Mid-Frequency Hearing Loss Is Characteristic Clinical Feature of OTOA-Associated Hearing Loss


 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Short Variant Analysis Including SNVs, Indels, and Splicing Variants
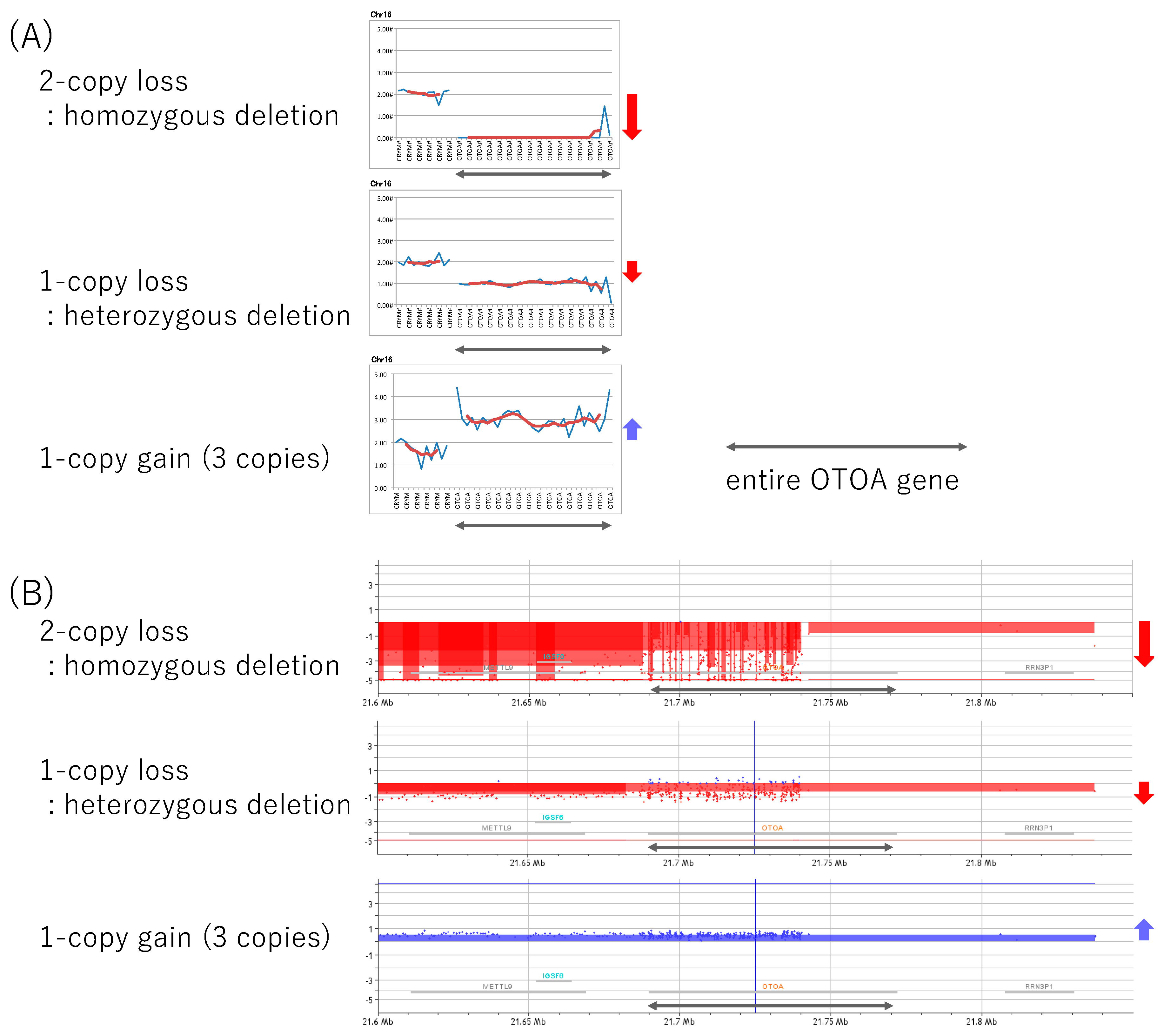
2.3. Copy Number Variations (CNVs) Analysis
2.4. Clinical Evaluations
3. Results
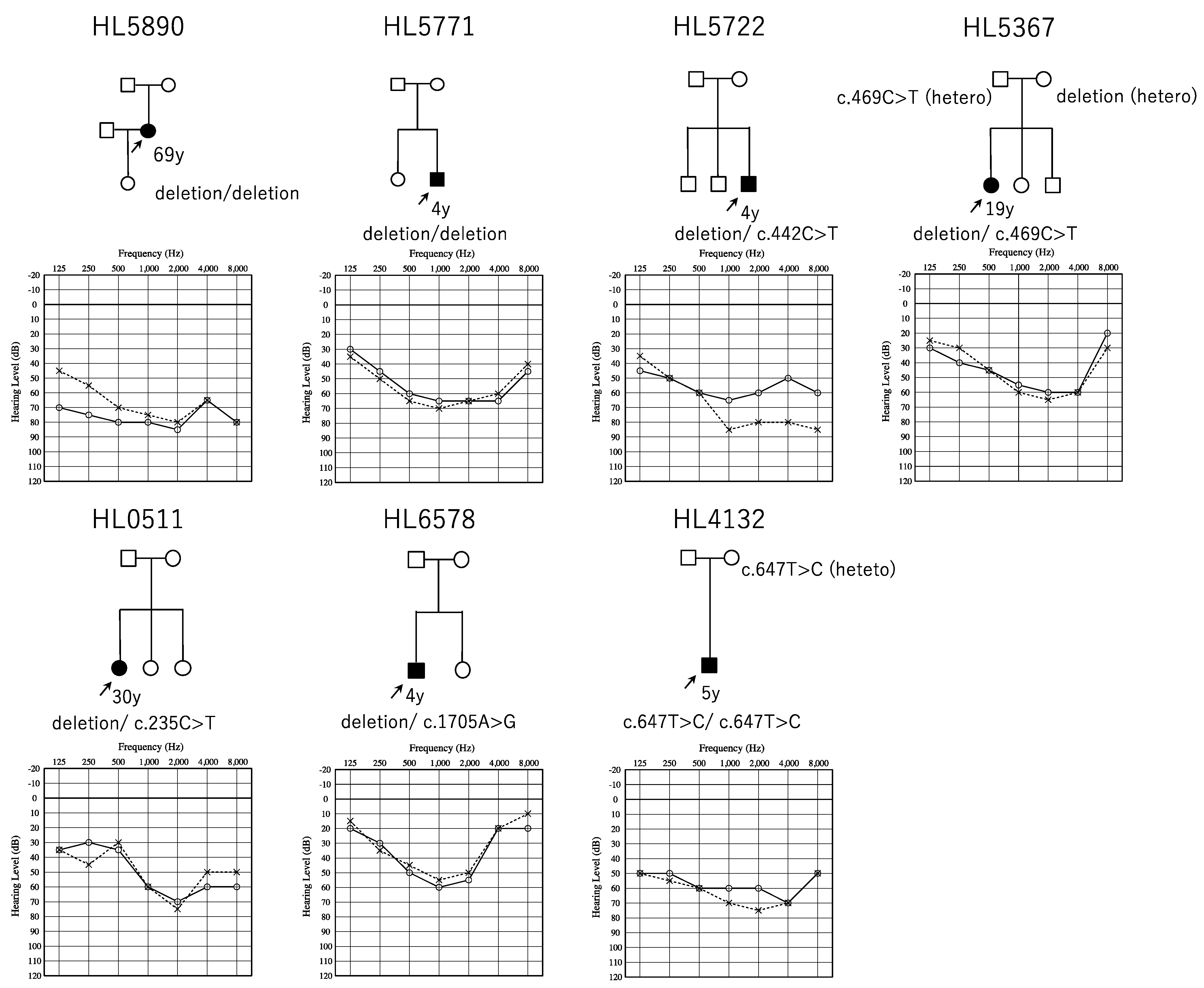
3.1. Identified OTOA Variants and Their Prevalence in Japanese ARSNHL Patients
3.2. Confirmation of CNVs and Short Variants, and The Pathogenic Interpretation of These Variants
3.3. Clinical Features of OTOA-Associated SNHL Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability Statement
References
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org (accessed on 13 May 2019).
