A Single Dose of Modified Vaccinia Ankara Expressing Lassa Virus-like Particles Protects Mice from Lethal Intra-cerebral Virus Challenge


 , ,
, ,
Abstract
1. Introduction
2. Results
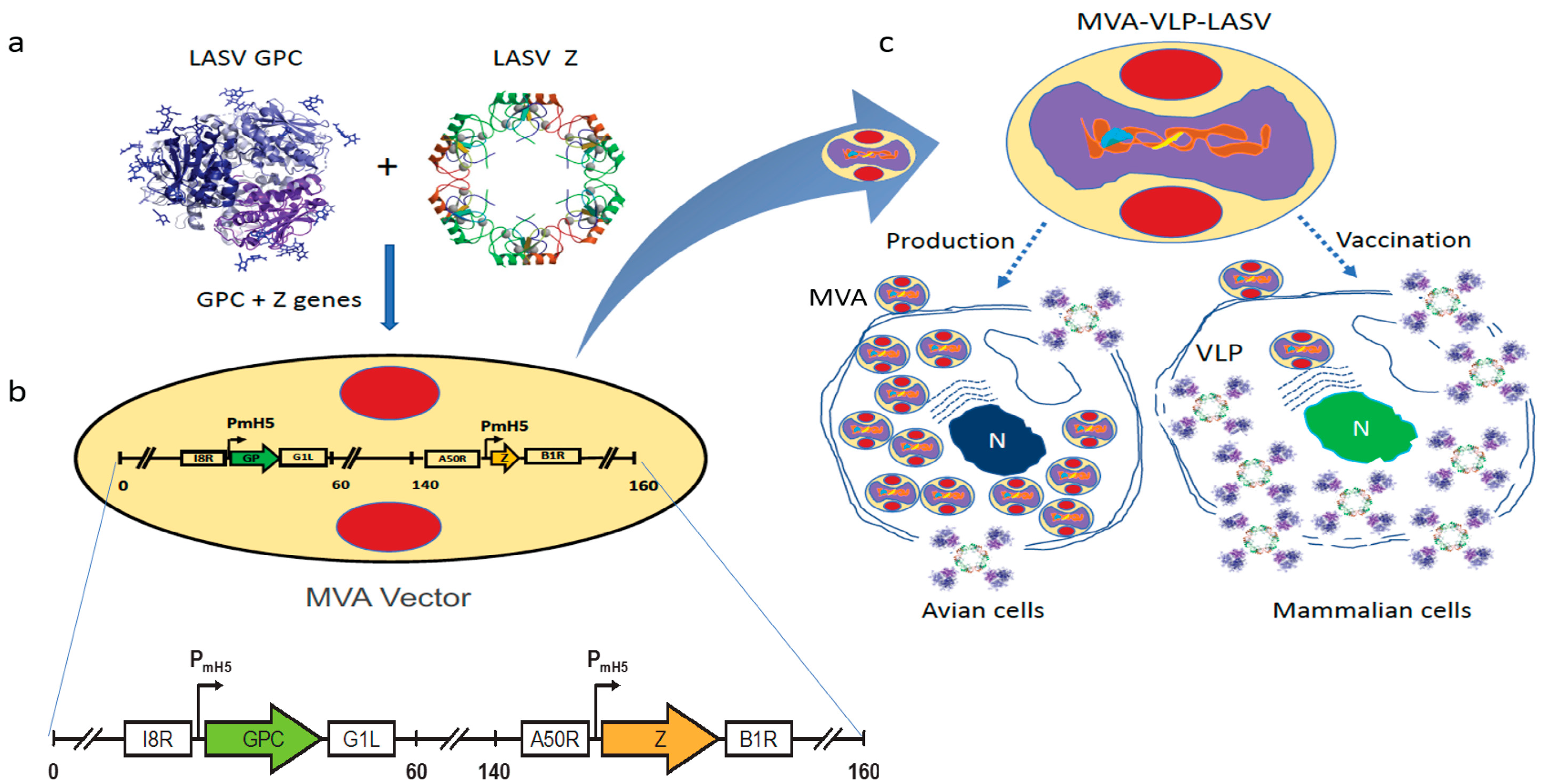
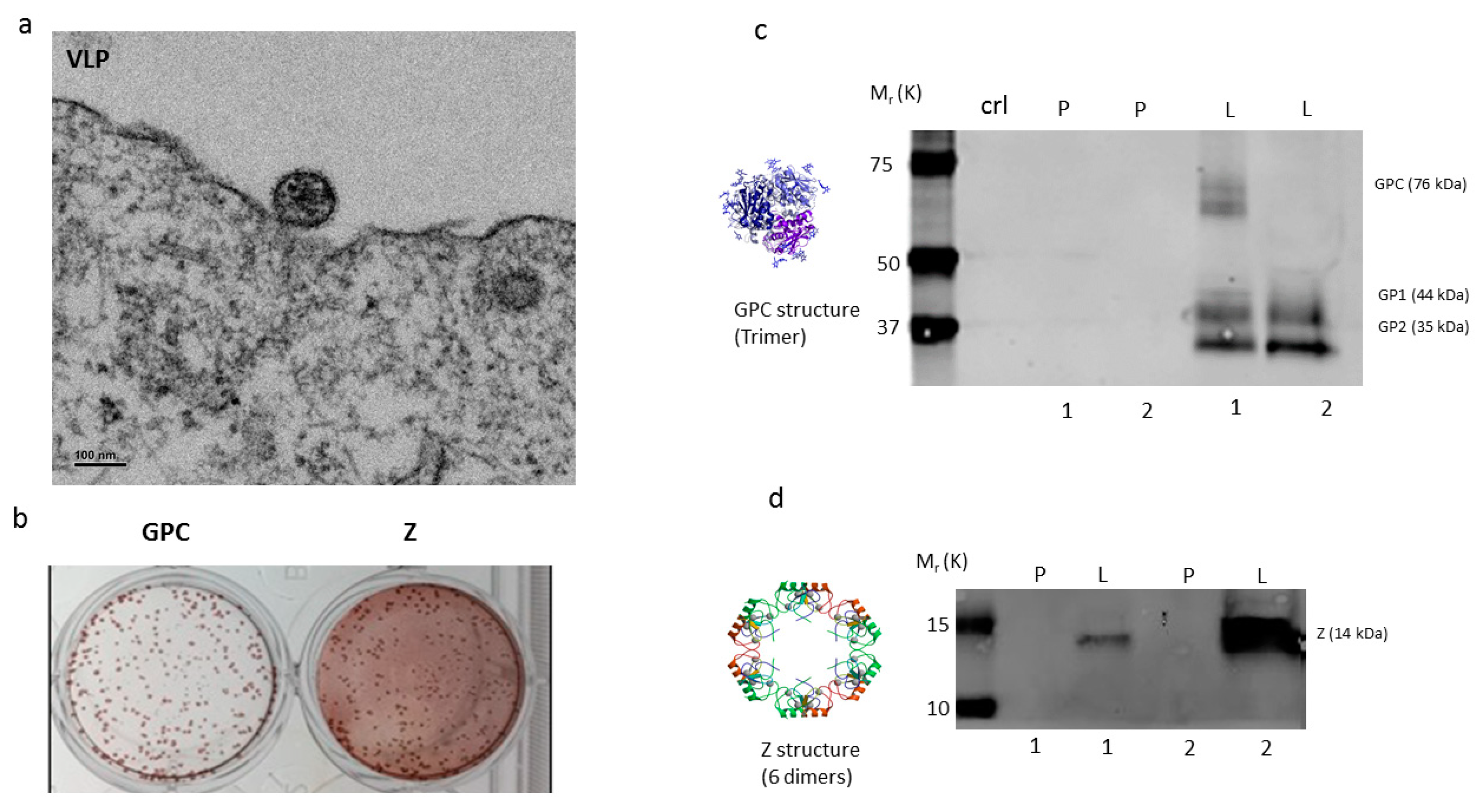
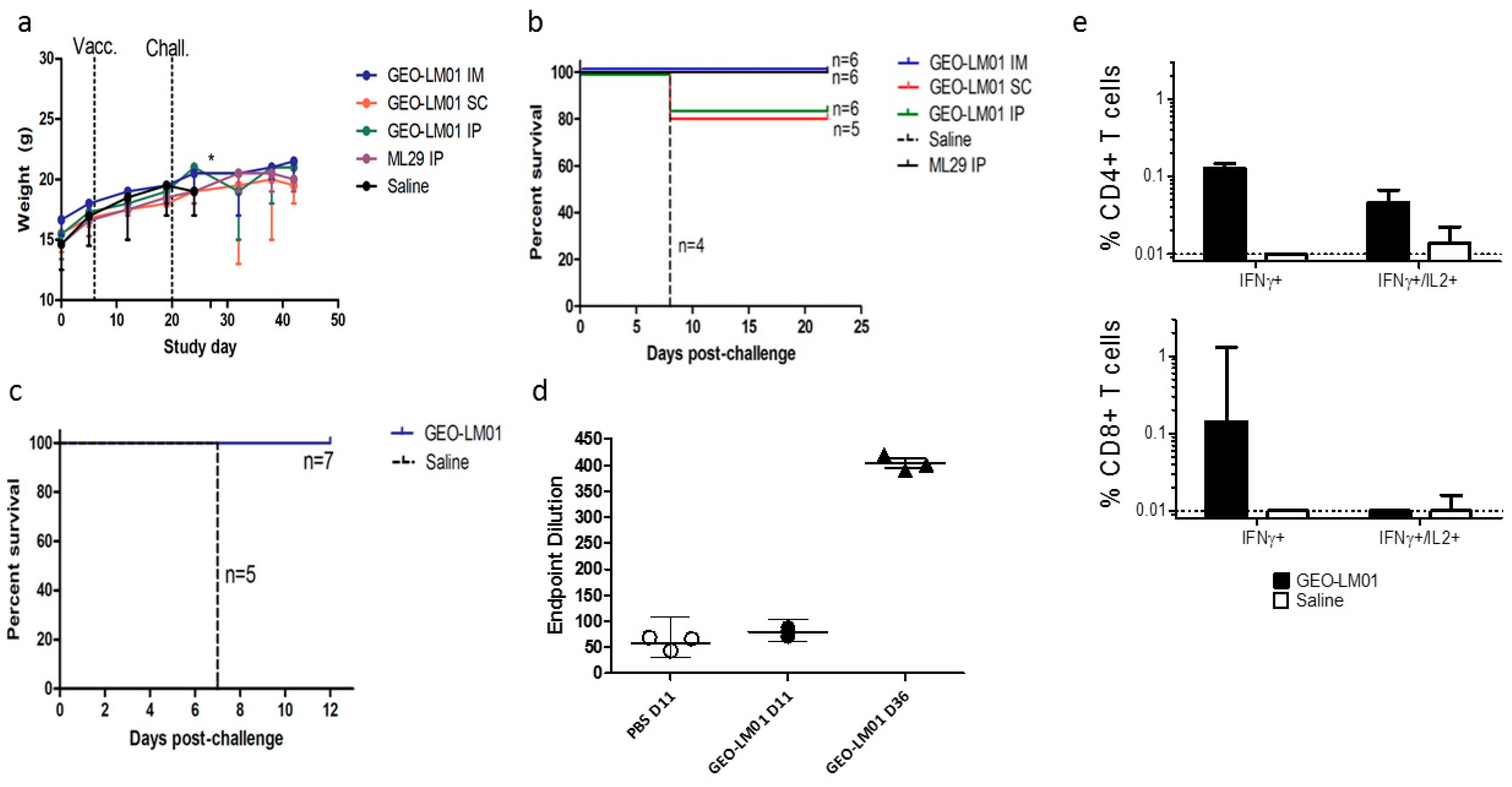
2.1. Vaccine Characterization
2.2. Vaccine Efficacy Testing in a ML29 Mouse Challenge Model
3. Discussion
4. Materials and Methods
4.1. Vaccine Construction, Seed Stock Preparation, VLP Formation, and Protein Expression
4.2. Mouse Immunogenicity and Efficacy Studies
4.2.1. Experiment 1. Determination of Vaccine Efficacy by Different Routes
4.2.2. Experiment 2. Confirmation of Route and Immune Response
4.3. Splenocyte Isolations and T Cell Assay
4.4. ELISA
4.5. Statistical Analyses
4.6. Regulatory Compliance
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Epidemic Focus: Lassa fever Weekly Epidemiological Record. Available online: https://www.who.int/wer/2016/wer9121.pdf?ua=1 (accessed on 27 May 2016).
