Abstract
Based on the density functional theory and crystal structure prediction approaches, we found a novel high-pressure structure of FeCO-. It is characterized by the presence of ethane-like OC–CO groups or so-called orthooxalate groups. The formation of such OC–CO groups has been proposed earlier in melts and aqueous carbonate solutions, but no such examples were known in inorganic crystalline materials. We found that this structure is dynamically and thermally stable at pressures of 50 GPa. Similar structures were also predicted to be dynamically stable for MnCO, NiCO, and CoCO. In addition, FeCO was found to transform into a similar structure with OC–CO orthooxalate groups at a pressure above 275 GPa. Additionally, for the first time, we describe the self-diffusion of metal atoms in carbonates at high pressure and at high temperatures. The prediction of novel carbonate structures extends the crystal chemistry of inorganic carbonates beyond the established ones with [CO] triangles, [CO] pyro-groups, and [CO] tetrahedra.
1. Introduction
Currently, theoretical methods for crystal structure predictions have proven to be an effective approach for discovering new compounds and their structures that precede real experiments. Recently, using these methods, it was shown that in the MO–CO systems (where M = Mg, Ca, Sr, Ba, and Pb), in addition to the well-known MCO carbonates, MCO, MCO, and MCO compounds became thermodynamically stable at high pressures [1,2,3,4,5]. Subsequently, the formation of the MgCO [6], CaCO [7], CaCO [8], SrCO [9], SrCO [10], SrCO [11], and PbCO [12] phases was experimentally confirmed.
For alkaline earth metals, the thermodynamic stabilization pressures of MCO structures (M = Mg, Ca, Sr, and Ba) were inversely proportional to the cation size, decreasing from 50–75 GPa for Mg [3,6] to 5 GPa for Ba [4]. CaCO, SrCO, and BaCO have the same structure , while MgCO has slightly different structure () [6]. The low-pressure thermodynamic stability limits for the crystal structures of MCO (M = Ca, Sr, and Ba) compounds vary in the range of 11–15 GPa, and these compounds are stable in the structure [4].
The structures of MCO and MCO are characterized by the presence of orthoanion groups of [CO] tetrahedra and form a class of orthocarbonates and oxy-orthocarbonates. The -hybridized carbon atoms in the structures of orthocarbonates are a key difference from the so-called “traditional” MCO carbonates, whose structures contain -hybridized carbon atoms only in triangular coordination at a pressure below 50 GPa. The structures of the MCO composition are characterized by the most diverse crystallochemistry, including [CO] pyro-groups (formed by two trigonal [CO] groups), complex tetrahedra, layers, and frameworks which consist of corner-shared [CO] tetrahedra [1,8,13]. In addition, at pressures above 70 GPa, traditional carbonates MgCO and CaCO are known to transform into structures that consist of corner-shared [CO] tetrahedra, forming chains or rings [14,15,16,17].
For iron, in addition to the traditional carbonate FeCO, FeCO and FeCO are also known [18]. Both carbonates contain tetrahedrally coordinated carbon units and are formed above ∼70 GPa [18]. However, these two carbonates contain iron in both 2+ and 3+ oxidation states and are beyond the FeO–CO system. Hence, to date, FeCO is the only known intermediate compound in this system. It should also be noted that, unlike MgCO and CaCO, iron carbonate does not undergo polymorphic phase transitions up to 110 GPa [18,19,20].
The aim of the present work is to reveal the possible stable structure of FeCO and FeCO using crystal structure prediction methods. The obtained results could extend the known crystal chemistry of inorganic carbonates and motivate future experimental studies of the FeO–CO system, which is complicated by the presence of iron in two oxidation states.
2. Computational Methods
Crystal structure prediction has been performed using evolutionary algorithms implemented in the USPEX package [21,22,23,24] and random structure searching implemented in the AIRSS package [25,26] at 25, 50, 75, and 100 GPa for fixed-composition FeCO and at 100, 200, and 300 GPa for FeCO for 1–4 formula units per unit cell.
The first generation in the USPEX calculations consisted of 65 structures created using a random symmetry operator. Furthermore, to obtain the next generation, 60% of the structures with the lowest enthalpy from the previous generation were selected. In all subsequent generations, structures were produced by heredity (35%), atomic mutation (20%), lattice permutation (10%), and random generator (35%) operators. On average, 32–36 generations were produced and relaxed at each pressure.
