Abstract
The rise of bat-associated zoonotic viruses necessitates a close monitoring of their natural hosts. Since the detection of severe acute respiratory syndrome coronavirus (SARS-CoV), it is evident that bats are vital reservoirs of coronaviruses (CoVs). In this study, we investigated the presence of CoVs in multiple bat species in Nigeria to identify viruses in bats at high-risk human contact interfaces. Four hundred and nine bats comprising four bat species close to human habitats were individually sampled from five states in Nigeria between 2019 and 2021. Coronavirus detection was done using broadly reactive consensus PCR primers targeting the RNA-dependent RNA polymerase (RdRp) gene of CoVs. Coronavirus RNA was detected in 39 samples (9.5%, CI 95%: [7.0, 12.8]), of which 29 were successfully sequenced. The identified CoVs in Nigerian bats were from the unclassified African alphacoronavirus lineage and betacoronavirus lineage D (Nobecovirus), with one sample from Hipposideros ruber coinfected with alphacoronavirus and betacoronavirus. Different bat species roosting in similar or other places had CoVs from the same genetic lineage. The phylogenetic and evolutionary dynamics data indicated a high CoV diversity in Nigeria, while host switching may have contributed to CoV evolution. Robust sentinel surveillance is recommended to enhance our knowledge of emerging and re-emerging coronaviruses.
1. Introduction
Bat (order Chiroptera) are mammals with over 1300 species across 20 families and 175 genera, accounting for over 20% of known mammalian species globally [1]. They are widely spread in nature and play a significant role in the biological diversity of various ecosystems [2]. Multiple studies have documented the role of bats as reservoirs of different viral agents of public health importance, including the progenitors of SARS-CoV and SARS-CoV-2, the causative agent for the COVID-19 pandemic [3,4,5]. The origin of SARS-CoV-2 and the possible role of intermediate animal host(s) in early animal-to-human transmission are unanswered questions associated with the COVID-19 pandemic. Several studies have revealed various CoVs in African bats [6,7,8,9]. However, there is a paucity of data on bat CoVs in Nigeria, where we only have information on betacoronavirus infection in bats from North-Central and South-West Nigeria [10,11].
Bats are hunted and eaten in some parts of Africa, including Nigeria [12,13,14,15,16]. Hunters in Ghana (Afram plains and Volta regions) have confessed to consuming Eidolon helvum bats with the decision to hunt bats based on their family tradition, further enhanced by economic necessity [15]. At the same time, Rousettus aegyptiacus is heavily hunted in eastern Nigeria, and several hunters in South-South Nigeria (Niger Delta region) were reported to hunt bats occasionally [16]. In southwestern Nigeria, the straw-coloured fruit bat is popular meat [12]. This close interaction between humans and bats may allow the large-scale emergence of novel virus types and species with unpredictable pathogenicity and clinical impacts. Thus, proactive measures, including surveillance and enhanced pathogen discovery techniques in emerging infectious disease “hotspots”, especially when there are no known epidemics, might improve the early recognition of potential outbreaks and the detection of novel pathogens. This research aimed to catalogue coronavirus diversity in Nigerian bats, a critical component for public health measures to prevent future outbreaks that other bat coronaviruses may cause.
2. Materials and Methods
2.1. Study Area and Sample Collection
Samples were collected from insectivorous and free-ranging fruit bats in five states in Nigeria between November 2019 and May 2021 (Figure 1). Bats were trapped around fruit trees and human dwellings using harp traps and mist nets. Each captured bat was assessed, and morphological characteristics such as weight (g), forearm and tibia length (mm), sex, reproductive state, and age were recorded to determine bat species. Oral and rectal swabs were collected and placed into tubes containing 1 mL of virus transport medium. A few of the trapped bats were humanely euthanised under a veterinarian’s supervision in full compliance with the local ethical and legal guidelines, and voucher specimens were collected. All experiments were conducted in a microbiological safety station with personal protective equipment, a mask, and a visor. All samples were immediately transferred to −20 °C containers before being transported to the laboratory at the African Centre of Excellence for Genomics of Infectious Diseases (ACEGID), Redeemer’s University, Nigeria and stored at −80 °C until processed.