- Shao, J.; Liang, Y.; Ly, H. Human Hemorrhagic Fever Causing Arenaviruses: Molecular Mechanisms Contributing to Virus Virulence and Disease Pathogenesis. Pathogens 2015, 4, 283–306. [Google Scholar] [CrossRef] [PubMed]
- Leski, T.A.; Stockelman, M.G.; Moses, L.M.; Park, M.; Stenger, D.A.; Ansumana, R.; Bausch, D.G.; Lin, B. Sequence Variability and Geographic Distribution of Lassa Virus, Sierra Leone. Emerg. Infect. Dis. 2015, 21, 609–618. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.B.; Webb, P.A.; Krebs, J.W.; Johnson, K.M.; Smith, E.S. A Prospective Study of the Epidemiology and Ecology of Lassa Fever. J. Infect. Dis. 1987, 155, 437–444. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.B. Lassa Fever, in Emergence and Control of Rodent-Borne Viral Diseases; Saluzzo, J.F., Dodet, B., Eds.; Elsevier: Amsterdam, The Nertherlands, 1999; pp. 177–195. [Google Scholar]
- Richmond, J.K.; Baglole, D.J. Lassa fever: Epidemiology, clinical features, and social consequences. BMJ 2003, 327, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Control, N.C.f.D. Disease Situation Reports. 2018. Available online: https://ncdc.gov.ng/diseases/sitreps (accessed on 2 September 2018).
- Olayemi, A.; Cadar, D.; Magassouba, N.F.; Obadare, A.; Kourouma, F.; Oyeyiola, A.; Fasogbon, S.; Igbokwe, J.; Rieger, T.; Bockholt, S.; et al. New Hosts of The Lassa Virus. Sci. Rep. 2016, 6, 25280. [Google Scholar] [CrossRef] [PubMed]
- Fisher-Hoch, S.P.; Tomori, O.; Nasidi, A.; I Perez-Oronoz, G.; Fakile, Y.; Hutwagner, L.; McCormick, J.B. Review of cases of nosocomial Lassa fever in Nigeria: The high price of poor medical practice. BMJ 1995, 311, 857–859. [Google Scholar] [CrossRef] [PubMed]
- Mylne, A.Q.N.; Pigott, D.M.; Longbottom, J.; Shearer, F.; Duda, K.A.; Messina, J.P.; Weiss, D.J.; Moyes, C.L.; Golding, N.; Hay, S.I. Mapping the zoonotic niche of Lassa fever in Africa. Trans. R. Soc. Trop. Med. Hyg. 2015, 109, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Bowen, M.D.; Rollin, P.E.; Ksiazek, T.G.; Hustad, H.L.; Bausch, D.G.; Demby, A.H.; Bajani, M.D.; Peters, C.J.; Nichol, S.T. Genetic Diversity among Lassa Virus Strains. J. Virol. 2000, 74, 6992–7004. [Google Scholar] [CrossRef] [PubMed]
- Hallam, H.J.; Hallam, S.; Rodriguez, S.E.; Barrett, A.D.T.; Beasley, D.W.C.; Chua, A.; Ksiazek, T.G.; Milligan, G.N.; Sathiyamoorthy, V.; Reece, L.M. Baseline mapping of Lassa fever virology, epidemiology and vaccine research and development. NPJ Vaccines 2018, 3, 11. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Bao, Y.; Buchmeier, M.J.; Charrel, R.N.; Clawson, A.N.; Clegg, C.S.; DeRisi, J.L.; Emonet, S.; Gonzalez, J.-P.; Kuhn, J.H.; et al. Past, present, and future of arenavirus taxonomy. Arch. Virol. 2015, 160, 1851–1874. [Google Scholar] [CrossRef] [PubMed]
- Djavani, M.; Lukashevich, I.S.; Sanchez, A.; Nichol, S.T.; Salvato, M.S. Completion of the Lassa fever virus sequence and identification of a RING finger ORF at the L RNA 5′ end. Virology 1997, 235, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Lukashevich, I.S.; Nichol, S.T.; Shapiro, K.; Ravkov, E.; Sanchez, A.; Djavani, M.; Salvato, M.S. The Lassa fever virus L gene: Nucleotide sequence, comparison, and precipitation of a predicted 250 kDa protein with monospecific antiserum. J. Gen. Virol. 