All calculations were performed within the VASP 5.4.4 package [27,28] based on density functional theory (DFT). The exchange-correlation effects were calculated via generalized gradient approximation (GGA) in Perdew–Burke–Ernzerhof (PBE) parameterization [29]. To describe core-valence interactions, we used the projector augmented wave basis set with energy cutoff of plane waves equal to 430 eV. The first Brillouin zone was sampled according to the Monkhorst–Pack scheme with a k-point mesh of 0.5 Å and Gaussian smearing with a parameter = 0.1 eV. The most promising predicted structures were relaxed with higher accuracy as follows: the cutoff energy was 600 eV, k-point sampling grid of spacing was 0.25 Å, and = 0.05 eV. Pseudopotentials with 34 (Fe), 22 (C), and 22 (O) electronic configurations were used.
It is well-known that standard DFT tends to over-delocalize the electronic states for transition metals, which prevents them from describing the systems with strongly localized d-electrons appropriately. In order to describe the properties of considered structures more accurately, Dudarev’s DFT+U method was used [30]. In our study, U was set to 2 eV, since in [31], it was found to give the best agreement with the experimental values for the spin transition pressure of iron in FeCO.
Calculations of the phonon spectra were performed using the PHONOPY program [32]. The real space force constants were calculated using supercell and finite displacement approaches, with 2 × 2 × 2 supercells for the considered structures. In this case, high-quality settings were used: the cutoff energy was 800 eV, k-point sampling grid of spacing was 0.2 Å, and = 0.05 eV.
To examine the thermal stability of FeCO, ab initio molecular dynamic (AIMD) simulations based on DFT were performed using a supercell with 255 atoms at 300, 1000, and 2000 K. The time step was set to 1 fs. In the simulations at 300 and 1000 K, the total simulation time was 10 ps, and in the simulations at 2000 K, it was 20 ps. The NPT ensemble (where N is the number of particles, P is pressure, and T is the temperature) were used. During the simulation, the frequency of the temperature oscillations was controlled by the Langevin thermostat.
Images of the structures were produced using VESTA software [33]. The MD simulation animation was made in OVITO software [34].
3. Results
Experiments performed for FeCO crystal structure predictions have revealed the known structures of CaCO- [2] and MgCO- (Olivine type) [3]. However, these structures are not the most favorable among the predicted ones. According to the results obtained, the structure with symmetry had the lowest enthalpy in the entire studied pressure range of 10–100 GPa (Figure 1a). The predicted FeCO- was dynamically stable, as evidenced by the absence of imaginary frequencies in the calculated phonon spectrum (Figure 1b).
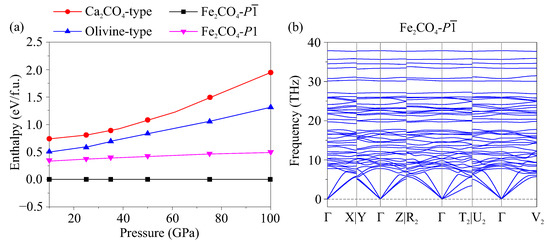
Figure 1.
(a) Relative enthalpy-pressure dependencies of FeCO modifications. (b) Phonon dispersion curves of FeCO- at 50 GPa.
In DFT+U calculations, the total energy of a phase strongly depends on the Hubbard correction U. Since U must vary for each compound, it makes no sense to compare the total energies obtained from the calculations with the same values of U, and it is incorrect to compare the energies obtained with different U values. Consequently, estimation of the pressure at which FeCO- became stable relative to decomposition into carbonate and oxide is currently difficult. For instance, we estimated the stability of FeCO- relative to FeO in the wustite structure and FeCO in the siderite structure without consideration of Hubbard correction (U = 0) and with U = 2 eV (Figure 2), respectively. In the first case, FeCO- stabilizes with respect to a mechanical mixture of FeO + FeCO above 40 GPa. In the second case, it stabilizes at sufficiently higher pressures above 100 GPa. Thus, the stability of FeCO- relative to FeO + FeCO remains an open question.