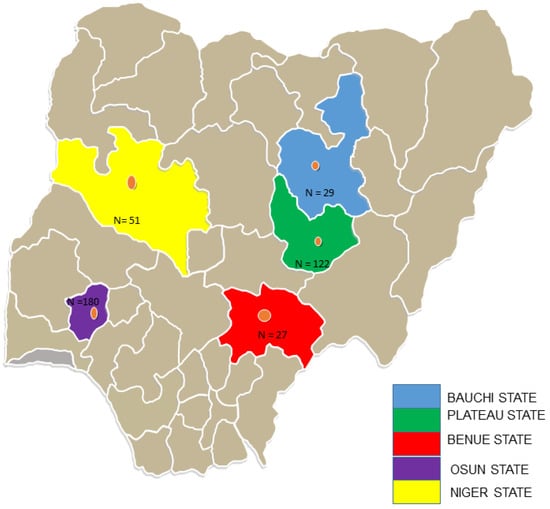
Figure 1.
Map of Nigeria showing the five states where bat samples were collected.
2.2. Nucleic Acid Extraction
Total RNA was extracted from oral and rectal swabs using the QIAamp® Viral RNA extraction kit (Qiagen®, Hilden, Germany) according to the manufacturer’s instructions with an elution volume of 60 μL. According to the manufacturer’s manual, DNA was isolated from faecal swabs using the DNeasy Blood and Tissue Kit (#69506; Qiagen).
2.3. Molecular Confirmation of Bat Species
Bat host species identification was confirmed for each bat in which coronavirus was detected by selectively amplifying segments of vertebrate mitochondrial cytochrome oxidase subunit 1 (COI) and cytochrome b (Cyt b) mtDNA [17]. Briefly, a fragment of approximately 700 bp of COI (primer pair COI_long-f 5′-AACCACAAAGACATTGGCAC-3′ and COI_long-r 5′-AAGAATCAGAATARGTGTTG-3′) and 520 bp of Cytb (primer pair Cytb-f 5′-GAGGMCAAATATCATTCTGAGG-3′ and Cytb-r 5′-TAGGGCVAGGACTCCTCCTAGT-3′) was amplified. PCR products were purified using the QIAquick® Gel and PCR Clean-up kit and sequenced directly using an automated ABI 3500xl DNA Sequencer at ACEGID, Redeemer’s University, Ede, Nigeria. Nucleotide sequences were edited using BioEdit Sequence Alignment Editor Version 7.2.6, and a BLASTn search was done to identify bat species. For samples with low similarity (<90%) hits with sequences in GenBank, an alignment with reference sequences was done using the MUSCLE program in MEGA 11 software with default settings [18,19], and phylogenetic trees were constructed using the maximum likelihood method.
2.4. RT-PCR Screening for Detection of Coronavirus RNA using Heminested Reverse-Transcription PCR (RT-PCR) and Sanger Sequencing
The detection of bat coronaviruses was done using heminested reverse-transcription PCR (RT-PCR) with broadly reactive consensus PCR primers targeting the RNA-dependent RNA polymerase (RdRp) gene of different CoVs as previously described [10]. The synthesis of cDNA was carried out using Superscript IV First-Strand Synthesis kit (Invitrogen) followed by the nested PCR. The amplified product of 328 bp was visualised using 2% agarose gel electrophoresis. The RdRp PCR products were purified using the QIAquickn® Gel and PCR Clean-up kit and sequenced directly using an automated ABI 3500xl DNA Sequencer available at ACEGID.
2.5. Phylogenetic Analysis
MAFFT online service [20] was used to align the sequences, and MEGA version 11 [19] was used to build a phylogenetic tree using the maximum likelihood with the gamma-distributed Hasegawa–Kishino–Yano (HKY+G) model [21], with 1000 bootstrap replications to assess phylogenetic robustness. The tree was visualised using Interactive Tree of Life (iTOL) v5 [22]. We aligned every unique pair of sequences, estimated sequence pairwise identity between sequences from our work, and published references using the Sequence Demarcation Tool (SDT) [23].
Phylogenetic trees were also generated by IQ-TREE version 1.6.12 5 [24] using a partition model [25] with ModelFinder [26] and ultrafast bootstrap (1000 replicates) [27] to explore the reconstruction of ancestral-state phylogeographic transmission across countries and host species. Coding genes were partitioned into first + second and third codon positions. Results were visualised using Microreact (https://microreact.org/ (accessed on 1 March 2022)) [28].
2.6. Statistical Analysis
The Wilson method [29] in the Epitools calculator (http://epitools.ausvet.com.au (accessed on 1 March 2022)) was used to calculate confidence intervals for prevalence.
3. Results
3.1. Bats Samples Collected and Prevalence of Coronavirus
From 2019 to 2021, 409 bat samples were collected from six roosting sites in five states in Nigeria (Table 1). After morphological inspection and sequence analysis of the Cyt b and COI mtDNA, these bats were classified into four species: E. helvum, Hipposideros ruber, Mops condylurus, and Chaerephon sp.

