Abstract
The feasibility of investigating the adsorption of n-octanohydroxamate collector on copper and iron oxide minerals with static secondary ion mass spectrometry has been assessed. Secondary ion mass spectra were determined for abraded surfaces of air-exposed copper metal, malachite, pseudomalachite and magnetite that had been conditioned in aqueous potassium hydrogen n-octanohydroxamate solution, as well as for the corresponding bulk CuII and FeIII complexes. In each case, the chemical species present at the solid/vacuum interface of a similarly prepared surface were established by X-ray photoelectron spectroscopy. The most abundant positive and negative metal-containing fragment ions identified for the bulk complexes were also found to be diagnostic secondary ions for the collector adsorbed on the oxide surfaces. The relative abundances of those diagnostic ions varied with, and could be rationalised by, the monolayer or multilayer coverage of the adsorbed collector. However, the precise mass values for the diagnostic ions were not able to corroborate the different bonding in the copper and iron hydroxamate systems that had been deduced from photoelectron and vibrational spectra. Parent secondary ions were able to provide supporting information on the co-adsorption of hydroxamic acid at each conditioned surface.
1. Introduction
Hydroxamate collectors are being applied increasingly in the concentration of oxide Cu and rare earth ores by flotation, with potassium hydrogen n-octanohydroxamate [1] in most common use [2]. However, there are also significant concentrations of Fe oxides such as magnetite or hematite in some of those ores, and to achieve an acceptable grade when Fe oxide gangue minerals are present, strategies to optimise the depression of Fe oxides may be required. To facilitate the development of such strategies, it is advantageous to have a detailed understanding of the interaction of hydroxamate collectors with those Fe oxides as well as with the more valuable minerals to be concentrated. The most relevant conditions for the interaction of potassium n-octanohydroxamate include a pH near 9.5, a collector concentration equivalent to a dose of up to 1 kg per tonne of ore, and a conditioning period less than 15 min.
The adsorption of n-octanohydroxamate (as hydroximate because of deprotonation of the N) on oxide Cu minerals at the collector’s unadjusted pH of ~9.5 is now reasonably well understood [3], and there is strong evidence from X-ray photoelectron spectroscopy (XPS) to suggest that the interaction of this collector with Fe oxides [4], rare earth oxides [5], and probably many other metal oxides, is substantially different. In the case of the Cu oxide minerals, it is believed that the collector initially chemisorbs parallel to the mineral surface so that the two oxygen atoms of each hydroximate anion bond to a Cu atom in the outermost layer of the mineral and the deprotonated N atom interacts with a second (adjacent) Cu atom in the oxide surface:
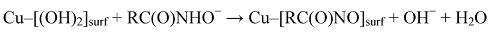
With increasing collector adsorption, it is surmised that the chemisorbed hydroximate progressively adopts an essentially upright orientation so that each deprotonated N atom can then interact with the metal atom of a Cu hydroximate molecule to start a multilayer patch (Figure 1).
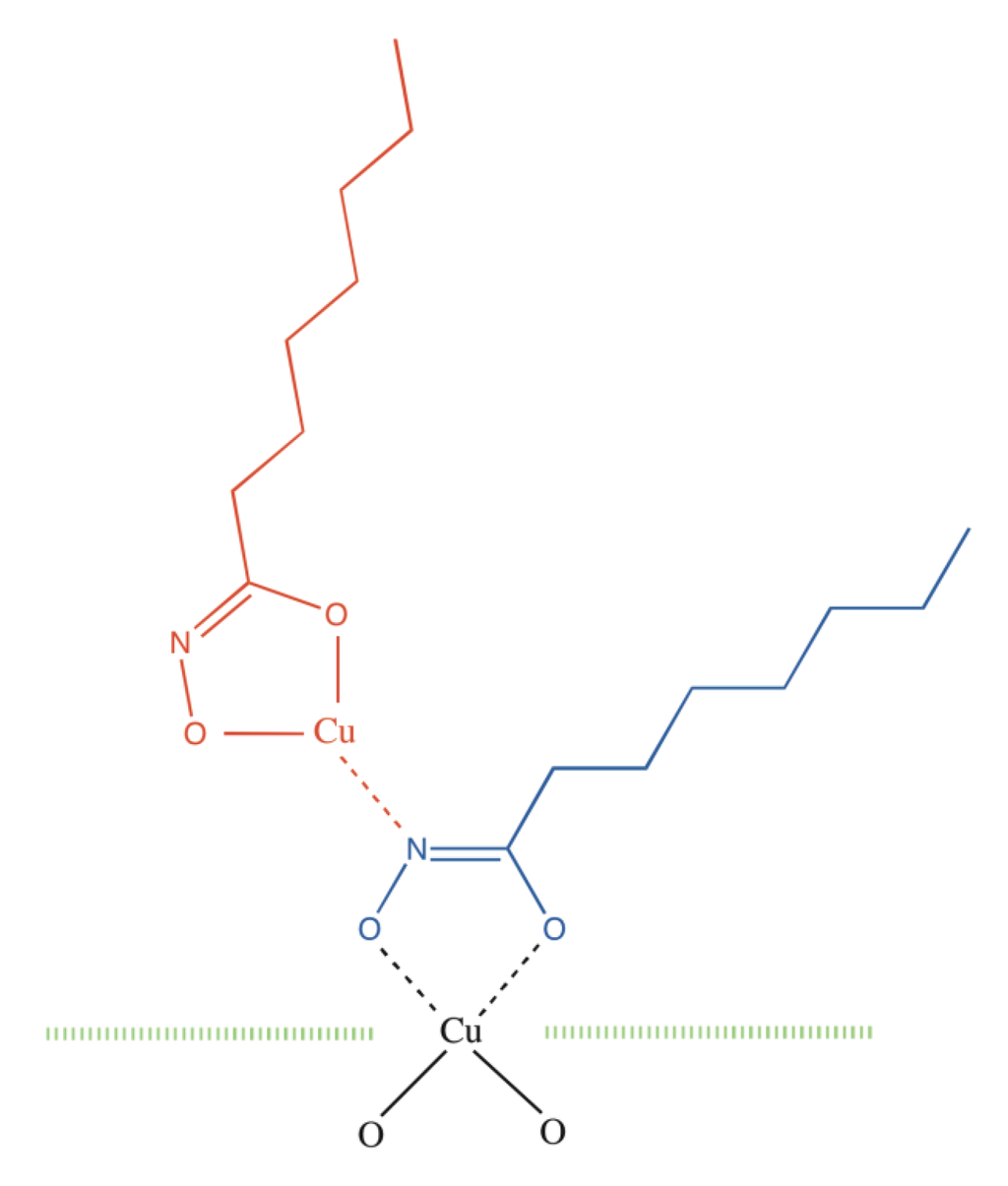
Figure 1.
Schematic representation of n-octanohydroximate (blue) chemisorbed to a Cu atom in a Cu oxide surface (green) and interacting with a Cu hydroximate molecule (red).
With the Fe oxides, on the other hand, it is believed that each collector anion initially chemisorbs through both its O atoms to an Fe atom in the mineral surface as bidentate hydroxamate and with its N remaining protonated. At greater adsorbate coverage, some collector anions are believed to interact via only one of their O atoms; in such a chemisorbed monodentate hydroxamate, the N would be deprotonated but doubly bonded to the adjacent C atom. No evidence for the formation of multilayer Fe hydroxamate on the Fe oxides under the aqueous conditions described above has been obtained by XPS or Raman spectroscopy for conditioning times of relevance to flotation.
Although the evidence for the different adsorption mechanisms outlined above is robust, the XPS findings have yet to be corroborated. Attempts to obtain surface enhanced Raman scattering (SERS) spectra from these systems via gold decoration of the conditioned oxide mineral surface have not yet been successful, although SERS spectra have been obtained for the oxidised layer on air-exposed pyrite [4]. Conventional Raman spectra have been consistent with the photoelectron spectra, in that multilayer hydroxamate collector adsorbed on oxide Cu minerals has appeared similar in structure to bulk Cu hydroximate, and hydroxamate adsorbed on Fe oxides has not been detected for conditioning times of relevance to flotation, an observation in accord with the no more than monolayer coverage expected. Nonetheless, given the importance of the interaction of this collector with Cu and rare earth oxide minerals on the one hand and Fe oxides on the other, positive corroboration of the different adsorption mechanisms rather than mere consistency would be desirable. It was for this principal reason that the application of static secondary ion mass spectrometry (SIMS), a surface analytical technique that is much more surface sensitive than conventional vibrational spectroscopy, was investigated to establish whether it could provide the definitive support sought. A secondary reason was to obtain a complementary diagnostic method for the presence of adsorbed Cu hydroximate or Fe hydroxamate at the surface of air-exposed Cu-Fe sulfide minerals.
The basis of the anticipation that static SIMS might be able to provide the required information was that the secondary ions detected should predominantly reflect substantial fragments of the chemical species actually present at the conditioned mineral surface prior to impact by the primary ion, rather than fragments of only a “scrambled” impact zone created by the primary ion. It is known that for some materials, stable secondary ions are detected that would not have been present as neutral species at the surface prior to primary ion impact, but it is generally assumed that in static SIMS, a significant proportion of secondary ions detected do reflect the bonding arrangement at the surface [6]. Nevertheless, countering this general assumption has been the limited ability of ToF-SIMS to independently differentiate monolayer and multilayer adsorption of thiol collectors on sulfide minerals [7].
In this investigation, sets of metal-containing diagnostic positive and negative secondary ions were sought for bulk Cu hydroximate and Fe hydroxamate complexes. If such sets could be established, the diagnostic ions should be the same—and have similar abundances—as those from the corresponding adsorbed multilayer, but not necessarily from the chemisorbed monolayer. Of particular interest was to ascertain whether the Cu- and Fe-containing diagnostic ions reflected the different bonding arrangements in the two bulk complexes. The secondary ion mass spectra for the bulk complexes were compared with those from surfaces of air-exposed Cu metal, malachite [Cu2CO3(OH)2], pseudomalachite [Cu5(PO4)2(OH)4], and magnetite [Fe3O4] conditioned in hydroxamate solution, to establish the presence of adsorbed collector and to determine whether monolayer and multilayer adsorption could be distinguished. Similarly treated surfaces were also characterised by XPS to confirm the adsorption of hydroxamate collector under the conditions used, and to establish the likely chemical composition at the solid/vacuum interface.