1997, 78, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Hastie, K.M.; Zandonatti, M.A.; Kleinfelter, L.M.; Heinrich, M.L.; Rowland, M.M.; Chandran, K.; Branco, L.M.; Robinson, J.E.; Garry, R.F.; Saphire, E.O. Structural basis for antibody-mediated neutralization of Lassa virus. Science 2017, 356, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Lenz, O.; Ter Meulen, J.; Klenk, H.-D.; Seidah, N.G.; Garten, W. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc. Natl. Acad. Sci. USA 2001, 98, 12701–12705. [Google Scholar] [CrossRef] [PubMed]
- Ölschläger, S.; Flatz, L. Vaccination Strategies against Highly Pathogenic Arenaviruses: The Next Steps toward Clinical Trials. PLoS Pathog. 2013, 9, e1003212. [Google Scholar] [CrossRef] [PubMed]
- Urata, S.; Yasuda, J. Cis- and cell-type-dependent trans-requirements for Lassa virus-like particle production. J. Gen. Virol. 2015, 96 Pt 7, 1626–1635. [Google Scholar] [CrossRef]
- Safronetz, D.; Mire, C.; Rosenke, K.; Feldmann, F.; Haddock, E.; Geisbert, T.; Feldmann, H. A Recombinant Vesicular Stomatitis Virus-Based Lassa Fever Vaccine Protects Guinea Pigs and Macaques against Challenge with Geographically and Genetically Distinct Lassa Viruses. PLoS Negl. Trop. Dis. 2015, 9, e0003736. [Google Scholar] [CrossRef]
- Verheust, C.; Goossens, M.; Pauwels, K.; Breyer, D. Biosafety aspects of modified vaccinia virus Ankara (MVA)-based vectors used for gene therapy or vaccination. Vaccine 2012, 30, 2623–2632. [Google Scholar] [CrossRef]
- Hastie, K.M.; Zandonatti, M.; Liu, T.; Li, S.; Woods, V.L.; Saphire, E.O. Crystal Structure of the Oligomeric Form of Lassa Virus Matrix Protein Z. J. Virol. 2016, 90, 4556–4562. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Patterson, J.; Carrion, R.; Moshkoff, D.; Ticer, A.; Zapata, J.; Brasky, K.; Geiger, R.; Hubbard, G.B.; Bryant, J.; et al. A Live Attenuated Vaccine for Lassa Fever Made by Reassortment of Lassa and Mopeia Viruses. J. Virol. 2005, 79, 13934–13942. [Google Scholar] [CrossRef]
- Goicochea, M.A.; Zapata, J.C.; Bryant, J.; Davis, H.; Salvato, M.S.; Lukashevich, I.S. Evaluation of Lassa virus vaccine immunogenicity in a CBA/JML29 mouse model. Vaccine 2012, 30, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Botten, J.; Alexander, J.; Pasquetto, V.; Sidney, J.; Barrowman, P.; Ting, J.; Peters, B.; Southwood, S.; Stewart, B.; Rodriguez-Carreno, M.P.; et al. Identification of Protective Lassa Virus Epitopes That Are Restricted by HLA-A2. J. Virol. 2006, 80, 8351–8361. [Google Scholar] [CrossRef]
- Boesen, A.; Sundar, K.; Coico, R. Lassa Fever Virus Peptides Predicted by Computational Analysis Induce Epitope-Specific Cytotoxic-T-Lymphocyte Responses in HLA-A2.1 Transgenic Mice. Clin. Vaccine Immunol. 2005, 12, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Kyei, N.N.; Abilba, M.M.; Kwawu, F.K.; Agbenohevi, P.G.; Bonney, J.H.; Agbemaple, T.K.; Nimo-Paintsil, S.C.; Ampofo, W.; Ohene, S.-A.; Nyarko, E.O. Imported Lassa fever: A report of 2 cases in Ghana. BMC Infect. Dis. 2015, 15, 217. [Google Scholar] [CrossRef] [PubMed]
- Mehand, M.S.; Al Shorbaji, F.; Millett, P.; Murgue, B. The WHO R&D Blueprint: 2018 review of emerging infectious diseases requiring urgent research and development efforts. Antivir. Res. 2018, 159, 63–67. [Google Scholar] [PubMed]
- CEPI. News. 2018. Available online: http://cepi.net/news (accessed on 2 September 2018).