Table 1.
Georeferenced location, percentage of samples positive for coronavirus RNA in various bat families, and species sampled in 2019–2021.
Using nested RT-PCR, coronaviruses were detected in 39 samples, giving an overall detection rate of 9.5% (CI 95%: 7.0–12.8). The 39 samples identified as CoV positive included 8 (6.6%) among the 122 E. helvum from the Jos roosting site, 3 (3.5%) among the 86 H. ruber from the Ife roosting site, 3 (3.2%) among the 93 E. helvum from the Ife roosting site, 6 (22.2%) among the 27 M. condylurus from the Gboko/Benue roosting site, and 19 (37.3%) among the 51 Chaerephon sp. from the Paiko/Niger roosting site (Table 1). Positive samples were detected in bats from all the roosting sites except E. helvum samples collected in 2019 and 2020 from Bauchi and Ede. Bats positive for CoVs were captured in May and June 2020, October–December 2020 and January–March 2021. Coronaviruses were detected in both male and female bats irrespective of age. Coronaviruses were also seen in the pooled rectal and oral swabs (in the case of samples with low volume) and unpooled rectal and oral swabs, respectively (Table 2).
Table 2.
Characteristics of bats infected with coronavirus.
3.2. Molecular Characterization of Identified CoVs and Estimation of Divergence Time
Of the 39 RT-PCR-positive bat samples screened, 29 bat samples were successfully sequenced, and the nucleotide sequences were compared to those in the public database using the “Blastn” algorithm of NCBI BLAST and SDT. Of the 29 newly identified CoVs, 18 belonged to alphacoronaviruses (α-CoV) and 10 to betacoronaviruses (β-CoV). One sample from H. ruber bat (CER024_NGR) was coinfected with alphacoronavirus and betacoronavirus (Table 2). According to the findings, all the nucleotide sequences from β-CoV detected in this study were 91.7–98.7% identical to the E. helvum coronavirus in lineage D (Nobecovirus) previously reported in E. helvum bats in Cameroon, Ghana, Tanzania, and Kenya (Figure 2a). The α-CoV nucleotide sequences were 95.94–98.61% identical to the Alphacoronavirus genus (Chaerephon bat coronavirus) (Figure 2b).
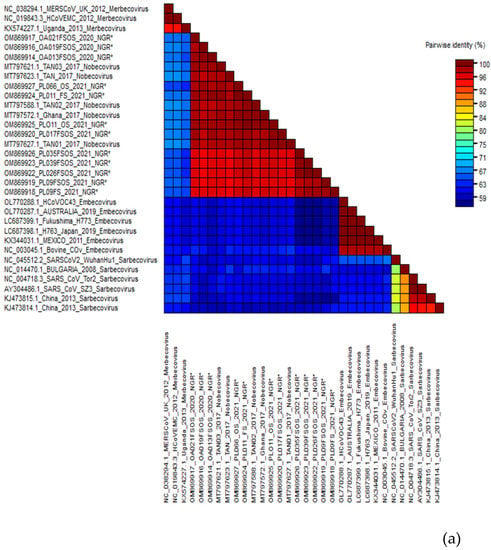
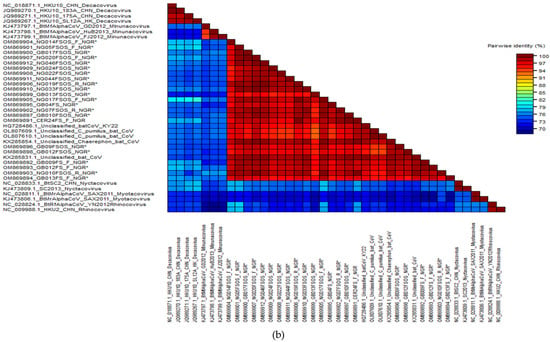
Figure 2.
(a) Alignment result of β-CoV sequences and estimated sequence pairwise identity between sequences from this study and published references (representative members from each β-CoV subgenera and close match from BLASTn search) using the Sequence Demarcation Tool. New sequences derived from the current study are denoted with an asterisk. (b) Alignment result of α-CoV sequences and estimated sequence pairwise identity between sequences from this study and published references (representative members from each α-CoV subgenera and close match from BLASTn search) using the Sequence Demarcation Tool. New sequences derived from the current study are denoted with an asterisk.