2. Results and Discussion
2.1. X-ray Photoelectron Spectra of Cu Hydroximate and Fe Hydroxamate
2.1.1. Cu Hydroximate XPS
The X-ray photoelectron spectra of the bulk Cu hydroximate, Cu(O2NCC7H15), characterised in the present investigation were similar to those reported previously for the complex prepared in a two-phase synthesis [8]. In particular, the N 1s spectrum determined at the start of the spectral suite (Figure 2a) comprised primarily a single component of width 1.4 eV at a binding energy (corrected for overcompensation by the flood-gun electrons) of 400.0 eV. A minor component near 402 eV accounting for less than 4% of the total N 1s intensity was required for an adequate fit to the spectrum. This small component might have been an artefact of non-uniform charge compensation by the flood-gun electrons. The spectrum determined at the end of the spectral suite was essentially unchanged, indicating that the bulk complex was not susceptible to alteration brought about by the low energy electrons from the flood-gun or by the secondary electrons inherent in the photoemission process. The principal N 1s component at 400 eV can be assigned to the deprotonated N of the hydroximate ligand that is interacting with a Cu atom in a neighbouring Cu hydroximate molecule in a bonding arrangement similar to that shown in Figure 1 [3,8]. Such a N chemical environment is also consistent with the Raman spectrum for the complex [8], but the crystal structure of Cu n-octanohydroximate has not been determined because a suitable solvent from which to grow crystals of the complex has yet to be found. Although Cu hydroximate is definitely a 1:1 Cu/ligand complex, it is expected that in the solid state, each CuII would be fully coordinated in a multinuclear or oligomeric structure.
2.1.2. Fe Hydroxamate XPS
The N 1s spectrum for the bulk Fe hydroxamate, Fe(O2NHCC7H15)3, prepared by Hope and co-workers [3] is shown in Figure 2b. This spectrum too primarily comprised a single component of width 1.5 eV, but at a corrected binding energy of 401.0 eV. Two minor components, each accounting for less than 5% of the N 1s intensity, were observed at 399.2 and 407.2 eV. The intensity of the component at 399.2 eV increased slightly with analysis time, whereas the 407.2 eV component, which probably arose from a minor impurity, remained unchanged. The minor 399.2 eV component would be consistent with an alteration product brought about by the secondary or flood-gun electrons, but the bulk complex was not significantly susceptible to such beam damage. The principal component at 401.0 eV can be assigned to protonated N in the hydroxamate ligands [9,10], and this N chemical environment is consistent with the Raman spectrum for the complex [3] and the crystal structure for the acetohydroxamate analog Fe(O2NHCCH3)3·1.5H2O [11]. The other photoelectron spectra from Fe hydroxamate will be reported elsewhere.
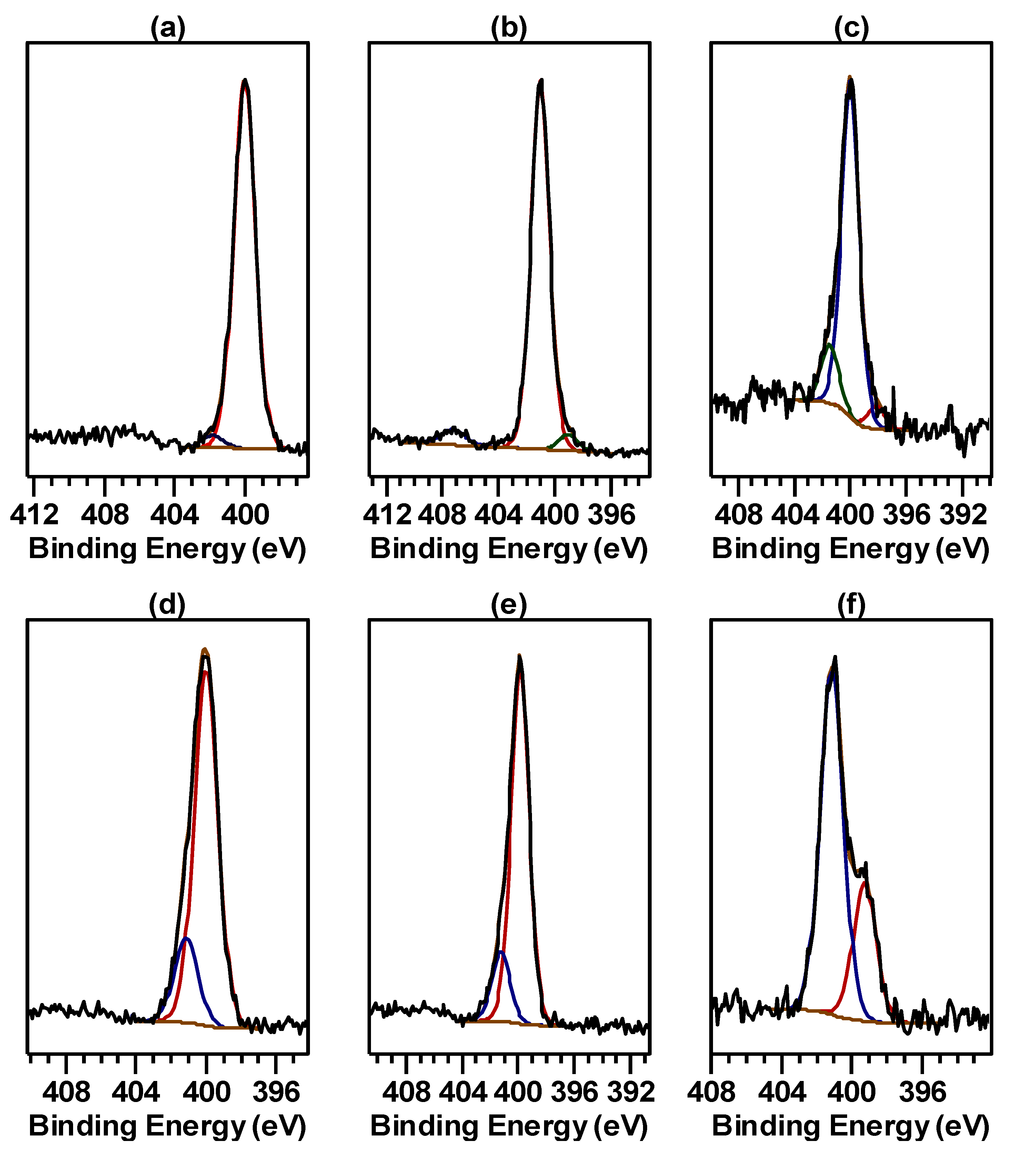
Figure 2.
N 1s spectrum determined at the start of the spectral suite for: (a) bulk Cu hydroximate; (b) bulk Fe hydroxamate; (c)–(f) surfaces conditioned in hydroxamate collector: (c) Cu metal; (d) malachite; (e) pseudomalachite; (f) magnetite.
2.2. Static Secondary Ion Mass Spectra of Cu Hydroximate and Fe Hydroxamate
2.2.1. Cu Hydroximate ToF-SIMS
Peaks of appreciable intensity (at least 0.5% of the 63Cu+ peak intensity) in the secondary ion mass spectra of Cu hydroximate extended beyond its molecular weight of 220 amu. In most cases, Cu-containing ions could be identified with high confidence from the 63Cu:65Cu stable isotopic ratio of 0.6915:0.3085, whereby ions containing one Cu gave rise to two peaks with intensity ratio approximately 2.2:1 and separated by 1.998 amu, while any ions containing two Cu atoms would have produced three peaks of intensity ratio ~2:3:1, each separated by ~2 amu. In this investigation, metal-containing secondary ions were of primary interest as they would be more likely to reflect the different bonding in Cu hydroximate and Fe hydroxamate and in the corresponding adsorbed collector systems. The observed m/z values for the most abundant Cu-containing secondary ions from Cu hydroximate are listed in Table 1 with their possible identification, together with selected organic fragment ions.
The abundances in Table 1 are reported relative to the integrated intensity of the Cu+ or Cu− peak, and also to the intensity of the C5H11+ or C3H3O2− peak to compare normalisation by metal ions and organic fragments. In Table 1, the peak used for normalisation is evident from the abundance of 1, 10 or 1000 for that ion. In each case the abundances for the eight 200 μm × 200 μm regions of the specimen surface characterised have been tabulated as a range (rather than a weighted mean) to show the variability. The intensity of the Cu− rather than the CuH− peak was used for normalisation, as although the latter was more intense than the former for Cu hydroximate, the situation was reversed for the spectra from the conditioned Cu metal and mineral surfaces. The m/z range shown is the variation in measured peak centroid for the eight 200 μm × 200 μm regions. A typical Cu-containing positive secondary ion doublet, tentatively assigned to CuNCH2+ rather than CuC2H4+, is shown in Figure 3. Broader range secondary ion mass spectra for Cu hydroximate are shown in Figure 4.
Some other positive Cu-containing ions are also listed in Table 1 because they corresponded to ions that were abundant for Fe hydroxamate (Section 2.2.2) even though they were of low abundance and hence not useful diagnostic ions for Cu hydroximate. Not listed in Table 1, but of interest when the static SIMS data for conditioned Cu metal and minerals are being considered, were CuH+ peaks with intensity ~1% of Cu+, CuO− peaks with intensity ~30% of Cu−, and CuO2− peaks with intensity ~25% of Cu− peaks.
