, M.; Gershon, A.A.; Steinberg, S.P.; Breuer, J. An evaluation of single nucleotide polymorphisms used to differentiate vaccine and wild type strains of varicella-zoster virus. J. Med. Virol. 2005, 75, 174–180. [Google Scholar] [CrossRef]
- Kim, J.I.; Jung, G.S.; Kim, Y.Y.; Ji, G.Y.; Kim, H.S.; Wang, W.D.; Lee, K.M.; Park, H.S.; Park, S.Y.; Kim, G.E.; et al. Sequencing and characterization of Varicella-zoster virus vaccine strain SuduVax. Virol. J. 2011, 8, 547. [Google Scholar] [CrossRef]
- Oh, S.H.; Choi, E.H.; Shin, S.H.; Kim, Y.K.; Chang, J.K.; Choi, K.M.; Lee, S.Y.; Hur, J.K.; Kim, K.-H.; Kim, J.Y.; et al. Varicella and varicella vaccination in South Korea. Clin. Vaccine Immunol. 2014, 21, 762–768. [Google Scholar] [CrossRef]
- Jeon, J.S.; Won, Y.H.; Kim, I.K.; Ahn, J.H.; Shin, O.S.; Kim, J.H.; Lee, C.H. Analysis of single nucleotide polymorphism among Varicella-Zoster Virus and identification of vaccine-specific sites. Virology 2016, 496, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Abendroth, A.; Kinchington, P.R.; Slobedman, B. Varicella zoster virus immune evasion strategies. Curr. Top. Microbiol. Immunol. 2010, 342, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Sen, N.; Oliver, S.L.; Arvin, A.M. Molecular mechanisms of varicella zoster virus pathogenesis. Nat. Rev. Microbiol. 2014, 12, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Park, S.K.; Seo, S.W.; Lee, C.H.; Shin, O.S. STING Is Involved in Antiviral Immune Response against VZV Infection via the Induction of Type I and III IFN Pathways. J. Investig. Dermatol. 2017, 137, 2101–2109. [Google Scholar] [CrossRef]
- Jones, M.; Dry, I.R.; Frampton, D.; Singh, M.; Kanda, R.K.; Yee, M.B.; Breuer, J.; Kellam, P.; Hollinshead, M.; Kinchington, P.R.; et al. RNA-seq analysis of host and viral gene expression highlights interaction between varicella zoster virus and keratinocyte differentiation. PLoS Pathog. 2014, 10, e1003896. [Google Scholar] [CrossRef]
- Baird, N.L.; Bowlin, J.L.; Cohrs, R.J.; Gilden, D.; Jones, K.L. Comparison of varicella-zoster virus RNA sequences in human neurons and fibroblasts. J. Virol. 2014, 88, 5877–5880. [Google Scholar] [CrossRef]
- Oh, S.J.; Choi, Y.K.; Shin, O.S. Systems biology-based platforms to accelerate research of emerging infectious diseases. Yonsei Med. J. 2018, 59, 176–186. [Google Scholar] [CrossRef]
- Kim, J.A.; Park, S.K.; Kumar, M.; Lee, C.H.; Shin, O.S. Insights into the role of immunosenescence during varicella zoster virus infection (shingles) in the aging cell model. Oncotarget 2015, 6, 35324–35343. [Google Scholar] [CrossRef][Green Version]
- Kim, S.C.; Won, Y.H.; Park, J.S.; Jeon, J.S.; Ahn, J.H.; Song, M.J.; Lee, C.H.; Shin, O.S. Vaccine-type mutations identified in Varicella zoster virus passaged in cell culture. Virus Res. 2018, 245, 62–68. [Google Scholar] [CrossRef]
- Won, Y.H.; Kim, J.I.; Kim, Y.Y.; Lee, C.H. Characterization of the Repeat Sequences of Varicella-Zoster Virus. J. Bacteriol. Virol. 2014, 44, 326–335. [Google Scholar] [CrossRef]
- Park, S.J.; Kumar, M.; Kwon, H.I.; Seong, R.K.; Han, K.; Song, J.M.; Shin, O.S.; Kim, C.-J.; Choi, Y.-K. Dynamic changes in host gene expression associated with H5N8 avian influenza virus infection in mice. Sci. Rep. 2015, 5, 16512. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 21 March 2018).