To determine the genetic relationships between the sequenced bat CoVs from this study and previously described CoVs, a phylogenetic analysis using the maximum likelihood technique was performed based on 327 bp RdRp truncated sequences. Phylogenetic analysis confirmed that all the β-CoV sequences from this study were in the Nobecovirus lineage, while the alphacoronavirus clustered within the unclassified African α-CoV lineage (Chaerephon bat coronavirus) (Figure 3).
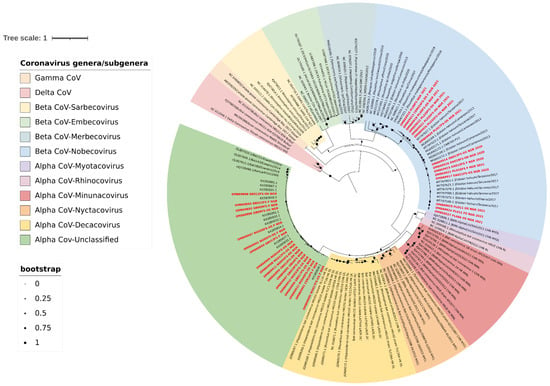
Figure 3.
Maximum likelihood tree with gamma-distributed Hasegawa–Kishino–Yano (HKY+G) model based on 327 bp RdRp sequences, with 1000 bootstrap replications. Sequences reported in this study are in bold red font. The tree was visualised using Interactive Tree of Life (iTOL) v5 with midpoint rooting and each coronavirus lineage colour-coded as shown in the legend.
The partition model tree with host trait revealed that virtually all the E. helvum infected bats were infected with Nobecovirus, with the subgenera circulating mainly in E. helvum, H. ruber, and Rousettu sp. bats in Africa. In contrast, the other subgenera were equally spread in diverse bat species. All the Sarbecovirus isolates were from human hosts and Rhinolophus sp. except a single bovine isolate (Figure 4).
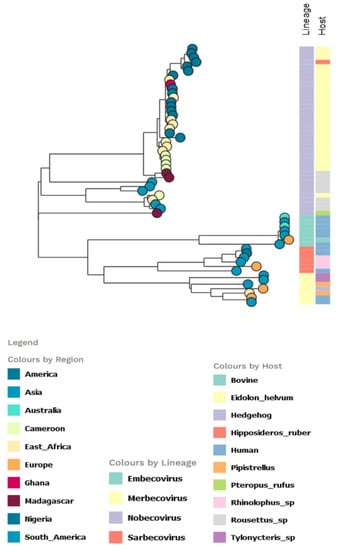
Figure 4.
Partitioned model tree to explore phylogeographic transmission across countries and host species of partial RdRp gene region of β-CoV. The taxa are represented by their countries of origin with the lineage and host equally shown.
For the AlphaCoV partition tree, the unclassified African lineage showed a more diversified host species distribution, with the Chaerephon sp. having the highest distribution, followed by M. condylurus among the Nigerian bat viruses (Figure 5). The lineage also infected other bat species in Africa, including Chaerephon pumilus (in Eswatini and Kenya).
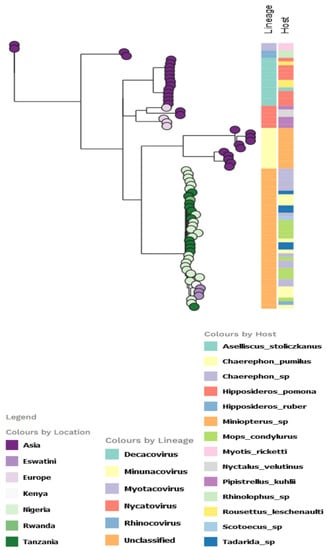
Figure 5.
Partitioned model tree to explore phylogeographic transmission across countries and host species of partial RdRp gene region of α-CoV. The taxa are represented by their countries of origin with the lineage and host equally shown.
4. Discussion
In this study, we analysed samples from 409 bats collected from five states in Nigeria. We detected coronavirus RNA in 9.5% (39) specimens sampled from all bat species and observed a high CoV diversity in Nigerian bats. Different bat species roosting in similar or other places had CoVs from the same genetic lineage, suggesting that host switching may contribute to CoV evolution in Nigeria. All the betacoronaviruses belonged to the E. helvum coronavirus in lineage D (Nobecovirus). In contrast, the alphacoronavirus belonged to the unclassified Chaerephon bat coronavirus lineage, which is the first report of this virus in Nigerian bats.