- Zapata, J.C.; Medina-Moreno, S.; Guzmán-Cardozo, C.; Salvato, M.S. Improving the Breadth of the Host’s Immune Response to Lassa Virus. Pathogens 2018, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Ter Meulen, V. Lassa fever: Implications of T-cell immunity for vaccine development. J. Biotechnol. 1999, 73, 207–212. [Google Scholar] [CrossRef]
- Domi, A.; Feldmann, F.; Basu, R.; McCurley, N.; Shifflett, K.; Emanuel, J.; Hellerstein, M.S.; Guirakhoo, F.; Orlandi, C.; Flinko, R.; et al. A Single Dose of Modified Vaccinia Ankara expressing Ebola Virus Like Particles Protects Nonhuman Primates from Lethal Ebola Virus Challenge. Sci. Rep. 2018, 8, 864. [Google Scholar] [CrossRef]
- Brault, A.C.; Domi, A.; McDonald, E.M.; Talmi-Frank, D.; McCurley, N.; Basu, R.; Robinson, H.L.; Hellerstein, M.; Duggal, N.K.; Bowen, R.A.; et al. A Zika Vaccine Targeting NS1 Protein Protects Immunocompetent Adult Mice in a Lethal Challenge Model. Sci. Rep. 2017, 7, 14769. [Google Scholar] [CrossRef]
- Djomand, G.; Quaye, S.; Sullivan, P.S. HIV epidemic among key populations in west Africa. Curr. Opin. HIV AIDS 2014, 9, 506–513. [Google Scholar] [CrossRef]
- Zapata, J.C.; Poonia, B.; Bryant, J.; Davis, H.; Ateh, E.; George, L.; Crasta, O.; Zhang, Y.; Slezak, T.; Jaing, C.; et al. An attenuated Lassa vaccine in SIV-infected rhesus macaques does not persist or cause arenavirus disease but does elicit Lassa virus-specific immunity. Virol. J. 2013, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Sutter, G.; Moss, B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc. Natl. Acad. Sci. USA 1992, 89, 10847–10851. [Google Scholar] [CrossRef] [PubMed]
- Overton, E.T.; Stapleton, J.; Frank, I.; Hassler, S.; Goepfert, P.A.; Barker, D.; Wagner, E.; Von Krempelhuber, A.; Virgin, G.; Weigl, J.; et al. Safety and Immunogenicity of Modified Vaccinia Ankara-Bavarian Nordic Smallpox Vaccine in Vaccinia-Naive and Experienced Human Immunodeficiency Virus-Infected Individuals: An Open-Label, Controlled Clinical Phase II Trial. Open Forum Infect. Dis. 2015, 2, ofv040. [Google Scholar] [CrossRef] [PubMed]
- Committee for Medicinal Products for Human Use (CHMP). Assessment Report, IMVANEX, Common Name: Modified Vaccinia Ankara Virus, Procedure No. EMEA/H/C/002596; Committee for Medicinal Products for Human Use (CHMP), Ed.; European Medicines Agency: London, UK, 2013. [Google Scholar]
- Bliss, C.M.; Bowyer, G.; Anagnostou, N.A.; Havelock, T.; Snudden, C.M.; Davies, H.; De Cassan, S.C.; Grobbelaar, A.; Lawrie, A.M.; Venkatraman, N.; et al. Assessment of novel vaccination regimens using viral vectored liver stage malaria vaccines encoding ME-TRAP. Sci. Rep. 2018, 8, 3390. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, P.A.; Elizaga, M.L.; Sato, A.; Qin, L.; Cardinali, M.; Hay, C.M.; Hural, J.; DeRosa, S.C.; Defawe, O.D.; Tomaras, G.D.; et al. Phase 1 Safety and Immunogenicity Testing of DNA and Recombinant Modified Vaccinia Ankara Vaccines Expressing HIV-1 Virus-like Particles. J. Infect. Dis. 2011, 203, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, P.A.; Elizaga, M.L.; Seaton, K.; Tomaras, G.D.; Montefiori, D.C.; Sato, A.; Hural, J.; DeRosa, S.C.; Kalams, S.A.; McElrath, M.J.; et al. Specificity and 6-Month Durability of Immune Responses Induced by DNA and Recombinant Modified Vaccinia Ankara Vaccines Expressing HIV-1 Virus-Like Particles. J. Infect. Dis. 2014, 210, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.; Heath, S.L.; Sweeton, B.; Williams, K.; Cunningham, P.; Keele, B.F.; Sen, S.; Palmer, B.E.; Chomont, N.; Xu, Y.; et al. DNA/MVA Vaccination of HIV-1 Infected Participants with Viral Suppression on Antiretroviral Therapy, followed by Treatment Interruption: Elicitation of Immune Responses without Control of Re-Emergent Virus. PLoS ONE 2016, 11, 0163164. [Google Scholar] [CrossRef]