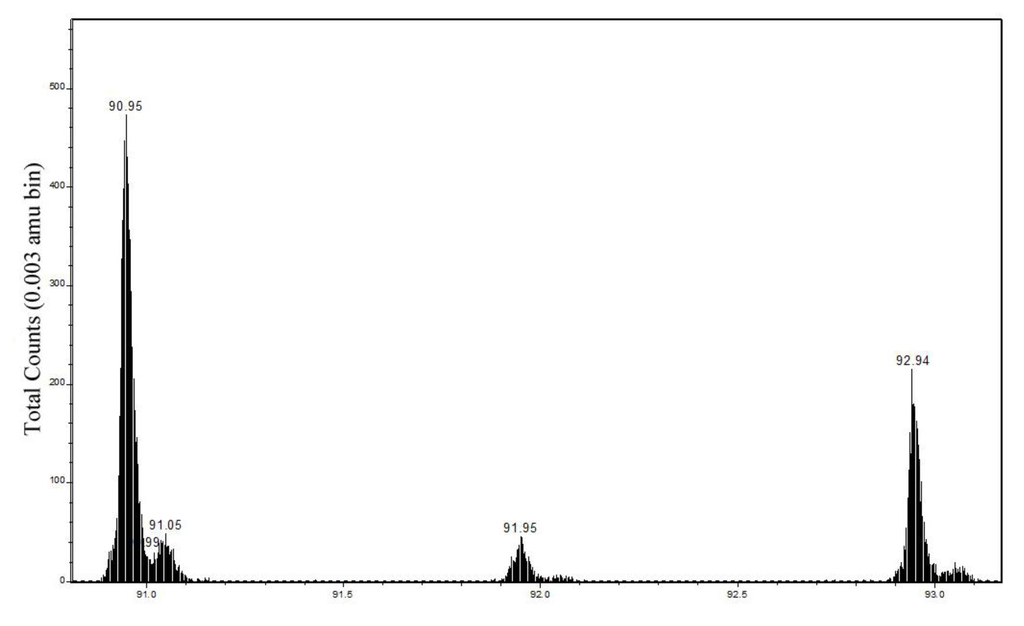
Figure 3.
Positive secondary ion mass spectrum in the m/z 91–93 region from bulk Cu hydroximate showing the doublet assigned to 63CuNCH2+ and 65CuNCH2+.
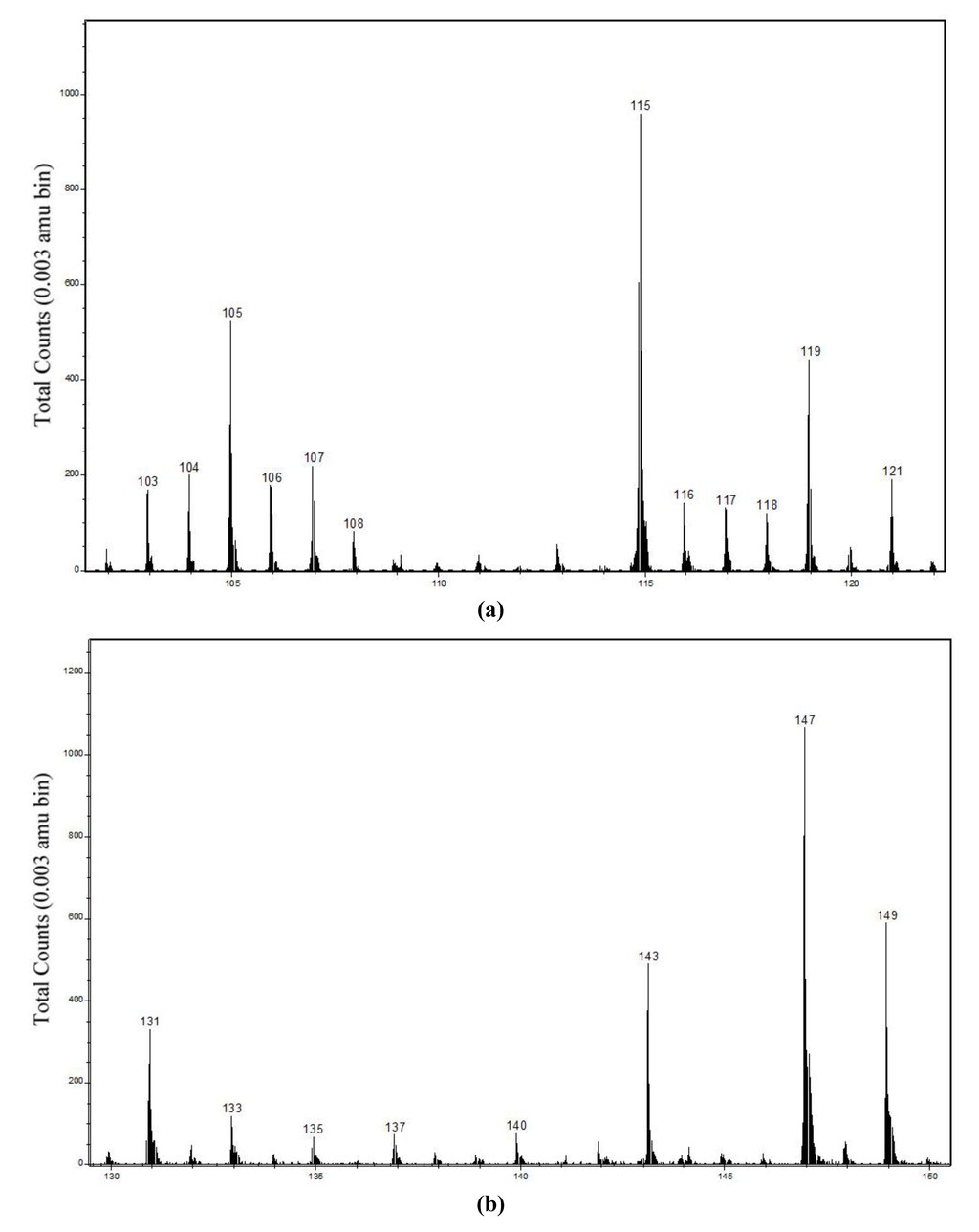
Figure 4.
(a) Positive secondary ion mass spectrum in m/z 102–122 region; and (b) negative secondary ion mass spectrum in m/z 130–150 region from bulk Cu hydroximate.

Table 1.
Diagnostic ions and their observed m/z and relative abundance ranges for bulk Cu hydroximate and conditioned, air-exposed Cu metal.
| Ion | Mass (amu) | Cu hydroximate observed m/z range | Cu hydroximate abundance (rel. Cu) | Cu hydroximate abundance (rel. organic) | Cu metal Observed m/z range | Cu metal abundance (rel. Cu) | Cu metal abundance (rel. organic) |
|---|---|---|---|---|---|---|---|
| 39K+ | 38.9637 | - | - | - | 38.964–38.965 | 12 ± 7 | 3.5 ± 2 |
| 40Ca+ | 39.9626 | - | - | - | 39.961–39.962 | 33 ± 6 | 9 ± 1.5 |
| 63Cu+ | 62.9296 | 62.920–62.932 | 1,000 | 350 ± 140 | 62.924–62.927 | 1000 | 275 ± 20 |
| C5H11+ | 71.086 | 71.083–71.088 | 2.9 ± 0.7 | 1 | 71.089–71.092 | 3.6 ± 0.4 | 1 |
| 63CuNCH2+ | 90.949 | 90.951–90.961 | 47 ± 4 | 18 ± 5 | 90.954–90.957 | 36 ± 2 | 10 ± 1 |
| 63CuC2H4+ | 90.962 | ||||||
| 63CuNC2H4+ | 104.964 | 104.964–104.974 | 10 ± 1 | 3.5 ± 1.5 | 104.969–104.973 | 6.1 ± 0.5 | 1.9 ± 0.3 |
| 63CuC3H6+ | 104.977 | ||||||
| 63CuNC3H6+ | 118.981 | 118.975–118.986 | 8.5 ± 0.5 | 3.5 ± 0.5 | 118.982–118.987 | 5.1 ± 0.2 | 1.45 ± 0.1 |
| 63CuC4H8+ | 118.994 | ||||||
| 63CuONC2H3+ | 119.951 | ~119.97 | <1 | <1 | ~119.98 | <1 | <1 |
| 63CuOC3H5+ | 119.964 | ||||||
| 63CuO2NCH2+ | 122.939 | ~122.96 | <1 | <1 | ~122.95 | <1 | <1 |
| 63CuO2C2H4+ | 122.951 | ||||||
| (hydroxamic acid)+ | 159.126 | 159.012–159.021 | 0.38 ± 0.03 | 0.16 ± 0.02 | 159.140–159.151 | 5 ± 4 | 1.1 ± 1 |
| 63Cu− | 62.9296 | 62.928–62.933 | 10 | 7.5 ± 4 | 62.930–62.932 | 10 | 1.8 ± 0.2 |
| 63CuH− | 63.9374 | 63.944–63.947 | 15 ± 0.5 | 12 ± 5 | 63.937–63.951 | 3 ± 1.5 | 3 ± 1.3 |
| C3H3O2− | 71.0133 | 71.017–71.022 | 2 ± 1 | 1 | 71.017–71.018 | 5 ± 1 | 1 |
| C4H9O− | 73.065 | 73.067–73.072 | 0.7 ± 0.2 | 0.7 ± 0.4 | 73.054–73.072 | 0.4 ± 0.05 | 0.07 ± 0.02 |
| 63CuONCH− | 105.935 | 105.939–105.944 | 6 ± 1 | 4.4 ± 2 | 105.940–105.943 | 6.8 ± 0.5 | 1.2 ± 0.2 |
| 63CuOC2H3− | 105.948 | ||||||
| 63CuO2NCH− | 121.930 | 121.934–121.939 | 10 ± 2 | 8.7 ± 3 | 121.937–121.938 | 14.5 ± 1 | 2.4 ± 0.2 |
| 63CuO2C2H3− | 121.943 | ||||||
| 63CuONC3H2− | 130.943 | 130.936–130.942 | 7 ± 0.8 | 6 ± 1.5 | 130.937–130.940 | 7.5 ± 0.5 | 1.2 ± 0.1 |
| 63CuOC4H4− | 130.956 | ||||||
| 63CuO2NC3H2− | 146.938 | 146.933–146.939 | 26 ± 5 | 24 ± 6 | 146.934–146.937 | 32 ± 2 | 5.5 ± 0.6 |
| 63CuO2C4H4− | 146.951 | ||||||
| (hydroxamic acid−H)− | 158.118 | 158.111–158.123 | 0.21 ± 0.01 | 0.20 ± 0.01 | 158.119–158.129 | 0.6 ± 0.1 | 0.10 ± 0.02 |
It is pertinent to note that the most abundant positive Cu-containing ions did not also contain O, whereas the most abundant negative Cu-containing ions all contained at least one O atom. The Cu analogs of the largest, abundant, Fe-containing positive ions observed for Fe hydroxamate (Section 2.2.2), viz, CuONC2H3+ (119.951 amu) or CuOC3H5+ (119.964 amu for 63Cu) and CuO2NCH2+ (122.939 amu) or CuO2C2H4+ (122.951 amu for 63Cu), were of very low abundance for Cu hydroximate.