- Miyagawa, M.; Naito, T.; Nishio, S.Y.; Kamatani, N.; Usami, S.I. Targeted Exon Sequencing Successfully Discovers Rare Causative Genes and Clarifies the Molecular Epidemiology of Japanese Deafness Patients. PLoS ONE 2013, 8, e71381. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.Y.; Usami, S.I. Deafness Gene Variations in a 1120 Nonsyndromic Hearing Loss Cohort: Molecular Epidemiology and Deafness Mutation Spectrum of Patients in Japan. Ann. Otol. Rhinol. Laryngol. 2015, 124, 49S–60S. [Google Scholar] [CrossRef] [PubMed]
- Kitano, T.; Miyagawa, M.; Nishio, S.-Y.; Moteki, H.; Oda, K.; Ohyama, K.; Miyazaki, H.; Hidaka, H.; Nakamura, K.-I.; Murata, T.; et al. POU4F3 mutation screening in Japanese hearing loss patients: Massively parallel DNA sequencing-based analysis identified novel variants associated with autosomal dominant hearing loss. PLoS ONE 2017, 12, e0177636. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Miyagawa, M.; Nishio, S.Y.; Moteki, H.; Fujikawa, T.; Ohyama, K.; Sakaguchi, H.; Miyanohara, I.; Sugaya, A.; Naito, Y.; et al. WFS1 mutation screening in a large series of Japanese hearing loss patients: Massively parallel DNA sequencing-based analysis. PLoS ONE 2018, 13, e0193359. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, Y.I.; Nishio, S.Y.; Sugaya, A.; Kataoka, Y.; Kanda, Y.; Taniguchi, M.; Nagai, K.; Naito, Y.; Ikezono, T.; Horie, R.; et al. OTOF mutation analysis with massively parallel DNA sequencing in 2265 Japanese sensorineural hearing loss patients. PLoS ONE 2019, 14, e0215932. [Google Scholar] [CrossRef] [PubMed]
- Yokota, Y.; Moteki, H.; Nishio, S.-Y.; Yamaguchi, T.; Wakui, K.; Kobayashi, Y.; Ohyama, K.; Miyazaki, H.; Matsuoka, R.; Abe, S.; et al. Frequency and clinical features of hearing loss caused by STRC deletions. Sci. Rep. 2019, 9, 4408. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.E.; Kolbe, D.L.; Azaiez, H.; Sloan, C.M.; Frees, K.L.; Weaver, A.E.; Clark, E.T.; Nishimura, C.J.; Black-Ziegelbein, E.A.; Smith, R.J.H. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 2014, 6, 37. [Google Scholar] [CrossRef]
- Zwaenepoel, I.; Mustapha, M.; Leibovici, M.; Verpy, E.; Goodyear, R.; Liu, X.Z.; Nouaille, S.; Nance, W.E.; Kanaan, M.; Avraham, K.B.; et al. Otoancorin, an inner ear protein restricted to the interface between the apical surface of sensory epithelia and their overlying acellular gels, is defective in autosomal recessive deafness DFNB22. Proc. Natl. Acad. Sci. USA 2002, 99, 6240–6245. [Google Scholar] [CrossRef]
- Lukashkin, A.N.; Legan, P.K.; Weddell, T.D.; Lukashkina, V.A.; Goodyear, R.J.; Welstead, L.J.; Petit, C.; Russell, I.J.; Richardson, G.P. A mouse model for human deafness DFNB22 reveals that hearing impairment is due to a loss of inner hair cell stimulation. Proc. Natl. Acad. Sci. USA 2012, 109, 19351–19356. [Google Scholar] [CrossRef]
- Kim, B.J.; Kim, D.-K.; Han, J.H.; Oh, J.; Kim, A.R.; Lee, C.; Kim, N.K.; Park, H.-R.; Kim, M.Y.; Lee, S.; et al. Clarification of glycosylphosphatidylinositol anchorage of OTOANCORIN and human OTOA variants associated with deafness. Hum. Mutat. 2019, 40, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Sloan-Heggen, C.M.; Babanejad, M.; Beheshtian, M.; Simpson, A.C.; Booth, K.T.; Ardalani, F.; Frees, K.L.; Mohseni, M.; Mozafari, R.; Mehrjoo, Z.; et al. Characterising the spectrum of autosomal recessive hereditary hearing loss in Iran. J. Med. Genet. 2015, 52, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Sommen, M.; Schrauwen, I.; Vandeweyer, G.; Boeckx, N.; Corneveaux, J.J.; Ende, J.V.D.; Boudewyns, A.; De Leenheer, E.; Janssens, S.; Claes, K.; et al. DNA Diagnostics of Hereditary Hearing Loss: A Targeted Resequencing Approach Combined with a Mutation Classification System. Hum. Mutat. 