The overall prevalence of CoV in bats of 9.5% (CI 95%: 7.0–12.8) in this study is consistent with reports of CoVs in bats from Ghana, Germany, and a recent global survey of coronaviruses in bats, rodents, and nonhuman primates [6,30,31]. However, in previous studies from Nigeria, infection rates were generally lower [10,11,32]. High rates were observed in two insectivorous bat species in the family Molossidae (6/27 M. condylurus and 19/51 Chaerephon sp.). However, it is probable that the high rates observed in these bat species may be related to the season in which the samples were obtained. Seasonal variations in infection prevalence are most likely influenced by density fluctuations during colony establishment or migration, affecting contact rates and disease dynamics. Furthermore, due to continuous interaction among individuals inside a maternity roost, virus transmission is more easily facilitated during the breeding season than at other times [30,33,34]. Additionally, as previously suggested [35,36,37], coronavirus transmission may be aided by the high colony density caused by the birth pulse. Subsequently, the seasonal surge of vulnerable juveniles could speed the spread of the virus throughout the colony, including the infection of adult bats. Thus, there is a need for data covering both breeding seasons and nonbreeding seasons to understand how coronaviruses are maintained in the Nigerian bat population.
Our study showed that Nigerian bats harboured phylogenetically structured CoVs, of both α-CoV and β-CoV subclades, clustering mostly by bat family. Host specificity has been widely reported for some bat CoVs subgenera; whereas β-D CoVs are mostly in Pteropodidae, β-C CoVs are associated mainly with Vespertilionidae [31,38]. Our findings of Nobecovirus (E. helvum bat coronavirus-like cluster) dominance among Pteropodidae are consistent with a prior study that found widespread Nobecovirus (Lineage D) circulation among fruit bats in some African countries [11,39]. The species-specific phylogenetic clustering observed among E. helvum bats suggests limited interspecies β-CoV transmission and host-specific evolution among these species of bats in Nigeria. However, detecting the virus in H. ruber in an abandoned basement around the E. helvum roosting site suggests the existence of potentially evolving virus strains, with a possible ability to cross the species barrier.
We observed a strong geographic influence on CoV diversity within the family Molossidae, which may have resulted from host switching. Host switching and coevolution have been reported as influential evolutionary mechanisms for African CoVs [7,8,31,40,41]. We found that genetically related CoVs were present in other bat species. For example, the Chaerephon bat coronavirus cluster was detected in Chaerephon sp. (Paiko, Niger state), M. condylurus (Gboko, Benue state) and H. ruber (CER/OAU-Ife/Osun state). Similar CoVs were seen in the same type of bat in different locations, as noted for E. helvum-like CoVs clusters detected in E. helvum from other sites, including Jos and Ife in Plateau and Osun states, respectively. These findings suggest that genetically diverse coronaviruses cocirculate among bats in various regions in Nigeria.
We observed an alpha- and betacoronavirus coinfection in a single H. ruber bat. Coinfections have previously been documented in African R. aegyptiacus bats, Hipposideros bat species and Asian insectivorous bats [8,42,43,44,45,46].
5. Conclusions
This study demonstrated high rates of CoVs in frugivorous and insectivorous bats in Nigeria, with significant genetic diversity. Phylogenetic and evolutionary dynamics data indicate a high CoV diversity in Nigeria, while host switching may contribute to CoV evolution. A robust sentinel surveillance is recommended to enhance our knowledge of emerging and re-emerging CoVs. Furthermore, future research should concentrate on locations and bat species where bat–human contact is expected or likely to become common due to climate change and other environmental factors.
Author Contributions
Conceptualization, U.G., O.G., I.K. and C.H.; methodology, U.G. and P.E.; sample collection, U.G., O.G., J.K., A.O., A.A., O.S., R.A., S.A. and N.S.; software and bioinformatics analysis, U.G. and B.M.; resources, U.G., O.F., A.H., I.K. and C.H.; writing—original draft preparation, U.G.; writing—review and editing, B.M., J.O., A.H., O.F., I.K. and C.H.; visualisation, U.G. and B.M.; supervision, O.F., A.H., I.K. and C.H.; funding acquisition, U.G. and C.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded partially by the 2020 ISID grant to U.G. This work was made possible by support from the ACEGID lab and a cohort of generous donors through TED’s Audacious Project, including the ELMA Foundation, MacKenzie Scott, the Skoll Foundation, and Open Philanthropy. This work was supported by grants from the National Institute of Allergy and Infectious Diseases (https://www.NIAID.NIH.gov (accessed on 1 March 2022)), NIH-H3Africa (https://h3africa.org (accessed on 1 March 2022)) (U01HG007480 and U54HG007480), the World Bank grants (project ACE-019 and ACE-IMPACT), the Rockefeller Foundation (Grant #2021 HTH), the Africa CDC through the African Society of Laboratory Medicine (ASLM) (Grant #INV018978), the Wellcome Trust (Project 216619/Z/19/Z), and the Science for Africa Foundation.