It can be seen from Table 1 that distinguishing ions containing N (14.003 amu) from those containing CH2 (14.016 amu) was not as straightforward as might have been expected given the spectral resolution of ~8000. The two main reasons for this uncertainty were the determination of m/z values outside the range that could be calibrated reliably, and the specimen surface roughness at the microscopic level (Section 3.3). Nevertheless, for the negative Cu-containing secondary ions in particular, assignment of the observed diagnostic peak to the N rather than CH2 species appeared to be justified. However, even if correct, that assignment would not have constituted support for the hydroximate structure and Cu–N interaction, as it will be seen below (Section 2.2.2) that the analogous Fe-containing negative ions from Fe hydroxamate also appeared to be the N rather than CH2 species. Therefore, it would appear that these secondary ions do not necessarily reflect the structure prior to impact by the primary ions.
Among the ions not containing Cu, there were no abundant ones near m/z 220 or 221 that might have been the parent Cu hydroximate ion or its protonated analog. No abundant secondary ions containing two Cu atoms were positively identified, although peaks at m/z 247.765, 249.763 and 251.764 were observed in the positive ion spectra with approximately the correct intensity ratio. One possible origin might have been (Cu–O–N–Cu–O2C5)+ ions, with the 63Cu-63Cu peak of mass 247.848 amu, but because those peaks were so far outside the calibration range, their assignment remains uncertain. Neither a positive ion peak near m/z 159.126, that for a surface conditioned in hydroxamate collector might have been assigned to adsorbed n-octanohydroxamic acid, nor a negative ion peak near m/z 158.118, that might have been assigned to (acid − H)− ions, was of appreciable abundance. Although abundances greater than zero have been shown for those ions in Table 1, the corresponding peak intensities were barely greater than noise level.
2.2.2. Fe Hydroxamate ToF-SIMS
Peaks of appreciable intensity in the secondary ion mass spectra from Fe hydroxamate extended to near its molecular weight of about 530 amu. The identification of Fe-containing ions was less definite than for Cu-containing ions, as 56Fe with mass 55.935 amu accounts for 91.75% of the stable isotopic composition of Fe, so that each Fe-containing ion gives rise to only a single substantial peak. The most abundant of the Fe-containing secondary ions, selected impurity element ions and organic fragment ions are listed in Table 2. Also listed in Table 2 are the relative abundances of negative ions of m/z ~139.95, that could be assigned to 56FeO2NC3H2− or 56FeO2C4H4−, not because they were useful diagnostic ions for Fe hydroxamate but because they corresponded to the strong 63Cu-containing diagnostic peak near m/z 146.9 for Cu hydroximate. Abundances were normalised by the integrated intensities of peaks from Fe+ or FeH− and C4H9+ or O2C2H2− ions, and the observed m/z ranges across the eight 200 μm × 200 μm regions characterised are tabulated. In Table 2, the peak used for normal- isation is evident from the abundance of 10, 100 or 1000 for that ion.
The largest of the most abundant Fe-containing positive secondary ions for Fe hydroxamate also contained one or two O atoms, unlike the positive diagnostic secondary ions for Cu hydroximate. As noted in Section 2.2.1, the Cu analogs of the largest, abundant, Fe-containing positive ions observed for Fe hydroxamate, viz, CuONC2H3+ (119.951 amu) or CuOC3H5+ (119.964 amu for 63Cu) and CuO2NCH2+ (122.939 amu) or CuO2C2H4+ (122.951 amu for 63Cu), were of very low abundance for Cu hydroximate. The metal-containing ions concerned would be consistent with the proposed structures for both Cu hydroximate and Fe hydroxamate, and the m/z values for the Cu-containing ions are only marginally higher than for the positive ions that were observed with appreciable abundance.
Table 2.
Diagnostic ions and their observed m/z and relative abundance ranges for bulk Fe hydroxamate and conditioned magnetite (Fe3O4).
| Ion | Mass (amu) | Fe hydroxamate Observed m/z range | Fe hydrox-amate Abundance (rel. Fe) | Fe hydrox-amate Abundance (rel. organic) | Magnetite Observed m/z range | Magnetite Abundance (rel. Fe) | Magnetite Abundance (rel. organic) |
|---|---|---|---|---|---|---|---|
| 39K+ | 38.9637 | - | - | - | 38.960-38.963 | 130 ± 45 | 145 ± 60 |
| 40Ca+ | 39.9626 | - | - | - | 39.954-39.960 | 156 ± 20 | 180 ± 45 |
| 56Fe+ | 55.9349 | 55.930–55.936 | 1000 | 675 ± 60 | 55.920–55.930 | 1000 | 1080 ± 100 |
| C4H9+ | 57.0704 | 57.071–57.072 | 158 ± 17 | 100 | 57.070–57.072 | 94 ± 9 | 100 |
| C5H11+ | 71.086 | 71.079–71.085 | 4.1 ± 0.5 | 4.1 ± 0.5 | 71.088–71.090 | 37 ± 3 | 40 ± 4.3 |
| 56FeNCH2+ | 70.946 | 70.953–70.959 | 41.5 ± 2.4 | 29.5 ± 1.5 | 70.945–70.954 | 16.8 ± 2.8 | 17.3 ± 3 |
| 56FeC2H4+ | 70.958 | ||||||
| 56FeNC2H4+ | 84.961 | 84.967–84.972 | 10.6 ± 0.8 | 10.6 ± 0.8 | 84.958–84.968 | 9 ± 0.8 | 9.5 ± 1.3 |
| 56FeC3H6+ | 84.974 | ||||||
| 56FeNC3H6+ | 96.961 | 96.965–96.970 | 12.6 ± 1.2 | 12.7 ± 1.2 | 96.959–96.968 | 12.8 ± 1.5 | 12.6 ± 2.5 |
| 56FeC4H8+ | 96.974 | ||||||
| 56FeONC2H3+ | 112.956 | 112.954–112.959 | 6.8 ± 0.8 | 6.8 ± 0.8 | 112.966–112.982 | 4 ± 0.8 | 4.3 ± 1.1 |
| 56FeOC3H4+ | 112.969 | ||||||
| 56FeO2NCH2+ | 115.943 | 115.937–115.941 | 8.3 ± 1 | 8.3 ± 1 | 115.933–115.939 | 1.4 ± 0.5 | 1.55 ± 0.65 |
| 56FeO2C2H4+ | 115.956 | ||||||
| (hydroxamic acid)+ | 159.126 | no peak | - | - | 159.137–159.141 | 82 ± 50 | 75 ± 44 |
| 56Fe− | 55.9349 | 55.929–55.937 | 3.4 ± 0.2 | 3.3 ± 0.7 | 55.927–55.932 | 3.1 ± 0.7 | 1.2 ± 0.3 |
| 56FeH− | 56.9427 | 56.936–56.945 | 10 | 9.6 ± 1.6 | 56.935–56.940 | 10 | 4.2 ± 0.9 |
| C2H2O2− | 58.005 | 57.994–58.000 | 10.8 ± 1.8 | 10 | 58.005–58.009 | 24 ± 7 | 10 |
| C3H3O2− | 71.0133 | 71.016–71.023 | 5.6 ± 1.2 | 4.5 ± 1.1 | 71.017–71.021 | 24 ± 7 | 10 ± 0.7 |
| 56FeONCH− | 98.941 | 98.938–98.950 | 24.5 ± 5.5 | 23.1 ± 1.7 | 98.948–98.952 | 5.6 ± 1.7 | 2.3 ± 0.3 |
| 56FeOC2H3− | 98.953 | ||||||
| 56FeO2NC− | 113.928 | 113.920–113.938 | 55 ± 11 | 49 ± 1.6 | 113.931–113.944 | 7 ± 2.6 | 2.7 ± 0.6 |
| 56FeO2C2H2− | 113.941 | ||||||
| 56FeO2NCH− | 114.936 | 114.931–114.948 | 91 ± 20 | 86 ± 7 | 114.940–114.952 | 9.3 ± 3.2 | 3.8 ± 0.7 |
| 56FeO2C2H3− | 114.949 | ||||||
| 56FeO2NC3H2− | 139.944 | 139.938–139.945 | 35 ± 6 | 36 ± 5 | 139.933–139.952 | 2.2 ± 1 | 0.8 ± 0.3 |
| 56FeO2C4H4− | 139.956 | ||||||
| (hydroxamic acid − H)− | 158.118 | 158.106–158.126 | 6.1 ± 1 | 5.9 ± 0.6 | 158.115–158.129 | 7.5 ± 2.4 | 2.7 ± 0.8 |
The difference can be partly rationalised by each Fe atom in Fe hydroxamate being coordinated by the six O atoms of three hydroxamate ligands, so that any moderately large Fe-containing secondary ion would be expected to contain at least one O. On the other hand, while each Cu atom in Cu hydroximate would be coordinated by the two O atoms of one ligand, it is believed to interact with the N atom of an adjacent ligand, and it might also have other ligand atoms as neighbours in its presumed oligomeric form in the solid state. Therefore not all large Cu-containing secondary ions would necessarily be expected to also contain O.
Apart from the difference in metal-containing positive ions discussed above, it can be seen from Table 1 and Table 2 that there is no obvious difference in the organic structure of the other diagnostic ions for the two bulk complexes. In particular, the smaller, abundant Fe-containing positive ions also contained either N or CH2, but not O. In other words, the most abundant fragment secondary ions did not clearly distinguish between the different bonding arrangements in the Cu and Fe complexes expected from spectroscopic and solubility data, and therefore do not corroborate the structures deduced from XPS and Raman spectroscopy. Because of those different structures of the two complexes, it might have been expected that secondary ions containing Cu and N but no O would have been observed, whereas ions containing Fe and N without O should not have been abundant unless rearrangement prior to ion ejection had occurred. In Cu hydroximate, there is intermolecular interaction between Cu and N atoms, whereas in Fe hydroxamate, the N atoms are protonated and the Fe atoms are fully coordinated by the six O atoms of three hydroxamate ligands. Given the unambiguous nature of the XPS and Raman spectroscopic information, it is highly likely that such rearrangement was common adjacent to the Au+ primary ion impact zone. It is just possible that the negative ion peak near m/z 146.94 amu for Cu hydroximate arose from 63CuO2NC3H2− rather than 63CuO2C4H4−, while that near 139.94 amu for Fe hydroxamate arose from 56FeO2C4H4− rather than 56FeO2NC3H2−, for example, but it is considered improbable.