2016, 37, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Fontana, P.; Morgutti, M.; Pecile, V.; Lenarduzzi, S.; Cappellani, S.; Falco, M.; Scarano, F.; Lonardo, F. A novel OTOA mutation in an Italian family with hearing loss. Gene Rep. 2017, 9, 111–114. [Google Scholar] [CrossRef]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Qual. Life Res. 2016, 135, 441–450. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Pang, X.; Liu, H.; Chai, Y.; Wu, H.; Yang, T. Targeted next-generation sequencing and parental genotyping in sporadic Chinese Han deaf patients. Clin. Genet. 2018, 93, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; Abu Rayan, A.; Abu Sa’Ed, J.; Shahin, H.; Shepshelovich, J.; Lee, M.K.; Hirschberg, K.; Tekin, M.; Salhab, W.; Avraham, K.B.; et al. Genomic analysis of a heterogeneous Mendelian phenotype: Multiple novel alleles for inherited hearing loss in the Palestinian population. Hum. Genom. 2006, 2, 203–211. [Google Scholar]
- Tsai, E.A.; Berman, M.A.; Conlin, L.K.; Rehm, H.L.; Francey, L.J.; Deardorff, M.A.; Holst, J.; Kaur, M.; Gallant, E.; Clark, D.M.; et al. PECONPI: A Novel Software for Uncovering Pathogenic Copy Number Variations in Non-Syndromic Sensorineural Hearing Loss and Other Genetically Heterogeneous Disorders. Am. J. Med. Genet. Part A 2013, 161, 2134–2147. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cabanillas, R.; Diñeiro, M.; Cifuentes, G.A.; Castillo, D.; Pruneda, P.C.; Álvarez, R.; Sánchez-Durán, N.; Capín, R.; Plasencia, A.; Viejo-Díaz, M.; et al. Comprehensive genomic diagnosis of non-syndromic and syndromic hereditary hearing loss in Spanish patients. BMC Med. Genomics 2018, 11, 58. [Google Scholar] [CrossRef]
- Lee, K.; Chiu, I.; Santos-Cortez, R.; Basit, S.; Khan, S.; Azeem, Z.; Andrade, P.; Kim, S.; Ahmad, W.; Leal, S. Novel OTOA mutations cause autosomal recessive non-syndromic hearing impairment in Pakistani families. Clin. Genet. 2013, 84, 294. [Google Scholar] [CrossRef]
- Ammar-Khodja, F.; Bonnet, C.; Dahmani, M.; Ouhab, S.; Lefevre, G.M.; Ibrahim, H.; Hardelin, J.-P.; Weil, D.; Louha, M.; Petit, C. Diversity of the causal genes in hearing impaired Algerian individuals identified by whole exome sequencing. Mol. Genet. Genom. Med. 2015, 3, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Shahin, H.; Walsh, T.; Rayyan, A.A.; Lee, M.K.; Higgins, J.; Dickel, D.; Lewis, K.; Thompson, J.; Baker, C.; Nord, A.S.; et al. Five novel loci for inherited hearing loss mapped by SNP-based homozygosity profiles in Palestinian families. Eur. J. Hum. Genet. 2010, 18, 407. [Google Scholar] [CrossRef] [PubMed]
- Bademci, G.; Diaz-Horta, O.; Guo, S.; Duman, D.; Van Booven, D.; Foster, J., II; Cengiz, F.B.; Blanton, S.; Tekin, M. Identification of Copy Number Variants Through Whole-Exome Sequencing in Autosomal Recessive Nonsyndromic Hearing Loss. Genet. Test. Mol. Biomarkers 2014, 18, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Alkowari, M.K.; Vozzi, D.; Bhagat, S.; Krishnamoorthy, N.; Morgan, A.; Hayder, Y.; Logendra, B.; Najjar, N.; Gandin, I.; Gasparini, P.; et al. Targeted sequencing identifies novel variants involved in autosomal recessive hereditary hearing loss in Qatari families. Mutat. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Nishio, S.Y.; Ikeda, T.; Fukushima, K.; Usami, S.I. Massively Parallel DNA Sequencing Successfully Identifies New Causative Mutations in Deafness Genes in Patients with Cochlear Implantation and EAS. PLoS ONE 2013, 8, e75793. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- The Exome Aggregation Consortium Database (ExAC). Available online: http://exac.broadinstitute.org/ (accessed on 13 May 2019).