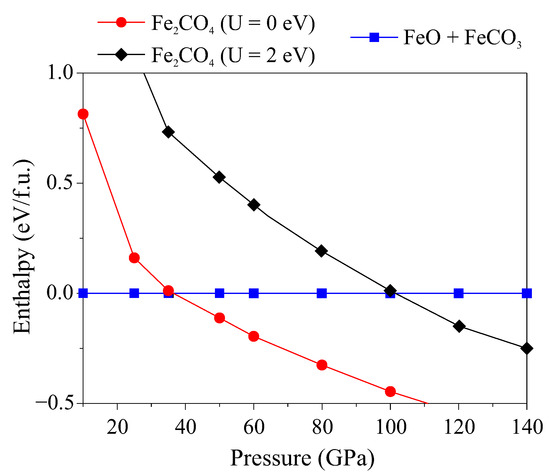
Figure 2.
The enthalpy-pressure dependency of FeCO- relative to the mechanical mixture of FeO + FeCO.
The structural data of the predicted FeCO- are given in Table 1. The structure was characterized by the presence of ethane-like [CO] groups, in which two carbon atoms were bonded together and, in addition, each of them bonded to three more oxygen atoms (Figure S1). By analogy with orthocarbonates, we will call such a group an orthooxalate group and label it as OC–CO. The found FeCO- is the first example of inorganic compounds with the OC–CO group. However, there exists one example of an organic compound with an orthooxalate group, namely a unique orthooxalate: 2,5,7,10,11,14-hexaoxa[4.4.4]propellane [35].

Table 1.
Structural data of predicted FeCO and FeCO modifications with the lowest enthalpy.
At 50 GPa, the C–C distances between two neighboring carbon atoms were equal to 1.53 Å, and the C–O distances varied in the range of 1.35–1.40 Å. These distances were almost equal to the bond lengths observed for the mentioned organic orthooxalate [35], in which the C–C bond length was 1.55 Å and the C–C bond was 1.395 Å. As expected, in the crystal structure of siderite FeCO-, the C–O distances were shorter, and they were equal to 1.27 Å at 50 GPa.
Similar to siderite, the crystal structure of FeCO- can be described as the filling of octahedral voids by cations in the close packing of anions. The array of oxygen atoms in it corresponds to a slightly distorted hcp structure. Half of the octahedral voids are filled with Fe atoms, with 1/8 of them being filled with covalently bonded carbon atoms which are part of orthooxalate groups. In the structure, slabs A and B of the partially filled close-packed octahedra of oxygen atoms can be distinguished (Figure 3a). In slab A, half of the octahedra were filled with Fe atoms. The FeO octahedra were connected in chains as shown in Figure 3b. In slab B, half of the octahedra were filled with Fe atoms, and one-fourth of the octahedra were filled with C–C groups (Figure 3c). Slabs A’ and B’ were similar to A and B in atomic arrangement but shifted in one octahedron, which could also be distinguished.
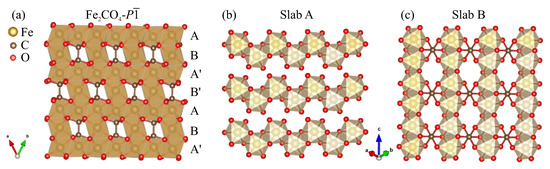
Figure 3.
Crystal structure of predicted FeCO-.
The atomic environment around the iron atoms had some similarities with the well-known ferrioxalate anion [Fe(CO)] [36] present in the tri-hydrated potassium tris(oxalato) ferrate(III) K[Fe(CO)]·3HO. Similar to this transition metal complex, in the found FeCO- structure, iron was in octahedral coordination with an Fe–O distance nearly equal to 2 Å. The ferrioxalate anion consisted of an [FeO] octahedron and three [CO] oxalate groups acting as bidentate ligands, while in the found FeCO structure, there was an [FeO] octahedron and two bidentate orthooxalate [CO] groups.
It should be noted that the second low-enthalpy phase FeCO- also had OC–CO orthooxalate groups in its structure (Figure 1). FeCO- and FeCO- were similar in the manner that they represented different fillings of octahedral voids in the array of oxygen with Fe atoms and C–C groups (Table S1 and Figure S2).