- Maciel, M.M., Jr.; Cruz, F.D.S.P.; Cordeiro, M.T.; da Motta, M.A.; de Melo Cassemiro, K.M.S.; Maia, R.D.C.C.; de Figueiredo, R.C.B.Q.; Galler, R.; da Silva Freire, M.; August, J.T.; et al. A DNA Vaccine against Yellow Fever Virus: Development and Evaluation. PLoS Negl. Trop. Dis. 2015, 9, e0003693. [Google Scholar] [CrossRef]
- Perdomo-Celis, F.; Salvato, M.S.; Medina-Moreno, S.; Zapata, J.C. T-Cell Response to Viral Hemorrhagic Fevers. Vaccines 2019, 7, 11. [Google Scholar] [CrossRef]
- Cross, R.W.; Mire, C.E.; Branco, L.M.; Geisbert, J.B.; Rowland, M.M.; Heinrich, M.L.; Goba, A.; Momoh, M.; Grant, D.S.; Fullah, M.; et al. Treatment of Lassa virus infection in outbred guinea pigs with first-in-class human monoclonal antibodies. Antivir. Res. 2016, 133, 218–222. [Google Scholar] [CrossRef]
- Mire, C.E.; Cross, R.W.; Geisbert, J.B.; Borisevich, V.; Agans, K.N.; Deer, D.J.; Heinrich, M.L.; Rowland, M.M.; Goba, A.; Momoh, M.; et al. Human-monoclonal-antibody therapy protects nonhuman primates against advanced Lassa fever. Nat. Med. 2017, 23, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Lukashevich, I.S.; Carrion, R.; Salvato, M.S.; Mansfield, K.; Brasky, K.; Zapata, J.; Cairo, C.; Goicochea, M.; Hoosien, G.E.; Ticer, A.; et al. Safety, immunogenicity, and efficacy of the ML29 reassortant vaccine for Lassa fever in small non-human primates. Vaccine 2008, 26, 5246–5254. [Google Scholar] [CrossRef] [PubMed]
- Kiley, M. Protection of Rhesus Monkeys from Lassa Virus by Immunisation With Closely Related Arenavirus. Lancet 1979, 314, 738. [Google Scholar] [CrossRef]
- Clegg, J.; Lloyd, G. Vaccinia Recombinant Expressing Lassa-Virus Internal Nucleocapsid Protein Protects Guinea pigs against Lassa Fever. Lancet 1987, 330, 186–188. [Google Scholar] [CrossRef]
- McCurley, N.P.; Domi, A.; Basu, R.; Saunders, K.O.; Labranche, C.C.; Montefiori, D.C.; Haynes, B.F.; Robinson, H.L. HIV transmitted/founder vaccines elicit autologous tier 2 neutralizing antibodies for the CD4 binding site. PLoS ONE 2017, 12, e0177863. [Google Scholar] [CrossRef] [PubMed]
- Sommerstein, R.; Flatz, L.; Remy, M.M.; Malinge, P.; Magistrelli, G.; Fischer, N.; Sahin, M.; Bergthaler, A.; Igonet, S.; Ter Meulen, J.; et al. Arenavirus Glycan Shield Promotes Neutralizing Antibody Evasion and Protracted Infection. PLoS Pathog. 2015, 11, e1005276. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.; Dowall, S.; Salguero, F.; Yeates, P.; Aram, M.; Hewson, R. A vaccine based on recombinant modified Vaccinia Ankara containing the nucleoprotein from Lassa virus protects against disease progression in a guinea pig model. Vaccine 2019, 37, 5404–5413. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.E.; Hastie, K.M.; Cross, R.W.; Yenni, R.E.; Elliott, D.H.; Rouelle, J.A.; Kannadka, C.B.; Smira, A.A.; Garry, C.E.; Bradley, B.T.; et al. Most neutralizing human monoclonal antibodies target novel epitopes requiring both Lassa virus glycoprotein subunits. Nat. Commun. 2016, 7, 11544. [Google Scholar] [CrossRef]
- Altenburg, A.F.; Kreijtz, J.H.C.M.; De Vries, R.D.; Song, F.; Fux, R.; Rimmelzwaan, G.F.; Sutter, G.; Volz, A. Modified Vaccinia Virus Ankara (MVA) as Production Platform for Vaccines against Influenza and Other Viral Respiratory Diseases. Viruses 2014, 6, 2735–2761. [Google Scholar] [CrossRef]