- The Genome Aggregation Database (gnomAD). Available online: https://gnomad.broadinstitute.org/ (accessed on 13 May 2019).
- Integrative Japanese Genome Variation Database (3.5KJPN). Available online: https://ijgvd.megabank.tohoku.ac.jp/statistics/statistics-3.5kjpn-all (accessed on 13 May 2019).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- The Human Gene Mutation Database professional (HGMD). Available online: http://www.hgmd.cf.ac.uk/ (accessed on 13 May 2019).
- Nishio, S.Y.; Moteki, H.; Usami, S.I. Simple and efficient germline copy number variant visualization method for the Ion AmpliSeqTM custom panel. Mol. Genet. Genomic Med. 2018, 6, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Moteki, H.; Azaiez, H.; Sloan-Heggen, C.M.; Booth, K.; Nishio, S.-Y.; Wakui, K.; Yamaguchi, T.; Kolbe, D.L.; Iwasa, Y.-I.; Shearer, A.E.; et al. Detection and confirmation of deafness-causing copy number variations in the STRC gene by massively parallel sequencing and comparative genomic hybridization. Ann. Otol. Rhinol. Laryngol. 2016, 125, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Mazzoli, M.G.V.C.; Van Camp, G.U.Y.; Newton, V.; Giarbini, N.; Declau, F.; Parving, A. Recommendations for the Description of Genetic and Audiological Data for Families with Nonsyndromic Hereditary Hearing Impairment. Audiol. Med. 2003, 1, 148–150. [Google Scholar]
- Tassano, E.; Ronchetto, P.; Calcagno, A.; Fiorio, P.; Gimelli, G.; Capra, V.; Scala, M. ‘Distal 16p12.2 microdeletion’ in a patient with autosomal recessive deafness-22. J. Genet. 2019, 98, 56. [Google Scholar] [CrossRef]
- Itsara, A.; Cooper, G.M.; Baker, C.; Girirajan, S.; Li, J.; Absher, D.; Krauss, R.M.; Myers, R.M.; Ridker, P.M.; Chasman, D.I.; et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2008, 84, 148–161. [Google Scholar] [CrossRef]
- Martin, J.; Han, C.; Gordon, L.A.; Terry, A.; Prabhakar, S.; She, X.; Xie, G.; Hellsten, U.; Chan, Y.M.; Altherr, M.; et al. The sequence and analysis of duplication-rich human chromosome 16. Nature 2004, 432, 988–994. [Google Scholar] [CrossRef]
- Mandelker, D.; Amr, S.S.; Pugh, T.; Gowrisankar, S.; Shakhbatyan, R.; Duffy, E.; Bowser, M.; Harrison, B.; Lafferty, K.; Mahanta, L.; et al. Comprehensive diagnostic testing for stereocilin: An approach for analyzing medically important genes with high homology. J. Mol. Diagnostics 2014, 16, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Moteki, H.; Nishio, S.-Y.; Hashimoto, S.; Takumi, Y.; Iwasaki, S.; Takeichi, N.; Fukuda, S.; Usami, S.-I. TECTA mutations in Japanese with mid-frequency hearing loss affected by zona pellucida domain protein secretion. J. Hum. Genet. 2012, 57, 587–592. [Google Scholar] [CrossRef] [PubMed]