Also because of the difference in the structures of the Cu and Fe complexes, especially the three hydroxamate ligands coordinating the Fe in the latter, it might be expected that ions containing CuOxNCyHz would not be observed for x > 4 but that those containing FeOxNCyHz might be observed for x > 4. This expectation was largely met, in that there were relatively intense peaks attributable to ions such as 56FeO5NC3H−, 56FeO6NC− and 56FeO6NC3H−, but no peaks that could have been assigned to the corresponding Cu ions such as 63CuO5NC3H− at m/z 193.916 or 63CuO6NC3H− at m/z 209.911. Although the Cu in a Cu hydroximate molecule is bonded to the two O atoms in a single hydroximate ligand, in the 3-dimensional and probably oligomeric structure of the bulk complex, each Cu atom might interact with up to four O atoms and two N atoms. Thus the observed secondary ions were consistent with the number of O nearest neighbours of the metal atoms in the two bulk complexes.
Notwithstanding the fact that the relatively abundant ions listed in Table 1 and Table 2 do not appear to reflect the different N bonding in the Cu and Fe complexes, they should nevertheless be diagnostic secondary ions for multilayer Cu hydroximate or Fe hydroxamate adsorbed on oxide Cu or Fe minerals. The same ions might also be diagnostic for hydroxamate collector chemisorbed to Cu or Fe atoms in a mineral surface, but that can only be established by characterising surfaces bearing such an adsorbate.
2.3. X-ray Photoelectron Spectra of Conditioned Cu and Fe Oxide Surfaces
2.3.1. Conditioned Cu Metal and Cu Mineral XPS
XPS data for surfaces of Cu metal, malachite and pseudomalachite that had been abraded in air prior to conditioning in nominally saturated aqueous solutions of potassium hydrogen n-octanohydroxamate have been reported previously [12]. It had been found that collector coverage on Cu metal depended on the extent of oxidation and conditioning times, but conditioning of the minerals in the collector solution for only short periods had resulted in the adsorption of multilayer Cu hydroximate. The Cu 2p spectra indicated that the multilayer Cu hydroximate had been only a few monolayers thick, and the N 1s spectra had suggested the presence of some co-adsorbed hydroxamic acid, as in addition to the principal N 1s component at 400.0 eV, about 15% of the N 1s intensity had been near 401.2 eV [12]. To confirm a similar type and extent of coverage on the specimens characterised by ToF-SIMS (Section 2.4), X-ray photoelectron spectra were determined for the same conditioning time (2 min) of Cu metal and mineral surfaces prepared in the same way.
The N 1s spectra for air-exposed Cu metal, malachite and pseudomalachite surfaces conditioned for 2 min in hydroxamate collector solution are shown in Figure 2c–e. As observed previously, in each case the principal component was at a binding energy of 400.0 eV, consistent with hydroxamate collector chemisorbed to Cu atoms in the oxide surface layer and, for the minerals, a thin layer of molecular Cu hydroximate covering the chemisorbed monolayer. In both monolayer and multilayer Cu hydroximate, each N atom would be deprotonated but interacting with a Cu atom. Also as observed previously, there was a minor unresolved component between 401.2 and 401.5 eV that would have arisen from protonated N. That protonated N would most probably have been in co-adsorbed hydroxamic acid. Such co-adsorption of acid would not be surprising as the pH of the potassium hydrogen n-octanohydroxamate solution would have been close to the pKa for n-octanohydroxamic acid, and indeed, co-adsorption of the acid had been proposed some years ago by Fuerstenau and Pradip [13]. For the air-exposed Cu metal surface, the N concentration (3.3 atom %) was about half that for the minerals, suggesting that the collector coverage of the former was predominantly monolayer only. In the N 1s spectrum obtained with the flood-gun off (Figure 2c), a component near 401.5 eV accounting for ~15% of the intensity was required for an adequate fit. However, under the influence of a beam of ~4.5 eV electrons from the flood-gun, but with the specimen still earthed, the principal component near 400 eV remained unshifted but the N 1s component previously observed at higher binding energy was shifted by about 4.5 eV to ~396.5 eV. This behaviour established that the species containing protonated hydroxamate N was in poor electrical contact with the air-exposed Cu substrate, and its N 1s component would probably have been charge-shifted by up to 0.5 eV above 401 eV in the absence of the low energy electron beam. Hence that species was much more likely to have been co-adsorbed hydroxamic acid than multilayer Cu hydroximate. The Cu 2p, C 1s and O 1s spectra determined with the flood-gun off and on were consistent with this conclusion. The low intensity component at 398.2 eV can be assigned to N in an hydroxamate decomposition product.
It is important to note that no K was observed at the conditioned Cu metal or oxide mineral surfaces. Any residual K from potassium hydrogen hydroxamate that had not been rinsed from the surface with water following the conditioning period would have been evident from a K 2p doublet near 293 and 296 eV. For the Cu metal, a Ca 2p doublet near 347 and 350.5 eV was not discernible, indicating that any Ca impurity would have had a surface concentration below 0.1 atom %. For the conditioned malachite, a low intensity Ca 2p doublet indicated a surface concentration of 0.4 atom % Ca and a barely detectable Mg 1s peak indicated a Mg concentration of no more than 0.1 atom %. For the conditioned pseudomalachite, a very low surface concentration (<0.1 atom %) of Ca was just discernible, but no other impurity elements were observed.
2.3.2. Conditioned Magnetite XPS
X-ray photoelectron spectra for a surface of magnetite abraded in air immediately prior to conditioning in hydroxamate solution for 5 min indicated the adsorption of no more than a monolayer of chemisorbed hydroxamate. A shorter conditioning time had been used for the Cu systems because of multilayer formation within 2 min in those systems. None of the spectra determined with or without a low energy electron beam from a flood-gun provided evidence for the presence of co-adsorbed hydroxamic acid. The Fe 2p spectrum revealed that some FeII remained within the depth analysed, but the spectrum was consistent with the FeII in the outermost layers having been oxidised to FeIII. The N 1s spectrum determined at the start of the spectral suite (without the use of a flood-gun) is shown in Figure 2f. The signal-to-noise of that spectrum is low both because of the low N surface concentration (2.5 atom %) and because the spectrum was determined as quickly as possible in order to monitor any beam-induced changes. The spectrum in Figure 2f could be fitted with two components of width 1.55 eV, the main one accounting for 70% of the N 1s intensity at 401.0 eV and a lower binding energy component at 399.2 eV. The component at 401.0 eV can be assigned to protonated N in bidentate hydroxamate chemisorbed through both its O atoms to an Fe atom in the oxide surface. A second N 1s spectrum determined at the end of the spectral suite could be fitted with components at the same binding energies, but the 401.0 eV component now accounted for only 64% of the intensity. The origin and increase in intensity of the 399.2 eV component will be explored in detail elsewhere, but it is tentatively proposed that the lower binding energy component arises from deprotonated N in chemisorbed monodentate hydroxamate. There is strong evidence to suggest that some of this monodentate species is present before any irradiation by the X-ray beam, but that its surface concentration increases moderately as a result of secondary electron alteration. Of most relevance to the present investigation is confirmation of no more than monolayer coverage of the hydroxamate collector arising from the 5 min conditioning period. In fact no more than monolayer coverage on magnetite has been observed for conditioning periods of up to 30 min under similar conditions. No residual K was detected at the conditioned and rinsed magnetite surfaces.
2.4. Static Secondary Ion Mass Spectra of Conditioned Cu and Fe Oxide Surfaces
In static SIMS, the sputtering of material from a surface results from a “collision cascade” initiated by the impact of the primary ion. For organic adsorbates, it is generally assumed that in the central impact region, mostly atomic and non-characteristic small organic fragments are generated, but immediately outside this impact region, more extensive fragments that may have undergone some structural rearrangement are produced. Further away from the impact region, the energy available for rearrangement is lower, so that larger and minimally rearranged fragment ions (and even parent ions) might be expected. On this basis, it might be anticipated that the larger diagnostic secondary ions should better reflect species present at the surface prior to the primary ion impact. However, as discussed in Section 3.3, the larger the secondary ion, the greater the need to extrapolate the mass range calibration; i.e., the more relevant the diagnostic ion, the more uncertain its measured m/z value.
2.4.1. Conditioned Cu Metal ToF-SIMS
For Cu metal surfaces that had been freshly abraded in air, then conditioned for 2 min in hydroxamate collector solution and subsequently rinsed with water, peaks of moderate intensity were observed in the positive secondary ion spectrum at m/z 38.964 and 39.961. The former could be assigned to K+ and the latter to Ca+ rather than KH+ (Table 1). In making that assignment, it should be noted that 39K with atomic mass 38.9637 accounts for more than 93.2% of the K stable isotopes, and 40Ca with atomic mass 39.9626 accounts for 96.9% of the Ca stable isotopes. Mostly because of its low (~4.3 eV) ionisation potential, K has one of the highest secondary ion yields, so that a very low surface concentration would be expected to give rise to a discernible peak at m/z 38.964. Ca has a somewhat higher (~6.1 eV) ionisation potential and hence lower secondary ion yield. Indeed, measured secondary ion yields from Au+ primary ions reported by King et al. [14] for selected elements in glass standards included 1400 for K, 154 for Ca and 45 for Fe. XPS analysis of Cu metal surfaces conditioned similarly to those characterised by ToF-SIMS showed that neither the Ca nor residual K surface concentration should have been significant (>0.1 atom %). Any Ca present below the XPS detection limit might have been an impurity introduced by the surface abrasion immediately before the specimen was conditioned in the collector solution or an impurity in the potassium hydrogen n-octanohydroxamate itself.