In order to analyze the bonding nature, we calculated the Bader charges, the charge density differences (crystal density minus superposition of isolated atomic densities), and electron localization functions (ELFs) of FeCO- at 50 GPa. The obtained Bader charge of C in FeCO- was lower than that of C in FeCO-, being +1.609 and +2.109 for C in FeCO- and in FeCO-, respectively. This indicates that part of the electrons which were used for the formation of C–O bonds within the [CO] triangle was spent on the formation of a C – C bond. The calculated Bader charges of other atoms are summarized in Table 2. As shown in Figure 4, the charge accumulation occurred halfway through the C–C and C–O vectors. The maximum ELF value between two carbon atoms was about 0.9, and between the carbon and oxygen atoms, it was about 0.7 (Figure 4b), which is indicative of the covalent bonding between these atoms. Thus, we can argue that there was covalent bonding between the carbon atoms, and the carbon in this structure was in the -hybridized state.
Table 2.
Effective Bader charges on each atom in the relaxed FeCO- and FeCO- at 50 GPa.
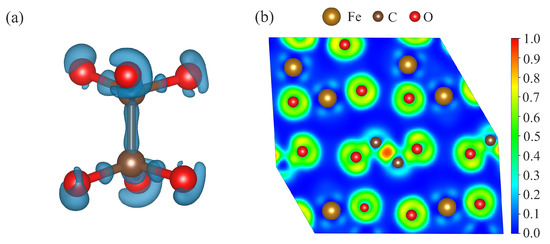
Figure 4.
(a) Charge density difference and (b) electron localization functions of FeCO- at 50 GPa.
To investigate the thermal stability of the predicted FeCO- structure, we performed AIMD calculations at 50 GPa and different temperatures. Although small shifts and Fe cation displacements were observed during the entire 10 ps AIMD simulations at temperatures of 300 and 1000 K, the covalent C–C and C–O bonds were not broken, and the Fe atoms were roughly in the same position. At 1000 K, the C–C and C–O distances varied from 1.35 to 1.74 Å and from 1.17 to 1.63 Å, respectively, confirming that there was no breaking of these bonds. At 2000 K, there was also no observed breaking of the C–C and C–O bonds. The C–C and C–O distances varied from 1.29 to 1.81 Å and from 1.11 to 1.72 Å through MD simulation. From Figure 5, it can clearly be seen that at 2000 K, the structure changed. Specifically, some Fe atoms migrated from one octahedral slab to another. Interestingly, with a more thorough analysis of the MD simulation performed at 2000 K, we can conclude that the process of migration of Fe atoms was continuous (Video S1). A similar self-diffusion of iron atoms was observed for the structure of bcc iron at pressures of the Earth’s inner core and high temperatures [37]. However, nothing similar was observed earlier for carbonate compounds, and only continuous rotation of [CO] groups (the so-called dynamical disorder) was described for alkaline earth carbonates [38].
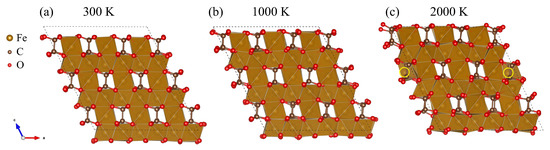
Figure 5.
Snapshots of FeCO at the end of the AIMD simulations at 50 GPa and (a) 300, (b) 1000, and (c) 2000 K. The migrated atoms are highlighted in yellow. The dashed line represents the simulated cell.
In addition, the thermal stability of the second low-enthalpy phase FeCO- was investigated. According to the results, it was also stable at 300 and 1000 K, and at 2000 K, a continuous migration of Fe cations was observed (Figure S3), but compared with FeCO-, it was more intense (Video S2).
In traditional carbonates, the cation can be replaced, and numerous alkaline earth and transition metal carbonates can be formed in the same calcite structure, among which are FeCO, MnCO, CoCO, NiCO, and ZnCO. Based on this, we performed preliminary calculations to examine the dynamic stability of MnCO, CoCO, NiCO, and ZnCO in the structure. No imaginary modes in the phonon spectra of MnCO-, CoCO-, and NiCO- were observed, and hence these orthocarbonates were dynamically stable (Figure 6). The relaxation of ZnCO- was accompanied by the rearrangement of atoms with the destruction of the C–C bonds (i.e., it was not stable against small atomic displacements). Only isolated CO triangles were observed in the final structure of ZnCO.