Institutional Review Board Statement
Sampling sites were proposed based on available information on bat roosts and foraging areas, especially in regions where bats are hunted, sold, and eaten. The Animal Care and Use Committee of the National Veterinary Research Institute (NVRI), Vom Nigeria, authorised this study and sample protocol (approval number AEC/03/65/19). We also obtained permission from the Cross-River State Health Research Ethics committee (approval reference CRS/MH/HREC/020/VOL.V1/207), Plateau State Health Research Ethics committee (approval reference PSSH/ADM/ETH.CO/2019/005), and Jos University Teaching Hospital Health Research Ethics committee (approval number JUTH/DCS/IREC/127/XXX/13). Village chiefs, heads of various facilities where bat roosting sites were located, and inhabitants gave verbal and written approval for setting traps in the different study areas.
Informed Consent Statement
Not applicable.
Data Availability Statement
Genome sequences of bat coronaviruses reported in this study have been deposited in GenBank, as shown in Table 2. Genome sequences from bat species identified in this study have been deposited in GenBank under accession numbers ON716456-ON716474, ON721268-ON721269, and ON748933-0N748941.
Acknowledgments
We would like to thank Moses Adewumi, Edyth Parker, Priscilla Abechi, Grace Chukwu, Ahmed Muhammed, Tobi Olanipekun, Joseph Ibekwe, Temitope Faleye, Nyor Delahan, Terhemen Ulaka, Gablong P. Letwak, and Jakawa B for their assistance during the sample collection, laboratory analysis, and technical guidance during data analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Burgin, C.J.; Colella, J.P.; Kahn, P.L.; Upham, N.S. How many species of mammals are there? J. Mammal. 2018, 99, 1–14. [Google Scholar] [CrossRef]
- Kunz, T.H.; Torrez, E.B.d.; Bauer, D.; Lobova, T.; Fleming, T.H. Ecosystem services provided by bats. Ann. N. Y. Acad. Sci. 2011, 1223, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats Are Natural Reservoirs of SARS-Like Coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global epidemiology of bat coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef]
- Wacharapluesadee, S.; Tan, C.W.; Maneeorn, P.; Duengkae, P.; Zhu, F.; Joyjinda, Y.; Kaewpom, T.; Ni Chia, W.; Ampoot, W.; Lim, B.L.; et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat. Commun. 2021, 12, 972. [Google Scholar] [CrossRef]
- Pfefferle, S.; Oppong, S.; Drexler, J.F.; Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Müller, M.A.; Annan, A.; Vallo, P.; Adu-Sarkodie, Y.; et al. Distant Relatives of Severe Acute Respiratory Syndrome Coronavirus and Close Relatives of Human Coronavirus 229E in Bats, Ghana. Emerg. Infect. Dis. 2009, 15, 1377–1384. [Google Scholar] [CrossRef]
- Maganga, G.D.; Pinto, A.; Mombo, I.M.; Madjitobaye, M.; Beyeme, A.M.M.; Boundenga, L.; Gouilh, M.A.; N’Dilimabaka, N.; Drexler, J.F.; Drosten, C.; et al. Genetic diversity and ecology of coronaviruses hosted by cave-dwelling bats in Gabon. Sci. Rep. 2020, 10, 7314. [Google Scholar] [CrossRef]
- Lacroix, A.; Vidal, N.; Keita, A.; Thaurignac, G.; Esteban, A.; De Nys, H.; Diallo, R.; Toure, A.; Goumou, S.; Soumah, A.; et al. Wide Diversity of Coronaviruses in Frugivorous and Insectivorous Bat Species: A Pilot Study in Guinea, West Africa. Viruses 2020, 12, 855. [Google Scholar] [CrossRef]
- Shapiro, J.T.; Mollerup, S.; Jensen, R.H.; Olofsson, J.K.; Nguyen, N.-P.D.; Hansen, T.A.; Vinner, L.; Monadjem, A.; McCleery, R.A.; Hansen, A.J. Metagenomic Analysis Reveals Previously Undescribed Bat Coronavirus Strains in Eswatini. EcoHealth 2021, 18, 421–428. [Google Scholar] [CrossRef]