Also in each positive secondary ion spectrum was a low intensity peak at m/z 159.151 (Table 1). This m/z value is well outside the calibration range, so it might have arisen from parent hydroxamic acid ions of mass 159.126 amu. There was an even weaker peak at m/z 160.151 that might be assigned to (acid + H)+ ions of mass 160.137 amu. In each corresponding negative secondary ion spectrum, there was no peak near m/z 159.1 and only very weak peaks at m/z 158.125 and 157.118 that might have arisen from (acid − H)− and (acid − 2H)− ions with mass 158.118 and 157.110 amu, respectively. Assuming those assignments were correct, it cannot be concluded that a low concentration of hydroxamic acid was necessarily present at the surface, as it is conceivable that chemisorbed hydroximate (157.110 amu) might have captured one or two protons prior to abstraction from the impact zone. Thus, while the secondary ion mass spectra do not provide unequivocal support for the presence of hydroxamic acid at the conditioned surface and hence are unable to definitely corroborate the XPS evidence for the co-adsorption of the acid, they are certainly consistent with such co-adsorption.
Secondary ion abundances associated with the major Cu-containing peaks are also listed in Table 1 as a range across the eight 200 μm × 200 μm regions characterised. 63Cu-containing positive secondary ion peaks near m/z 119.95 and 122.95 were of very low intensity, as they were for bulk Cu hydroximate. The major secondary ion peaks for conditioned (air-exposed) Cu metal were the same as those for Cu hydroximate, despite the fact that for the Cu metal, very little multilayer Cu hydroximate, as distinct from the monolayer (chemisorbed) collector, would have been present at the solid/vacuum interface. Thus, the diagnostic ions appear to be the same for both the monolayer and Cu hydroximate. However, for peak intensities relative to those for Cu+ and Cu−, while the abundances of the positive diagnostic secondary ions were consistently lower for the adsorbed layer than for Cu hydroximate, the abundances of the negative ions were consistently higher for the adsorbed layer (Table 3). Such a clear difference can be rationalised, if not predicted, by recognising that the negative diagnostic Cu-containing ions all contain one or two O atoms whereas the positive diagnostic ions contain no O atoms. The spectra indicated that for predominantly a chemisorbed monolayer, Cu-containing ions also including O were more abundant relative to Cu−, while those not including O were less abundant relative to Cu+ than they were for bulk Cu hydroximate.
Table 3.
Intensities of Cu-containing diagnostic secondary ion peaks from conditioned surfaces of Cu metal, malachite and pseudomalachite compared with those from bulk Cu hydroximate (L: lower, H: higher); for each surface, peak intensities have been normalised relative to the intensities of both Cu ion and organic fragment ion peaks as in Table 1 and Table 4.
| Peak m/z | Oxide surface/normalisation | |||||
|---|---|---|---|---|---|---|
| Cu metal/ | Cu metal/ | Malachite/ | Malachite/ | Pseudomalachite/ | Pseudomalachite/ | |
| Cu | organic | Cu | organic | Cu | organic | |
| +90.9 | 23% L | 44% L | 6% L | 39% H | 11% L | 17% H |
| +104.9 | 39% L | 46% L | 13% L | 43% H | 16% L | 17% H |
| +118.9 | 40% L | 59% L | 18% L | 29% H | 24% L | 11% L |
| −105.9 | 13% H | 73% L | 20% L | 57% L | 5% L | 53% L |
| −121.9 | 45% H | 72% L | 1% H | 55% L | 50% H | 39% L |
| −130.9 | 7% H | 80% L | 21% L | 63% L | 3% L | 58% L |
| −146.9 | 23% H | 77% L | 16% L | 65% L | 8% H | 57% L |
For normalisation by the integrated intensities of the organic secondary ion peaks, both the positive and negative secondary ions were less abundant for the conditioned Cu metal than for Cu hydroximate (Table 3). Different abundances for those two specimens would be consistent with the absence of a uniform multilayer of Cu hydroximate on the conditioned Cu metal surface. However, it is not immediately obvious whether the particular differences observed indicated a uniform chemisorbed monolayer, patches of chemisorbed monolayer, or patches of chemisorbed monolayer plus physically co-adsorbed hydroxamic acid on the conditioned Cu metal surface. As noted above, the positive secondary ion spectra were consistent with the presence of co-adsorbed hydroxamic acid, as the peak assigned to the acid parent positive ion (159.126 amu) was of barely detectable intensity for bulk Cu hydroximate but an order of magnitude greater for the conditioned Cu metal surface.
Peaks from CuO− and CuO2− were observed, but typically with intensities only ~27% and 8%, respectively, of the Cu− peaks. These intensities were lower than the normalised values observed for Cu hydroximate, and hence do not provide support for the possibility that oxidised Cu not covered by at least a monolayer of collector was a major surface species. Low intensity peaks near m/z 78.94 and 80.94 from Cu-containing positive secondary ions were observed, but these could be assigned to CuCH2+ and there was no evidence for CuO2+ ions of appreciable abundance.
As canvassed in Section 2.4, it is the diagnostic secondary ion of highest mass that might be expected to best represent the species present at the surface prior to the primary ion impact. For conditioned oxide Cu surfaces, it is the negative ion of m/z near 146.9 that meets this criterion, and the abundance of this ion also happens to be one of the largest of the diagnostic secondary ions. Relative to the Cu− peak intensity, the m/z 146.9 peak was significantly more abundant than for Cu hydroximate, whereas relative to organic fragment peak intensities it was much less abundant than for Cu hydroximate. One possible interpretation of these observations is that the Cu metal native oxide was covered predominantly by chemisorbed hydroximate and possibly also co-adsorbed hydroxamic acid rather than multilayer Cu hydroximate. In other words, there would have been few Cu atoms at the solid/vacuum interface, so that most of the Cu atoms ejected would have been those to which the overlying hydroximate had chemisorbed (through its O atoms) whereas in bulk or multilayer Cu hydroximate, some Cu atoms would have been present at the solid/vacuum interface. By contrast, because of the surface excess of chemisorbed hydroximate and co-adsorbed acid, relative to organic fragment secondary ions, Cu-containing fragments would have been less abundant. The positive Cu-containing secondary ions might also be expected to have been relatively less abundant regardless of the normalisation because the positive ions do not contain O, so that only Cu atoms interacting with the N of any “horizontally” oriented hydroximate would have contributed to secondary ions that had undergone minimal fragmentation (whereas in bulk Cu hydroximate essentially all Cu atoms interact with N atoms).
In summary, for conditioned Cu metal, while there is no static SIMS evidence to support a uniform coverage of multilayer Cu hydroximate or uncovered native oxide, the secondary ion spectra are consistent with monolayer coverage of chemisorbed hydroximate and co-adsorbed hydroxamic acid.
2.4.2. Conditioned Malachite and Pseudomalachite ToF-SIMS
The secondary ion mass spectra for the conditioned minerals were not markedly more complicated than those for the bulk Cu complex, probably because of the presence of multilayer Cu hydroximate at the mineral/vacuum interface. However, for the pseudomalachite, 31P− (30.974 amu) and 31PO2− (62.964 amu) peaks were observed, indicating that some P would have been near the solid/vacuum interface and consequently that the multilayer Cu hydroximate would have been in patches. The multilayer might also have been in patches on malachite. For each mineral, the abundance of 39K+ ions and 40Ca+ ions was only slightly greater than for the conditioned Cu metal surface (Table 4). Peaks from hydroxamic acid ions were of slightly lower intensity than those for the conditioned Cu metal, consistent with slightly higher co-adsorbed acid on the conditioned Cu metal. It can be seen from Table 1 and Table 4 that the abundances of the Cu-containing diagnostic secondary ions were comparable for both minerals, but somewhat different from those for Cu hydroximate and the conditioned Cu metal.
For both minerals, the positive Cu-containing ions were moderately (≈15%) less abundant than for bulk Cu hydroximate relative to Cu+ ions, but relative to C5H11+ ions, the Cu-containing diagnostic ions were ≈35% more abundant for malachite and ≈15% more abundant for pseudomalachite. These observations would be broadly consistent with patches of adsorbed hydroximate on both minerals (allowing Cu species other than Cu hydroximate at the solid/vacuum interface), but fewer or smaller multilayer patches than monolayer patches on pseudomalachite, where a higher concentration of chemisorbed hydroximate relative to multilayer Cu hydroximate would result in a higher concentration of the organic ligand oriented towards the solid/vacuum interface.
Table 4.
Diagnostic ions and their observed m/z and relative abundance ranges for conditioned malachite and pseudomalachite surfaces.