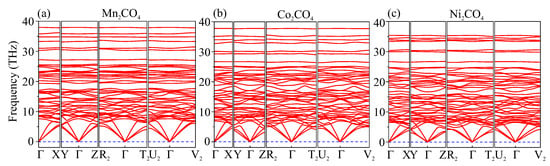
Figure 6.
Phonon dispersion curves of (a) MnCO, (b) CoCO, and (c) NiCO in structure at 50 GPa.
Stable structures of CaCO with [CO] tetrahedra are known to appear at pressures above 75 GPa [16,39], while CaCO with [CO] tetrahedra become stable starting from a pressure of 15 GPa [2,7]. Based on this tendency, according to which the structure in the MCO composition appears at much lower pressures than in the MCO composition, we assumed that there was a stable structure of FeCO with OC–CO orthooxalate groups at a pressure several times higher than that for FeCO. To examine this assumption, we performed crystal structure prediction calculations at pressures of 100–300 GPa. At 100 and 200 GPa, the siderite structure (FeCO-) was predicted to be the most favorable one. At 300 GPa, the new structure of FeCO with the symmetry was predicted to be the most favorable one. As expected, the new FeCO- was characterized by the presence of orthooxalate groups. According to our calculations, the phase transition from FeCO- to this structure occurred at a pressure of 275 GPa (Figure 7a). From the calculated phonon spectra, it is clear that FeCO- is dynamically stable (Figure 7b). It should be noted that the previously known structures MgCO-, -, -, and - [15] with [CO] tetraherda were also considered in the calculations. However, they were energetically unfavorable in the entire considered pressure range.
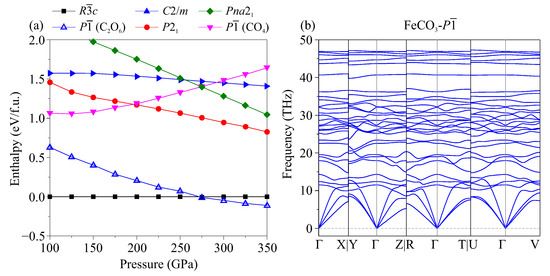
Figure 7.
(a) Relative enthalpy-pressure dependencies of FeCO modifications. (b) Phonon dispersion curves of FeCO- with [CO] groups at 300 GPa.
The structural data of the new phase FeCO- are given in Table 1. Similar to FeCO-, it can be described as an array of oxygen atoms in which half of the octahedral voids were filled: 1/3 was filled with Fe atoms, and 1/6 was filled with C–C groups. The A, A’, A", and B octahedral slabs can be distinguished (Figure 8a). In slab A, all octahedral voids were filled with Fe and C–C groups, as shown in Figure 8b, while in slab B, all octahedra were empty. Layers A’ and A" were filled in the same way as layer A but shifted relative to each other by one octahedron.
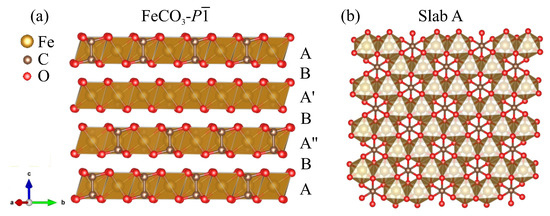
Figure 8.
Crystal structure of predicted FeCO-.
4. Discussion
The formation of OC–CO orthooxalate groups in melts and aqueous carbonate solutions was proposed earlier by AIMD simulations [40,41]. Solomatova et al. [40] reported that in pyrolite melts (NaCaFeMgAlSiO) with 6.48 and 9.82 wt% of CO at a pressure of 65 GPa, in addition to [CO], [CO], and [CO], other species with polymerized carbon were also formed. Among the polymerized carbon species, the most observed one was ethane-like [CO] (i.e., OC–CO groups). With increasing pressure, the relative number of OC–CO groups in the melts increases. This could mean that with increasing pressure, OC–CO groups become more favorable over other polymerized (CO) species. Kuang and Tse [41] investigated the reactions of hydrogen and calcium carbonate melts at high pressures. It was shown that a calcium carbonate melt contains only [CO] and [CO] species, while after reaction with H, the formation of complex oxo-carbon polymers ([CO]) displaying C–C bonds, including OC–CO groups, can be observed. Our results demonstrate the first example of possible solid structures with the same OC–CO groups.