- De Heer, A.R.; Pauw, R.J.; Huygen, P.L.M.; Collin, R.W.J.; Kremer, H.; Cremers, C.W.R.J. Flat threshold and mid-frequency hearing impairment in a dutch DFNA8/12 family with a novel mutation in TECTA: Some evidence for protection of the inner ear. Audiol. Neurotol. 2009, 14, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Pelayo, M.A.; Del Castillo, I.; Villamar, M.; Romero, L.; Hernández-Calvín, F.J.; Herraiz, C.; Barberá, R.; Navas, C.; Moreno, F. A cysteine substitution in the zona pellucida domain of α-tectorin results in autosomal dominant, postlingual, progressive, mid frequency hearing loss in a Spanish family. J. Med Genet. 2001, 38, e13. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Newborn | Average | Audiometric | Hearing | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hearing | Hearing Level | Age at | Configuration | Loss | Vertigo/ | |||||||
| ID | Zygosity | Allele #1 | Allele #2 | Onset | Screening R/L | R/L (dB) | Audiogram | R/L | Progression | Dizziness | ||
| HL5771 | homo | whole gene deletion | whole gene deletion | 3y | N/A | 58.75/62.5 | 4y | MF/MF | - | |||
| HL5890 | homo | whole gene deletion | whole gene deletion | childhood | N/A | 77.5/72.5 | 69y | Flat/MF | progressive | + | ||
| HL0511 | compound hetero | whole gene deletion | c.235C>T | p.(Arg79Trp) | 7y | N/A | 56.25/55 | 30y | HF/MF | progressive | - | |
| HL5722 | compound hetero | whole gene deletion | c.442C>T | p.(Arg148*) | 0m | refer/refer | 58.75/76.25 | 7y | Flat/HF | - | ||
| HL5367 | compound hetero | whole gene deletion | c.469C>T | p.(Arg157Cys) | 5y | N/A | 55/57.5 | 19y | MF/MF | progressive | - | |
| HL6578 | compound hetero | whole gene deletion | c.1705A>G | p.(Lys569Glu) | 0m | refer/refer | 46.25/42.5 | 4y | MF/MF | - | ||
| HL4132 | homo | c.647T>C | p.(Phe216Ser) | c.647T>C | p.(Phe216Ser) | 0m | refer/refer | 62.5/68.75 | 5y | Flat/MF | - |
| Prediction Score | Allele Frequency in Controls | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino | |||||||||||||
| Nucleotide | Acid | PolyPhen | Mut_ | Mut_ | ACMG | ||||||||
| Changes | Change | SIFT * | 2_HVAR * | LRT * | Taster * | Assessor * | REVEL * | Cadd | Exac | Gnomad | 3.5kJPN | Guidelines | |
| c.235C>T | p.(Arg79Trp) | D(0.4) | B(0.166) | N(0.132) | N(0.09) | M(0.552) | 0.21 | 23.6 | 0.00000824 | 0.00000812 | N/A | Uncertain Significance | PM2,PM3 |
| c.442C>T | p.(Arg148*) | - | - | N(0.225) | A(0.81) | - | - | 35 | 0.0000247 | 0.0000163 | N/A | Likely Pathogenic | PVS1, PM2 |
| c.469C>T | p.(Arg157Cys) | D(0.912) | D(0.916) | D(0.629) | D(0.548) | M(0.752) | 0.285 | 34 | 0.0000165 | 0.0000203 | N/A | Uncertain Significance | PM2,PM3 |
| c.1705A>G | p.(Lys569Glu) | D(0.427) | D(0.875) | D(0.629) | D(0.441) | M(0.567) | 0.598 | 31 | N/A | N/A | N/A | Uncertain Significance | PM2,PM3 |
| c.647T>C | p.(Phe 216Ser) | D(0.721) | D(0.764) | D(0.629) | D(0.412) | M(0.741) | 0.326 | 24.3 | N/A | N/A | N/A | Uncertain Significance | PM2 |
| Allele Frequency | Prediction Score | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | Amino Acid | GnomAD | Polyphen2 | Mut | Mut | |||||||
| Change | Change | Exon | Exac03 | Exome | Sift * | _Hvar * | LRT * | Taster * | Assessor * | Revel * | CADD | Reference |