- Moss, B.; Carroll, M.W.; Wyatt, L.S.; Bennink, J.R.; Hirsch, V.M.; Goldstein, S.; Elkins, W.R.; Fuerst, T.R.; Lifson, J.D.; Piatak, M.; et al. Host Range Restricted, Non-Replicating Vaccinia Virus Vectors as Vaccine Candidates. Results Probl. Cell Differ. 1996, 397, 7–13. [Google Scholar]
- Wyatt, L.S.; Earl, P.L.; Vogt, J.; Eller, L.A.; Chandran, D.; Liu, J.; Robinson, H.L.; Moss, B. Correlation of immunogenicities and in vitro expression levels of recombinant modified vaccinia virus Ankara HIV vaccines. Vaccine 2008, 26, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Earl, P.L.; Xiao, W.; Americo, J.L.; Cotter, C.A.; Vogt, J.; Moss, B. Elucidating and Minimizing the Loss by Recombinant Vaccinia Virus of Human Immunodeficiency Virus Gene Expression Resulting from Spontaneous Mutations and Positive Selection. J. Virol. 2009, 83, 7176–7184. [Google Scholar] [CrossRef] [PubMed]
- Earl, P.L.; Cotter, C.; Moss, B.; VanCott, T.; Currier, J.; Eller, L.A.; McCutchan, F.; Birx, D.L.; Michael, N.L.; Marovich, M.A.; et al. Design and evaluation of multi-gene, multi-clade HIV-1 MVA vaccines. Vaccine 2009, 27, 5885–5895. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hellerstein, M.; Xu, Y.; Marino, T.; Lu, S.; Yi, H.; Wright, E.R.; Robinson, H.L. Co-expression of HIV-1 virus-like particles and granulocyte-macrophage colony stimulating factor by GEO-D03 DNA vaccine. Hum. Vaccines Immunother. 2012, 8, 1654–1658. [Google Scholar] [CrossRef] [PubMed]
- Salvato, M.S.; Lukashevich, I.S.; Medina-Moreno, S.; Zapata, J.C. Diagnostics for Lassa Fever: Detecting Host Antibody Responses, in Hemorrhagic Fever Viruses: Methods and Protocols; Humana Press: New York, NY, USA, 2018. [Google Scholar]
- Lázaro-Frías, A.; Gomez-Medina, S.; Sánchez-Sampedro, L.; Ljungberg, K.; Ustav, M.; Liljeström, P.; Muñoz-Fontela, C.; Esteban, M.; García-Arriaza, J. Distinct Immunogenicity and Efficacy of Poxvirus-Based Vaccine Candidates against Ebola Virus Expressing GP and VP40 Proteins. J. Virol. 2018, 92, e00363–e00418. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Mice | Number of Mice | Survivors after 2nd Challenge |
|---|---|---|
| Survivors of 1st challenge | 4 | 4 |
| Control mice (naïve adult CBA/J) | 4 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvato, M.S.; Domi, A.; Guzmán-Cardozo, C.; Medina-Moreno, S.; Zapata, J.C.; Hsu, H.; McCurley, N.; Basu, R.; Hauser, M.; Hellerstein, M.; et al. A Single Dose of Modified Vaccinia Ankara Expressing Lassa Virus-like Particles Protects Mice from Lethal Intra-cerebral Virus Challenge. Pathogens 2019, 8, 133. https://doi.org/10.3390/pathogens8030133
Salvato MS, Domi A, Guzmán-Cardozo C, Medina-Moreno S, Zapata JC, Hsu H, McCurley N, Basu R, Hauser M, Hellerstein M, et al. A Single Dose of Modified Vaccinia Ankara Expressing Lassa Virus-like Particles Protects Mice from Lethal Intra-cerebral Virus Challenge. Pathogens. 2019; 8(3):133. https://doi.org/10.3390/pathogens8030133
Chicago/Turabian StyleSalvato, Maria S., Arban Domi, Camila Guzmán-Cardozo, Sandra Medina-Moreno, Juan Carlos Zapata, Haoting Hsu, Nathanael McCurley, Rahul Basu, Mary Hauser, Michael Hellerstein, and et al. 2019. "A Single Dose of Modified Vaccinia Ankara Expressing Lassa Virus-like Particles Protects Mice from Lethal Intra-cerebral Virus Challenge" Pathogens 8, no. 3: 133. https://doi.org/10.3390/pathogens8030133
APA StyleSalvato, M. S., Domi, A., Guzmán-Cardozo, C., Medina-Moreno, S., Zapata, J. C., Hsu, H., McCurley, N., Basu, R., Hauser, M., Hellerstein, M., & Guirakhoo, F. (2019). A Single Dose of Modified Vaccinia Ankara Expressing Lassa Virus-like Particles Protects Mice from Lethal Intra-cerebral Virus Challenge. Pathogens, 8(3), 133. https://doi.org/10.3390/pathogens8030133