For FeCO, several phases with a similar structure were predicted, which differed only in the distribution of Fe and C–C atoms in the oxygen octahedrons. It is possible that, in reality, FeCO has a structure in which the iron and C–C atoms are statistically distributed through the octahedral voids, especially at high temperatures.
Due to the fact that both FeCO (1FeO*1CO) and FeCO (2FeO*1CO) presented different filling of octahedral voids in the hcp array of oxygen atoms, it can be assumed that the enthalpies of the other structure of intermediate compositions (xFeO*yCO) will be close to the energetic convex hull, and some of them will be thermodynamically stable.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/sym15020421/s1. Figure S1: Orthooxalate group with C–O and C–C bond distances. Figure S2: Crystal structure of predicted FeCO-. Figure S3: Relaxed structure and snapshots of FeCO- at the end of the AIMD simulations at 50 GPa and 300, 1000, and 2000 K. Table S1: Structural data of predicted metastable FeCO-. Video S1: AIMD simulation of FeCO- at 50 GPa and 2000 K. Video S2: AIMD simulation of metastable FeCO- at 50 GPa and 2000 K.
Author Contributions
Conceptualization, P.N.G. and K.D.L.; methodology, N.E.S. and D.N.S.; validation, N.E.S. and P.N.G.; formal analysis, N.E.S. and D.N.S.; investigation, N.E.S. and D.N.S.; writing—original draft preparation, N.E.S., P.N.G., and D.N.S.; writing—review and editing, K.D.L.; visualization, N.E.S. and D.N.S.; supervision, P.N.G. and K.D.L.; funding acquisition, P.N.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the RFBR under research project no. 20-03-00774 and the state assignment of the IGM SB RAS.
Data Availability Statement
Not applicable.
Acknowledgments
The computations were performed using resources provided by the Novosibirsk State University Supercomputer Center.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yao, X.; Xie, C.; Dong, X.; Oganov, A.R.; Zeng, Q. Novel high-pressure calcium carbonates. Phys. Rev. B 2018, 98, 014108. [Google Scholar] [CrossRef]
- Sagatova, D.; Shatskiy, A.; Sagatov, N.; Gavryushkin, P.N.; Litasov, K.D. Calcium orthocarbonate, Ca2CO4-Pnma: A potential host for subducting carbon in the transition zone and lower mantle. Lithos 2020, 370, 105637. [Google Scholar] [CrossRef]
- Gavryushkin, P.N.; Sagatova, D.N.; Sagatov, N.; Litasov, K.D. Formation of Mg-Orthocarbonate through the Reaction MgCO3+ MgO= Mg2CO4 at Earth’s Lower Mantle P–T Conditions. Cryst. Growth Des. 2021, 21, 2986–2992. [Google Scholar] [CrossRef]
- Gavryushkin, P.N.; Sagatova, D.N.; Sagatov, N.; Litasov, K.D. Orthocarbonates of Ca, Sr, and Ba—The Appearance of sp3-Hybridized Carbon at a Low Pressure of 5 GPa and Dynamic Stability at Ambient Pressure. ACS Earth Space Chem. 2021, 5, 1948–1957. [Google Scholar] [CrossRef]
- Banaev, M.V.; Sagatov, N.E.; Sagatova, D.N.; Gavryushkin, P.N. High-Pressure Crystal Structures of Pb2CO4 and PbC2O5 with Tetrahedral [CO4] and Pyrocarbonate [C2O5] atomic groups. ChemistrySelect 2022, 7, e202201940. [Google Scholar] [CrossRef]
- Gavryushkin, P.; Martirosyan, N.; Rashchenko, S.; Sagatova, D.; Sagatov, N.; Semerikova, A.; Fedotenko, T.; Litasov, K. First Experimental Synthesis of Mg Orthocarbonate by the MgCO3 + MgO= Mg2CO4 Reaction at Pressures of the Earth’s Lower Mantle. JETP Lett. 2022, 116, 477–484. [Google Scholar] [CrossRef]