| Ion | Mass (amu) | Malachite Observed m/z range | Malachite Abundance (rel. Cu) | Malachite Abundance (rel. organic) | Pseudo-malachite Observed m/z range | Pseudo-malachite Abundance (rel. Cu) | Pseudo-malachite Abundance (rel. organic) |
|---|---|---|---|---|---|---|---|
| 31P | 30.974 | - | - | - | 30.969–30.974 | 0.25 ± 0.1 | 0.1 ± 0.05 |
| 39K+ | 38.9637 | 38.963–38.965 | 15 ± 3 | 12 ± 6 | 38.961–38.966 | 22 ± 6 | 11.5 ± 3 |
| 40Ca+ | 39.9626 | 39.958–39.961 | 39 ± 4 | 22 ± 8 | 39.959–39.965 | 40 ± 15 | 18 ± 6 |
| 63Cu+ | 62.9296 | 62.919–62.926 | 1000 | 550 ± 160 | 62.923–62.933 | 1000 | 485 ± 90 |
| C5H11+ | 71.086 | 71.084–71.090 | 2 ± 0.6 | 1 | 71.087–71.091 | 2 ± 0.5 | 1 |
| 63CuNCH2+ | 90.949 | 90.951–90.957 | 44 ± 2 | 25 ± 8 | 90.953–90.961 | 42 ± 3.5 | 21 ± 4 |
| 63CuC2H4+ | 90.962 | ||||||
| 63CuNC2H4+ | 104.964 | 104.963–104.970 | 8.7 ± 0.6 | 5 ± 1.5 | 104.967–104.974 | 8.4 ± 0.6 | 4.1 ± 0.8 |
| 63CuC3H6+ | 104.977 | ||||||
| 63CuNC3H6+ | 118.981 | 118.973–118.986 | 7 ± 1.5 | 4.5 ± 1.5 | 118.982–118.988 | 6.5 ± 1 | 3.1 ± 0.7 |
| 63CuC4H8+ | 118.994 | ||||||
| (hydroxamic acid)+ | 159.126 | 159.148–159.156 | 2.5 ± 1.5 | 2.4 ± 1.7 | 159.136–159.156 | 2 ± 1.6 | 1 ± 0.8 |
| 31P− | 30.974 | - | - | - | 30.972–30.973 | 3 ± 2 | 1 ± 0.7 |
| 63Cu− | 62.9296 | 62.928–62.933 | 10 | 3.6 ± 0.7 | 62.928–62.931 | 10 | 4 ± 1.4 |
| PO2− | 62.964 | - | - | - | 62.965–62.967 | 19 ± 11 | 5.6 ± 2.3 |
| C3H3O2− | 71.0133 | 71.014–71.021 | 2.8 ± 0.5 | 1 | 71.014–71.023 | 2.8 ± 1 | 1 |
| 63CuONCH− | 105.935 | 105.940–105.949 | 4.8 ± 0.6 | 1.9 ± 0.3 | 105.943–105.948 | 5.7 ± 1.5 | 2.05 ± 0.25 |
| 63CuOC2H3− | 105.948 | ||||||
| 63CuO2NCH− | 121.930 | 121.936–121.945 | 10.1 ± 1.4 | 3.9 ± 0.9 | 121.941–121.947 | 15 ± 3.5 | 5.3 ± 1.2 |
| 63CuO2C2H3− | 121.943 | ||||||
| 63CuONC3H2− | 130.943 | 130.936–130.946 | 5.5 ± 1 | 2.2 ± 0.8 | 130.942–130.949 | 6.8 ± 1 | 2.5 ± 0.6 |
| 63CuOC4H4− | 130.956 | ||||||
| 63CuO2NC3H2− | 146.938 | 146.931–146.946 | 21.8 ± 2.5 | 8.5 ± 2 | 146.940–146.948 | 28 ± 5 | 10.3 ± 2.2 |
| 63CuO2C4H4− | 146.951 | ||||||
| (hydroxamic acid − H)− | 158.118 | 158.119–158.126 | 0.55 ± 0.15 | 0.22 ± 0.08 | 158.128–158.139 | 0.9 ± 0.2 | 0.4 ± 0.1 |
Relative to Cu− ions, for malachite the negative Cu-containing ions were moderately less abundant than for Cu hydroximate, whereas for pseudomalachite they were comparable or even more abundant. Relative to C3H3O2− ions, for both minerals the negative Cu-containing ions were appreciably less abundant than for bulk Cu hydroximate. For the conditioned Cu metal surface (Section 2.4.1), it was found that relative to Cu− ions, the negative Cu-containing diagnostic ions (which also contain one or two O atoms) were more abundant than for Cu hydroximate. These observations can be rationalised by extensive monolayer coverage with multilayer patches on malachite, but not much more than monolayer patches on pseudomalachite.
2.4.3. Conditioned Magnetite ToF-SIMS
Abundant Fe-containing and selected organic fragment secondary ions are listed in Table 2 for comparison with the data for Fe hydroxamate. There was no obvious correlation between the intensity of the K+ peak and the intensity of the hydroxamic acid or hydroxamate ion peaks; in fact there was almost an inverse relationship, confirming that the K that was observed was unlikely to have been from residual collector solution that had not been rinsed from the surface at the end of the conditioning period. Also, there was no obvious correlation between the intensities of the peaks at m/z 38.96 and 39.96, so that the peak assigned to Ca+ ions (39.963 amu) was unlikely to have arisen instead solely from KH+. However, some of the m/z 39.96 peak intensity might have been due to KH+ ions (39.972 amu), as otherwise Ca+ ions would have been more abundant than expected. For the conditioned magnetite, (hydroxamic acid)+ ions were relatively abundant (compared with not observed for Fe hydroxamate), but the (acid)− and (acid − H)− abundances were comparable with those for Fe hydroxamate. As for Fe hydroxamate, FeH− ions were more abundant that Fe−, unlike the relative abundance of CuH− and Cu− for the analogous conditioned oxide Cu surfaces. As for conditioned oxide Cu surfaces, there was no clear indication that the metal-containing diagnostic ions also contained N rather than CH2 or vice versa.
Differences in the Fe-containing diagnostic ion abundances for the conditioned magnetite surface relative to those for Fe hydroxamate are listed in Table 5. It can be seen that the difference in abundance for each positive diagnostic ion is in fairly close agreement regardless of whether the peak intensities had been normalised by the Fe+ or C4H9+ peak intensities. With one exception, the difference for each negative diagnostic ion is also in close agreement for normalisation by FeH− or O2C2H2− peak intensities.
Table 5.
Intensities of Fe-containing diagnostic secondary ion peaks from conditioned magnetite surfaces compared with those from bulk Fe hydroxamate (L: lower, H: higher); peak intensities have been normalised relative to the intensities of both Fe ion and organic fragment ion peaks as in Table 2.
| Peak m/z | Normalisation | |
|---|---|---|
| Fe | Organic | |
| +70.9 | 60% L | 41% L |
| +84.9 | 15% L | 10% L |
| +96.9 | 2% H | 1% L |
| +112.9 | 41% L | 37% L |
| +115.9 | 83% L | 81% L |
| −98.9 | 77% L | 90% L |
| −113.9 | 87% L | 94% L |
| −114.9 | 90% L | 54% L |
| −139.9 | 94% L | 98% L |
The mid-range m/z Fe-containing positive diagnostic ions were of comparable abundance to those of bulk Fe hydroxamate, but the lower and higher m/z positive, and all the negative, diagnostic ions were up to an order of magnitude lower than for bulk Fe hydroxamate. Therefore, the secondary ion spectra indicated that the adsorbate on magnetite could not have been similar to a uniform Fe hydroxamate multilayer, but they were consistent with no greater than monolayer coverage or possibly sparse patches of multilayer Fe hydroxamate.
It might have been expected that FeOx(NCH) ions (where x > 4) would be more abundant for bulk Fe hydroxamate than for the adsorbed hydroxamate monolayer. For bulk Fe hydroxamate, relatively intense peaks attributable to ions such as 56FeO5NC3H−, 56FeO6NC− and 56FeO6NC3H− were observed with abundances ~45, 30 and 155, respectively, normalised by the FeH− intensity, whereas for conditioned magnetite the corresponding values were less than 1, 1.5 and 2.5.
3. Experimental Details
3.1. Materials and Surfaces Characterised
Cupric hydroximate, Cu(O2CNC7H15), was prepared by the addition of an aqueous Cu sulfate solution to a nominally saturated aqueous solution of potassium hydrogen n-octanohydroxamate, and the precipitated complex washed thoroughly with distilled water. The ferric hydroxamate, Fe(O2CNHC7H15)3, was prepared as described previously [3].
After being freshly abraded in air, surfaces of Cu metal, malachite from Morocco, pseudomalachite from Tottenham, NSW, Australia and magnetite from Mineville, NY, USA were conditioned in a nominally saturated (~0.02 M) aqueous solution of potassium hydrogen n-octanohydroxamate at its unadjusted pH (~9.5) for 2 min (Cu systems) or 5 min (magnetite). Following the conditioning period, the surfaces were rinsed thoroughly with distilled water and dried under a stream if nitrogen. In each case, the conditioned surface appeared to be hydrophobic, so that retention of any weakly adhering collector solution should not have been a problem.
3.2. X-ray Photoelectron Spectroscopy
X-ray photoelectron spectra were collected from Cu hydroximate and Fe hydroxamate powder pressed into the surface of freshly exposed indium, and from conditioned single piece specimens of Cu metal, malachite and pseudomalachite of size approximately 5 mm × 5 mm × 1 mm. Each Cu metal or mineral surface, that had been abraded until relatively smooth to the unaided eye, was rinsed with pure water at its unadjusted pH after conditioning in collector solution and before characterisation by XPS at ambient temperature.
XPS data were obtained on an ESCALAB 250Xi spectrometer located within the UNSW Analytical Centre using monochromatised Al Kα X-rays focused to a spot size of 0.5 mm and an electron analyser pass energy of 20 eV for narrow range scans. Included in the binding energies employed for calibration were 83.96 eV for Au 4f7/2 of metallic gold and 932.6 eV for Cu 2p3/2 of Cu metal. The pressure in the analysis chamber was better than 5 × 10−9 mbar during spectral acquisition. The data were collected and processed under Thermo Scientific Avantage 4.58 and 4.54 software.
Because of the low conductivity of the bulk complexes and Cu minerals, their spectra were obtained under the influence of low energy electrons from an in-lens flood-gun. In minimising peak broadening from a non-uniform potential within the specimen surface layer, the low energy electrons “over-compensate” for charging and the measured photoelectron binding energies are typically lower than their correct energies by a value approaching the energy of the electrons (~4.6 eV). The measured binding energies are corrected by assuming that they will all be affected by the same energy, and adjusting them so that the hydrocarbon C 1s binding energy is 285.0 eV. Whenever the flood-gun was used, the possibility of beam damage or alteration by the low energy electrons was monitored. In particular, N 1s spectra were obtained at the outset and again at the end of the spectral suite to quantify the extent of any alteration. However, in all cases, spectra were obtained as quickly as possible, at the expense of signal-to-noise, in order to minimise any damage arising from the secondary electrons associated with the X-ray photoemission. The magnetic lens mode of the analyser was used in all cases, so that the core of the flood-gun electron beam would have been of less than 1 mm diameter with the tail extending to no more than 3 mm diameter. When several specimens were mounted on the same stub, they were separated by more than 1 cm. Spectra from the conditioned Cu metal and magnetite were obtained without the need for a flood-gun.
3.3. Static SIMS
The static SIMS data were obtained using 30 keV Au+ primary ions in a PHI TRIFT V nanoTOF spectrometer located at the IWRI, University of South Australia. For each region analysed, the pulsed 0.5 nA primary ion beam from a liquid metal ion gun was rastered over a 200 μm × 200 μm area to provide an ion dose no greater than 2.5 × 1011 ions cm−2. Secondary ions were accumulated for a period of 1 min with a 100 μm aperture at an m/z resolution of ~8 000. Dual charge neutralisation was effected by an interlaced pulsed beam of 10 eV electrons and 10 eV Ar+ ions. Secondary ion spectra were obtained and processed under WinCadenceN version 1.8.1.3 software.