- Criscuolo, A.; Brisse, S. AlienTrimmer removes adapter oligonucleotides with high sensitivity in short-insert paired-end reads. Commentary on Turner (2014) Assessment of insert sizes and adapter content in FASTQ data from NexteraXT libraries. Front. Genet. 2014, 5, 130. [Google Scholar] [CrossRef] [PubMed]
- Flicek, P.; Ahmed, I.; Amode, M.R.; Barrell, D.; Beal, K.; Brent, S.; Fitzgerald, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; et al. Ensembl 2013. Nucleic Acids Res. 2013, 41, D48–D55. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S. miRBase: The microRNA sequence database. Methods Mol. Biol. 2006, 342, 129–138. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Jung, D.; Kim, B.; Freishtat, R.J.; Giri, M.; Hoffman, E.; Seo, J. miRTarVis: An interactive visual analysis tool for microRNA-mRNA expression profile data. BMC Proc. 2015, 9, S2. [Google Scholar] [CrossRef]
- Ko, H.; Lee, G.M.; Shin, O.S.; Song, M.J.; Lee, C.H.; Kim, Y.E.; Ahn, J.H. Analysis of IE62 mutations found in Varicella-Zoster virus vaccine strains for transactivation activity. J. Microbiol. 2018, 56, 441–448. [Google Scholar] [CrossRef]
- Sen, N.; Sommer, M.; Che, X.; White, K.; Ruyechan, W.T.; Arvin, A.M. Varicella-zoster virus immediate-early protein 62 blocks interferon regulatory factor 3 (IRF3) phosphorylation at key serine residues: A novel mechanism of IRF3 inhibition among herpesviruses. J. Virol. 2010, 84, 9240–9253. [Google Scholar] [CrossRef]
- Li, X.; Huang, Y.; Zhang, Y.; He, N. Evaluation of microRNA expression in patients with herpes zoster. Viruses 2016, 8, 326. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Zhu, Z.; Shi, Z.; Ge, Y.; Zhao, K.; Zhou, M.; Cui, L. Dysregulated microRNA expression in serum of non-vaccinated children with varicella. Viruses 2014, 6, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Terrier, O.; Textoris, J.; Carron, C.; Marcel, V.; Bourdon, J.C.; Rosa-Calatrava, M. Host microRNA molecular signatures associated with human H1N1 and H3N2 influenza A viruses reveal an unanticipated antiviral activity for miR-146a. J. Gen. Virol. 2013, 94, 985–995. [Google Scholar] [CrossRef]
- Wu, J.; Wei, B.; Wang, L. Host miR-146a promotes replication of human cytomegalovirus by suppressing type I IFN response in MRC-5 cells. Int. J. Clin. Exp. Pathol. 2016, 9, 10044–10051. [Google Scholar]
- Stewart, C.R.; Marsh, G.A.; Jenkins, K.A.; Gantier, M.P.; Tizard, M.L.; Middleton, D.; Deffrasnes, C.; Lowenthal, J.W.; Haining, J.; Izzard, L.; et al. Promotion of Hendra virus replication by microRNA 146a. J. Virol. 2013, 87, 3782–3791. [Google Scholar] [CrossRef]
- Bandiera, S.; Pernot, S.; El Saghire, H.; Durand, S.C.; Thumann, C.; Crouchet, E.; Schuster, C.; Ye, T.; Fofana, I.; Oudot, M.A.; et al. Hepatitis C virus-induced upregulation of MicroRNA miR-146a-5p in hepatocytes promotes viral infection and deregulates metabolic pathways associated with liver disease pathogenesis. J. Virol. 2016, 90, 6387–6400. [Google Scholar] [CrossRef]
- Gowrishankar, K.; Slobedman, B.; Cunningham, A.L.; Miranda-Saksena, M.; Boadle, R.A.; Abendroth, A. Productive varicella-zoster virus infection of cultured intact human ganglia. J. Virol. 2007, 81, 6752–6756. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | DEGs upregulated only in wildtype VZV | DEGs upregulated only in attenuated VZV |
|---|---|---|
| 1 | AKNAD1 | BST2 |
| 2 | ANKRD26P1 | CD74 |
| 3 | DNAH12 | CFB |
| 4 | FAM71F1 | COX6B2 |
| 5 | GAL3ST1 | CTSS |
| 6 | GALNT3 | DDX58 |
| 7 | GRIP2 | DLL4 |
| 8 | IFIT2 | ICAM1 |
| 9 | INPP5J | IFI44 |
| 10 | KBTBD8 | IFIH1 |
| 11 | LAT2 | IFITM1 |
| 12 | MYO5C | IL8 |
| 13 | NEB | ISG20 |
| 14 | PLEKHA6 | LINC00475 |
| 15 | SEMA4D | LYPD3 |
| 16 | SLC6A13 | ODF3B |
| 17 | ZBTB20 | PLSCR1 |
| 18 | RTP4 | |
| 19 | TNFAIP6 | |
| 20 | TYMP |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, S.-J.; Lim, S.; Song, M.J.; Ahn, J.H.; Lee, C.H.; Shin, O.S. Whole Transcriptome Analyses Reveal Differential mRNA and microRNA Expression Profiles in Primary Human Dermal Fibroblasts Infected with Clinical or Vaccine Strains of Varicella Zoster Virus. Pathogens 2019, 8, 183. https://doi.org/10.3390/pathogens8040183
Oh S-J, Lim S, Song MJ, Ahn JH, Lee CH, Shin OS. Whole Transcriptome Analyses Reveal Differential mRNA and microRNA Expression Profiles in Primary Human Dermal Fibroblasts Infected with Clinical or Vaccine Strains of Varicella Zoster Virus. Pathogens. 2019; 8(4):183. https://doi.org/10.3390/pathogens8040183
Chicago/Turabian StyleOh, Soo-Jin, Sooyeon Lim, Moon Jung Song, Jin Hyun Ahn, Chan Hee Lee, and Ok Sarah Shin. 2019. "Whole Transcriptome Analyses Reveal Differential mRNA and microRNA Expression Profiles in Primary Human Dermal Fibroblasts Infected with Clinical or Vaccine Strains of Varicella Zoster Virus" Pathogens 8, no. 4: 183. https://doi.org/10.3390/pathogens8040183
APA StyleOh, S.-J., Lim, S., Song, M. J., Ahn, J. H., Lee, C. H., & Shin, O. S. (2019). Whole Transcriptome Analyses Reveal Differential mRNA and microRNA Expression Profiles in Primary Human Dermal Fibroblasts Infected with Clinical or Vaccine Strains of Varicella Zoster Virus. Pathogens, 8(4), 183. https://doi.org/10.3390/pathogens8040183