- Quan, P.-L.; Firth, C.; Street, C.; Henriquez, J.A.; Petrosov, A.; Tashmukhamedova, A.; Hutchison, S.K.; Egholm, M.; Osinubi, M.O.V.; Niezgoda, M.; et al. Identification of a Severe Acute Respiratory Syndrome Coronavirus-Like Virus in a Leaf-Nosed Bat in Nigeria. mBio 2010, 1, e00208-10. [Google Scholar] [CrossRef]
- Leopardi, S.; Oluwayelu, D.; Meseko, C.; Marciano, S.; Tassoni, L.; Bakarey, S.; Monne, I.; Cattoli, G.; Benedictis, P.D. The close genetic relationship of lineage D Betacoronavirus from Nigerian and Kenyan straw-coloured fruit bats (Eidolon helvum) is consistent with the existence of a single epidemiological unit across sub-Saharan Africa. Virus Genes 2016, 52, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Funmilayo, O. Fruit Bats for Meat: Are Too Many Taken? Oryx 1978, 14, 377–378. [Google Scholar] [CrossRef]
- Halstead, L.B. Fruit bats—An example of wildlife management. Niger. Field 1977, 42, 50–56. [Google Scholar]
- Mickleburgh, S.; Waylen, K.; Racey, P. Bats as bushmeat: A global review. Oryx 2009, 43, 217–234. [Google Scholar] [CrossRef]
- Kamins, A.O.; Restif, O.; Ntiamoa-Baidu, Y.; Suu-Iret, R.; Hayman, D.; Cunningham, A.; Wood, J.; Rowcliffe, J. Uncovering the fruit bat bushmeat commodity chain and the true extent of fruit bat hunting in Ghana, West Africa. Biol. Conserv. 2011, 144, 3000–3008. [Google Scholar] [CrossRef]
- Friant, S.; Paige, S.B.; Goldberg, T.L. Drivers of Bushmeat Hunting and Perceptions of Zoonoses in Nigerian Hunting Communities. PLOS Negl. Trop. Dis. 2015, 9, e0003792. [Google Scholar] [CrossRef]
- Townzen, J.S.; Brower, A.V.; Judd, D.D. Identification of mosquito bloodmeals using mitochondrial cytochrome oxidase subunit I and cytochrome b gene sequences. Med. Vet. Entomol 2008, 22, 386–393. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Hasegawa, M.; Kishino, H.; Yano, T.-A. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Chernomor, O.; von Haeseler, A.; Minh, B.Q. Terrace Aware Data Structure for Phylogenomic Inference from Supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Argimón, S.; AbuDahab, K.; Goater, R.J.E.; Fedosejev, A.; Bhai, J.; Glasner, C.; Feil, E.J.; Holden, M.T.G.; Yeats, C.A.; Grundmann, H.; et al. Microreact: Visualizing and sharing data for genomic epidemiology and phylogeography. Microb. Genom. 2016, 2, e000093. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.D.; Cai, T.T.; DasGupta, A. Interval estimation for a binomial proportion. Stat. Sci. 2001, 16, 101–133. [Google Scholar] [CrossRef]
- Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Göttsche, M.; Panning, M.; Drexler, J.F.; Petersen, N.; Annan, A.; Grywna, K.; Müller, M.; et al. Detection and Prevalence Patterns of Group I Coronaviruses in Bats, Northern Germany. Emerg. Infect. Dis. 2008, 14, 626–631. [Google Scholar] [CrossRef]
- Anthony, S.J.; Johnson, C.K.; Greig, D.J.; Kramer, S.; Che, X.; Wells, H.; Hicks, A.L.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; et al. Global patterns in coronavirus diversity. Virus Evol. 2017, 3, vex012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kia, G.S.N.; Tao, Y.; Umoh, J.U.; Kwaga, J.K.P.; Tong, S. Identification of Coronaviruses, Paramyxoviruses, Reoviruses, and Rotaviruses among Bats in Nigeria. Am. J. Trop. Med. Hyg. 2021, 104, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Wegner, T.; Tateno, A.F.; Zerbinati, R.M.; Gloza-Rausch, F.; Seebens, A.; Müller, M.A.; Drosten, C. Amplification of Emerging Viruses in a Bat Colony. Emerg. Infect. Dis. 2011, 17, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, M.; Wilkinson, D.A.; Benlali, A.; Lagadec, E.; Ramasindrazana, B.; Dellagi, K.; Tortosa, P. Leptospira and paramyxovirus infection dynamics in a bat maternity enlightens pathogen maintenance in wildlife. Environ. Microbiol. 