- Binck, J.; Laniel, D.; Bayarjargal, L.; Khandarkhaeva, S.; Fedotenko, T.; Aslandukov, A.; Glazyrin, K.; Milman, V.; Chariton, S.; Prakapenka, V.B.; et al. Synthesis of calcium orthocarbonate, Ca2CO4-Pnma at P-T conditions of Earth’s transition zone and lower mantle. Am. Mineral. 2022, 107, 336–342. [Google Scholar] [CrossRef]
- König, J.; Spahr, D.; Bayarjargal, L.; Gavryushkin, P.N.; Sagatova, D.; Sagatov, N.; Milman, V.; Liermann, H.P.; Winkler, B. Novel Calcium sp3 Carbonate CaC2O5-I4d May Be a Carbon Host in Earth’s Lower Mantle. ACS Earth Space Chem. 2022, 6, 73–80. [Google Scholar] [CrossRef]
- Laniel, D.; Binck, J.; Winkler, B.; Vogel, S.; Fedotenko, T.; Chariton, S.; Prakapenka, V.; Milman, V.; Schnick, W.; Dubrovinsky, L.; et al. Synthesis, crystal structure and structure–property relations of strontium orthocarbonate, Sr2CO4. Acta Crystallogr. Sect. B 2021, 77, 131–137. [Google Scholar] [CrossRef]
- Spahr, D.; König, J.; Bayarjargal, L.; Gavryushkin, P.N.; Milman, V.; Liermann, H.P.; Winkler, B. Sr3[CO4]O Antiperovskite with Tetrahedrally Coordinated sp3-Hybridized Carbon and OSr6 Octahedra. Inorg. Chem. 2021, 60, 14504–14508. [Google Scholar] [CrossRef]
- Spahr, D.; König, J.; Bayarjargal, L.; Milman, V.; Perlov, A.; Liermann, H.P.; Winkler, B. Sr[C2O5] is an Inorganic Pyrocarbonate Salt with [C2O5]2– Complex Anions. J. Am. Chem. Soc. 2022, 144, 2899–2904. [Google Scholar] [CrossRef] [PubMed]
- Spahr, D.; König, J.; Bayarjargal, L.; Luchitskaia, R.; Milman, V.; Perlov, A.; Liermann, H.P.; Winkler, B. Synthesis and Structure of Pb[C2O5]: An Inorganic Pyrocarbonate Salt. Inorg. Chem. 2022, 61, 9855–9859. [Google Scholar] [CrossRef] [PubMed]
- Sagatova, D.N.; Gavryushkin, P.N.; Sagatov, N.E.; Banaev, M.V. High-pressure transformations of CaC2O5—A full structural trend from double [CO3] triangles through the isolated group of [CO4] tetrahedra to framework and layered structures. Phys. Chem. Chem. Phys. 2022, 24, 23578–23586. [Google Scholar] [CrossRef] [PubMed]
- Oganov, A.R.; Glass, C.W.; Ono, S. High-pressure phases of CaCO3: Crystal structure prediction and experiment. Earth Planet. Sci. Lett. 2006, 241, 95–103. [Google Scholar] [CrossRef]
- Pickard, C.J.; Needs, R.J. Structures and stability of calcium and magnesium carbonates at mantle pressures. Phys. Rev. B 2015, 91, 104101. [Google Scholar] [CrossRef]
- Smith, D.; Lawler, K.V.; Martinez-Canales, M.; Daykin, A.W.; Fussell, Z.; Smith, G.A.; Childs, C.; Smith, J.S.; Pickard, C.J.; Salamat, A. Postaragonite phases of CaCO3 at lower mantle pressures. Phys. Rev. Mater. 2018, 2, 013605. [Google Scholar] [CrossRef]
- Binck, J.; Bayarjargal, L.; Lobanov, S.S.; Morgenroth, W.; Luchitskaia, R.; Pickard, C.J.; Milman, V.; Refson, K.; Jochym, D.B.; Byrne, P.; et al. Phase stabilities of MgCO3 and MgCO3-II studied by Raman spectroscopy, x-ray diffraction, and density functional theory calculations. Phys. Rev. Mater. 2020, 4, 055001. [Google Scholar] [CrossRef]