Calibration of the m/z scale was particularly important in this investigation as differentiation of species containing N (mass 14.003) or CH2 (mass 14.016) was sought. Such a difference in mass would not be an issue with electron ionised vapour phase mass spectrometers, but is marginal with static SIMS [15]. For calibration of the m/z scale, peaks from CH3+, C2H5+ and C3H7+ or CH−, C2H− and extraneous Cl− ions were used in positive or negative mode, respectively, and checked with peaks from C4H9+ and O2C2H2−. Peaks from C4H9+ (57.070), C5H7+ (67.055), C5H9+ (69.070) and C6H5+ (77.039) ions were observed within 2, 3, 1 and 9 mamu, respectively, of the expected values. Positive secondary ion integrated peak intensities were normalised by the intensities of peaks from Cu+ and C5H11+ for Cu systems or Fe+ and C4H9+ for Fe hydroxamate and magnetite. Negative secondary ion peak intensities were normalised by the intensities of peaks from Cu− and O2C3H3− for Cu systems or FeH− and O2C2H2− for Fe hydroxamate and magnetite.
For calibration of the mass scale, it is generally accepted that either all inorganic or all organic secondary ions should be used, and with the latter, it is better to avoid the inclusion of ions that would have undergone significant rearrangement. If molecular ions are of primary interest, it is contended that it is better not to use atomic ions in the calibration procedure. Other difficulties typically encountered in determining an accurate m/z value for a secondary ion in ToF-SIMS have been discussed by Green et al. [15]. Included in those problems for specimens with a surface that is rough on the atomic scale is that small differences in specimen height can result in significant differences in time-of-flight for a secondary ion species. As the aperture is decreased, peak shape improves considerably but the intensity decreases; while the shape of a secondary ion peak is important, adequate intensity is required for determining a precise m/z value. All these measurement difficulties are in addition to the fundamental issue of whether a significant proportion of secondary ions reflect the species present at the surface prior to impact by the primary ion.
4. Conclusions
Related sets of diagnostic metal-containing secondary ions were identified for bulk Cu hydroximate and Fe hydroxamate. The most abundant Cu-containing positive ions for Cu hydroximate did not also contain O, whereas the most abundant Fe-containing positive ions for Fe hydroxamate did contain O. The most abundant negative Cu-containing ions all contained at least one O atom. The Cu analogs of the largest, abundant, Fe-containing positive ions observed for Fe hydroxamate were of very low abundance for Cu hydroximate. Apart from that difference in metal-containing positive ions, there was no obvious difference in the organic structure of those ions from the two bulk complexes. In particular, the smaller, abundant Fe-containing positive ions also contained either N or CH2, but not O. In other words, the most abundant fragment secondary ions did not clearly distinguish between the different bonding arrangements in the Cu and Fe complexes expected from spectroscopic and solubility data, and therefore did not corroborate the structures deduced from XPS and vibrational spectroscopy. Because of the different structures of the two complexes, it might have been expected that secondary ions containing Cu and N but no O would have been observed, whereas ions containing Fe and N without O should not have been abundant unless rearrangement prior to ion ejection had occurred.
Distinguishing ions containing N from those containing CH2 was not as straightforward as might have been expected given the high spectral resolution, principally because of the uncertainty in determining m/z values outside the range that could be calibrated reliably, and the specimen surface roughness at the microscopic level. Notwithstanding this uncertainty, for the negative Cu-containing secondary ions in particular, assignment of the observed diagnostic peak to the N rather than CH2 species appeared to be justified. However, the analogous Fe-containing negative ions from Fe hydroxamate also appeared to be the N rather than CH2 species. Therefore, it would appear that these secondary ions do not necessarily reflect the structure, especially the different N bonding arrangement, prior to impact by the primary ions. Nevertheless, the abundant secondary ions were consistent with the number of oxygen nearest neighbours in the two complexes, and they should also be diagnostic secondary ions for multilayer Cu hydroximate or Fe hydroxamate adsorbed on oxide Cu or Fe minerals.
For the conditioned oxide Cu surfaces, the photoelectron and secondary ion mass spectra were in agreement as far as collector coverage was concerned. The diagnostic ions appeared to be the same for both the monolayer adsorbed on air-exposed Cu metal and for Cu hydroximate. However, for peak intensities relative to those for Cu+ or Cu−, while the abundances of the positive diagnostic secondary ions were consistently lower for the adsorbed layer than for Cu hydroximate, the abundances of the negative ions were consistently higher for the adsorbed layer. The positive secondary ion spectra were consistent with the presence of co-adsorbed hydroxamic acid. For collector coverage on conditioned magnetite too, the static SIMS and XPS data were in agreement. The secondary ion spectra indicated that the adsorbate was certainly different from a uniform Fe hydroxamate multilayer, and were consistent with no greater than monolayer coverage or possibly sparse patches of multilayer Fe hydroxamate.
Although it proved too difficult to obtain sufficiently reliable m/z values for the larger diagnostic secondary ions, that would have been more likely to reflect the surface chemical structure prior to primary ion impact, static SIMS can provide useful additional information to complement XPS data for hydroxamate conditioned oxide mineral surfaces. When compared with the abundances of the metal-containing secondary ions for the bulk complexes, the relative abundances of those diagnostic ions for the conditioned mineral surfaces varied with, and could be rationalised by, the monolayer or multilayer coverage of the adsorbed collector. Furthermore, parent secondary ions were able to provide supporting information on the co-adsorption of hydroxamic acid at each conditioned surface. Nevertheless, while the secondary ion spectra complemented the XPS data, they would not have been able to provide stand-alone information as an alternative to the photoelectron spectra.
Acknowledgments
This research was supported under the Australian Research Council’s Linkage Project funding scheme (project No. LP 0990404) with industry partner Australian Metallurgical Services Pty Ltd. (established by Axis House). The authors acknowledge the assistance of the AMMRF at the South Australian Regional Facility, University of South Australia, a facility funded by the University, and State and Federal Governments. The authors are grateful to Craig Klauber (CSIRO) for the sample of magnetite and to Bill Gong (UNSW Analytical Centre) for assistance in obtaining the XPS data.
References
- Hope, G.A.; Woods, R.; Buckley, A.N.; White, J.M.; McLean, J. Spectroscopic characterisation of n-octanohydroxamic acid and potassium hydrogen n-octanohydroxamate. Inorg. Chim. Acta 2010, 363, 935–943. [Google Scholar] [CrossRef]
- Liu, W.; Wang, B.; Dai, S.; Ma, A.; Wei, D. Current application and development prospect of hydroxamic acid in flotation. Non-Ferr. Min. Metall. 2006, 22, 25–27. [Google Scholar]
- Parker, G.K.; Hope, G.A.; Woods, R.; Numprasanthai, A.; Buckley, A.N.; McLean, J. Investigation of n-octanohydroxamate reagent interaction with the surface of oxide copper minerals and copper metal. In Separation Technologies for Minerals, Coal, and Earth Resources; Young, C.A., Luttrell, G.H., Eds.; SME Inc.: Englewood, CO, USA, 2012; pp. 497–508. [Google Scholar]
- Parker, G.K.; Buckley, A.N.; Woods, R.; Hope, G.A. The interaction of the flotation reagent, n-octanohydroxamate, with sulfide minerals. Miner. Eng. 2012, 36–38, 81–90. [Google Scholar] [CrossRef]
- Cui, J.; Hope, G.A.; Buckley, A.N. Spectroscopic investigation of the interaction of hydroxamate with bastnaesite (cerium) and rare earth oxides. Miner. Eng. 2012, 36–38, 91–99. [Google Scholar] [CrossRef]
- Vickerman, J.C. Molecular SIMS—A journey from single crystal to biological surface studies. Surf. Sci. 2009, 603, 1926–1936. [Google Scholar]
- Goh, S.W.; Buckley, A.N.; Lamb, R.N.; Woods, R. The ability of static secondary ion mass spectrometry to discriminate submonolayer from multilayer adsorption of thiol collectors. Miner. Eng. 2006, 19, 571–581. [Google Scholar] [CrossRef]
- Hope, G.A.; Woods, R.; Parker, G.K.; Buckley, A.N.; McLean, J. Spectroscopic characterisation of copper acetohydroxamate and copper n-octanohydroxamate. Inorg. Chim. Acta 2011, 365, 65–70. [Google Scholar]
- Folkers, J.P.; Gorman, C.B.; Laibinis, P.E.; Buchholz, S.; Whitesides, G.M.; Nuzzo, R.G. Self-assembled monolayers of long-chain hydroxamic acids on the native oxides of metals. Langmuir 1995, 11, 813–824. [Google Scholar] [CrossRef]
- Keszthelyi, T.; Paszti, Z.; Rigo, T.; Hakkel, O.; Telegdi, J.; Guczi, L. Investigation of solid surfaces modified by Langmuir-Blodgett monolayers using sum-frequency vibrational spectroscopy and X-ray photoelectron spectroscopy. J. Phys. Chem. B 2006, 110, 8701–8714. [Google Scholar]
- Failes, T.W.; Hambley, T.W. Crystal structures of tris(hydroxamato) complexes of iron(III). Aust. J. Chem. 2000, 53, 879–881. [Google Scholar] [CrossRef]
- Hope, G.A.; Buckley, A.N.; Parker, G.K.; Numprasanthi, A.; Woods, R.; McLean, J. The interaction of n-octanohydroxamate with chrysocolla and oxide copper surfaces. Miner. Eng. 2012, 36–38, 2–11. [Google Scholar]
- Fuerstenau, D.W.; Pradip. Mineral flotation with hydroxamate collectors. In Reagents in the Minerals Industry; Jones, M.J., Oblatt, R., Eds.; Institution of Mining Metallurgy: London, UK, 1984; pp. 161–168. [Google Scholar]
- King, A.; Henkel, T.; Rost, D.; Lyon, I.C. Determination of relative sensitivity factors during secondary ion sputtering of silicate glasses by Au+, Au2+ and Au3+ ions. Rapid Commun. Mass Spectrom. 2010, 24, 15–20. [Google Scholar]
- Green, F.M.; Gilmore, I.S.; Seah, M.P. TOF-SIMS: Accurate mass scale calibration. J. Am. Soc. Mass Spectrom. 2006, 17, 514–523. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).