2015, 17, 4280–4289. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.; Cryan, P.M.; O’Shea, T.J.; Oko, L.M.; Ndaluka, C.; Calisher, C.H.; Berglund, A.D.; Klavetter, M.L.; Bowen, R.A.; Holmes, K.V.; et al. Alphacoronaviruses in New World Bats: Prevalence, Persistence, Phylogeny, and Potential for Interaction with Humans. PLoS ONE 2011, 6, e19156. [Google Scholar] [CrossRef]
- Jeong, J.; Smith, C.S.; Peel, A.J.; Plowright, R.K.; Kerlin, D.H.; Mcbroom, J.; Mccallum, H. Persistent infections support maintenance of a coronavirus in a population of Australian bats (Myotis macropus). Epidemiol. Infect. 2017, 145, 2053–2061. [Google Scholar] [CrossRef]
- Montecino-Latorre, D.; Goldstein, T.; Gilardi, K.; Wolking, D.; Van Wormer, E.; Kazwala, R.; Ssebide, B.; Nziza, J.; Sijali, Z.; Cranfield, M.; et al. Reproduction of East-African bats may guide risk mitigation for coronavirus spillover. One Health Outlook 2020, 2, 2. [Google Scholar] [CrossRef]
- Cui, J.; Han, N.; Streicker, D.; Li, G.; Tang, X.; Shi, Z.; Hu, Z.; Zhao, G.; Fontanet, A.; Guan, Y.; et al. Evolutionary Relationships between Bat Coronaviruses and Their Hosts. Emerg. Infect. Dis. 2007, 13, 1526–1532. [Google Scholar] [CrossRef]
- Yinda, C.K.; Ghogomu, S.M.; Conceição-Neto, N.; Beller, L.; Deboutte, W.; VanHulle, E.; Maes, P.; Van Ranst, M.; Matthijnssens, J. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. 2018, 4, vey008. [Google Scholar] [CrossRef]
- Tong, S.; Conrardy, C.; Ruone, S.; Kuzmin, I.V.; Guo, X.; Tao, Y.; Niezgoda, M.; Haynes, L.; Agwanda, B.; Breiman, R.F.; et al. Detection of Novel SARS-like and Other Coronaviruses in Bats from Kenya. Emerg. Infect. Dis. 2009, 15, 482–485. [Google Scholar] [CrossRef]
- Joffrin, L.; Goodman, S.M.; Wilkinson, D.A.; Ramasindrazana, B.; Lagadec, E.; Gomard, Y.; Le Minter, G.; Dos Santos, A.; Schoeman, M.C.; Sookhareea, R.; et al. Bat coronavirus phylogeography in the Western Indian Ocean. Sci. Rep. 2020, 10, 6873. [Google Scholar] [CrossRef] [Green Version]
- Latinne, A.; Hu, B.; Olival, K.J.; Zhu, G.; Zhang, L.; Li, H.; Chmura, A.A.; Field, H.E.; Zambrana-Torrelio, C.; Epstein, J.H.; et al. Origin and cross-species transmission of bat coronaviruses in China. Nat. Commun. 2020, 11, 4235. [Google Scholar] [CrossRef]
- Bourgarel, M.; Pfukenyi, D.M.; Boué, V.; Talignani, L.; Chiweshe, N.; Diop, F.; Caron, A.; Matope, G.; Missé, D.; Liégeois, F. Circulation of alphacoronavirus, betacoronavirus and paramyxovirus in hipposideros bat species in Zimbabwe. Infect. Genet. Evol. 2018, 58, 253–257. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Peiris, J.S.M.; Chen, H.; Guan, Y.; Poon, L.L.M. Genomic characterizations of bat coronaviruses (1A, 1B and HKU8) and evidence for co-infections in Miniopterus bats. J. Gen. Virol. 2008, 89, 1282–1287. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Ren, X.; Zhang, J.; Yang, F.; Zhang, S.; Jin, Q. ORF8-related genetic evidence for Chinese horseshoe bats as the source of human severe acute respiratory syndrome coronavirus. J. Infect. Dis. 2016, 213, 579–583. [Google Scholar] [CrossRef]
- Valitutto, M.T.; Aung, O.; Tun, K.Y.N.; Vodzak, M.E.; Zimmerman, D.; Yu, J.H.; Win, Y.T.; Maw, M.T.; Thein, W.Z.; Win, H.H.; et al. detection of novel coronaviruses in bats in Myanmar. PLoS ONE 2020, 15, e0230802. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).