- Cerantola, V.; Bykova, E.; Kupenko, I.; Merlini, M.; Ismailova, L.; McCammon, C.; Bykov, M.; Chumakov, A.I.; Petitgirard, S.; Kantor, I.; et al. Stability of iron-bearing carbonates in the deep Earth’s interior. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Nagai, T.; Ishido, T.; Seto, Y.; Nishio-Hamane, D.; Sata, N.; Fujino, K. Pressure-induced spin transition in FeCO3-siderite studied by X-ray diffraction measurements. J. Phys. Conf. Ser. 2010, 215, 012002. [Google Scholar] [CrossRef]
- Zhao, C.; Xu, L.; Gui, W.; Liu, J. Phase Stability and Vibrational Properties of Iron-Bearing Carbonates at High Pressure. Minerals 2020, 10, 1142. [Google Scholar] [CrossRef]
- Oganov, A.R.; Glass, C.W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 2006, 124, 244704. [Google Scholar] [CrossRef] [PubMed]
- Oganov, A.R.; Lyakhov, A.O.; Valle, M. How Evolutionary Crystal Structure Prediction Works–and Why. Accounts Chem. Res. 2011, 44, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Lyakhov, A.O.; Oganov, A.R.; Stokes, H.T.; Zhu, Q. New developments in evolutionary structure prediction algorithm USPEX. Comput. Phys. Commun. 2013, 184, 1172–1182. [Google Scholar] [CrossRef]
- Bushlanov, P.V.; Blatov, V.A.; Oganov, A.R. Topology-based crystal structure generator. Comput. Phys. Commun. 2019, 236, 1–7. [Google Scholar] [CrossRef]
- Pickard, C.J.; Needs, R.J. High-Pressure Phases of Silane. Phys. Rev. Lett. 2006, 97, 045504. [Google Scholar] [CrossRef] [PubMed]
- Pickard, C.J.; Needs, R.J. Ab initio random structure searching. J. Phys. Condens. Matter 2011, 23, 053201. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Li, Z.; Stackhouse, S. Iron-rich carbonates stabilized by magnetic entropy at lower mantle conditions. Earth Planet. Sci. Lett. 2020, 531, 115959. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Aped, P.; Fuchs, B.; Goldberg, I.; Senderowitz, H.; Tartakovsky, E.; Weinman, S. Structure and conformation of heterocycles. 21. Probing the anomeric effect in orthoesters. Structure, conformation, and dynamic behavior of a unique orthooxalate: 2, 5, 7, 10, 11, 14-hexaoxa [4.4. 4] propellane. J. Am. Chem. Soc. 1992, 114, 5585–5590. [Google Scholar] [CrossRef]
- Junk, P.C. Supramolecular interactions in the X-ray crystal structure of potassium tris (oxalato) ferrate (III) trihydrate. J. Coord. Chem. 2005, 58, 355–361. [Google Scholar] [CrossRef]
- Belonoshko, A.B.; Lukinov, T.; Fu, J.; Zhao, J.; Davis, S.; Simak, S.I. Stabilization of body-centred cubic iron under inner-core conditions. Nat. Geosci. 2017, 10, 312–316. [Google Scholar] [CrossRef]
- Ishizawa, N. Calcite V: A hundred-year-old mystery has been solved. Powder Diffr. 2014, 29, S19–S23. [Google Scholar] [CrossRef]
- Gavryushkin, P.N.; Martirosyan, N.S.; Inerbaev, T.M.; Popov, Z.I.; Rashchenko, S.V.; Likhacheva, A.Y.; Lobanov, S.S.; Goncharov, A.F.; Prakapenka, V.B.; Litasov, K.D. Aragonite-II and CaCO3-VII: New High-Pressure, High-Temperature Polymorphs of CaCO3. Cryst. Growth Des. 2017, 17, 6291–6296. [Google Scholar] [CrossRef]
- Solomatova, N.V.; Caracas, R.; Manning, C.E. Carbon sequestration during core formation implied by complex carbon polymerization. Nat. Commun. 2019, 10, 789. [Google Scholar] [CrossRef]
- Kuang, H.; Tse, J.S. High-Temperature, High-Pressure Reactions of H2 with CaCO3 Melts. Phys. Status Solidi B 2022, 259, 2100644. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).