| missense/nonsense variant | ||||||||||||
| c.131T>C | p.(Ile44Thr) | 3 | 0.0000494 | 0.0000731 | D | P | D | D | M | N/A | 23.8 | Christina M. Sloan-Heggen, 2016 [16] |
| c.235C>T | p.(Arg79Trp) | 5 | 0.00000824 | 0.00000812 | D(0.4) | B(0.166) | N(0.132) | N(0.09) | M(0.552) | 0.21 | 23.6 | this study |
| c.313A>T | p.(Lys105*) | 6 | N/A | N/A | - | - | - | - | - | - | - | Christina M. Sloan-Heggen, 2016 [16] |
| c.442C>T | p.(Arg148*) | 7 | 0.0000247 | 0.0000163 | - | - | N(0.225) | A(0.81) | - | - | 35 | this study |
| c.446C>A | p.(Ala149Asp) | 7 | 0.000016 | N/A | D | D | N | P | - | - | 28.8 | Shearer, 2014 [9] |
| c.469C>T | p.(Arg157Cys) | 7 | 0.0000165 | 0.0000203 | D(0.912) | D(0.916) | D(0.629) | D(0.548) | M(0.752) | 0.285 | 34 | this study |
| c.647T>C | p.(Phe216Ser) | 8 | N/A | N/A | D(0.721) | D(0.764) | D(0.629) | D(0.412) | M(0.741) | 0.326 | 24.3 | this study |
| c.878A>G | p.(Gln293Arg) | 10 | N/A | N/A | D | P | D | D | M | - | 24.2 | L. He, 2018 [17] |
| c.1025A>T | p.(Asp342Val) | 11 | N/A | N/A | D(0.784) | D(0.719) | N(0.388) | D(0.81) | M(0.552) | 0.453 | 26.7 | Walsh, 2006 [18] |
| c.1249C>T | p.(Leu417Phe) | 12 | 0.0000165 | 0.0000163 | D | P | D | D | M | - | 28.6 | Tsai, 2013 [19] |
| c.1282G>T | p.(Val428Phe) | 12 | N/A | N/A | D | P | N | P | L | - | 24.7 | Cabanillas, 2018 [20] |
| c.1352G>A | p.(Gly451Asp) | 13 | 0.00000824 | 0.00000407 | D(0.912) | D(0.971) | D(0.439) | D(0.524) | M(0.567) | 0.768 | 24.8 | K Lee, 2013 [21] |
| c.1705A>G | p.(Lys569Glu) | 16 | N/A | N/A | D(0.427) | D(0.875) | D(0.629) | D(0.441) | M(0.567) | 0.598 | 31 | this study |
| c.1728T>G | p.(Ile576Met) | 16 | 0.000033 | 0.0000284 | D | P | D | D | M | - | 23.8 | Christina M. Sloan-Heggen, 2016 [16] |
| c.1865T>A | p.(Leu622His) | 17 | 0.000008 | N/A | D | P | D | D | - | - | 29.1 | P Fontana, 2017 [15] |
| c.1807G>T | p.(Val603Phe) | 16 | N/A | 0.00000406 | T | P | N | D | M | - | 26.6 | Ammar-Khodja, 2015 [22]; Christina M. Sloan-Heggen, 2016 [16] |
| c.1814G>C | p.(Cys605Ser) | 17 | N/A | N/A | T | P | D | D | M | - | 26.8 | Christina M. Sloan-Heggen, 2016 [16] |
| c.1879C>T | p.(Pro627Ser) | 17 | 0.000033 | 0.0000366 | D(0.496) | D(0.916) | D(0.629) | D(0.548) | M(0.567) | 0.446 | 31 | K Lee, 2013 [21]; Christina M. Sloan-Heggen, 2015 [13] |
| c.1939G > C | p.(Gly647Arg) | 18 | N/A | 0.0000122 | T(0.363) | P(0.604) | D(0.629) | D(0.478) | M(0.567) | 0.813 | 23.6 | Christina M. Sloan-Heggen, 2015 [13] |
| c.2201A>G | p.(Gln734Arg) | 19 | 0.00000824 | 0.00000407 | T(0.330) | B(0.339) | N(0.229) | D(0.330) | M(0.723) | 0.079 | 8.163 | Christina M. Sloan-Heggen, 2015 [13] |
| splicing variant | ||||||||||||
| c.151+1G>A | N/A | N/A | - | - | - | D(0.81) | - | - | 26.3 | Christina M. Sloan-Heggen, 2015 [13] | ||
| c.1320+2T>C | N/A | N/A | - | - | - | D(0.81) | - | - | 24.2 | Zwaenepoel, 2002 [10] | ||
| c.1320+5G>C | N/A | 0.00001 | - | - | - | D | - | - | 21.7 | Bong Jik Kim, 2019 [12] | ||
| c.2208−1G>A | 0.000036 | N/A | - | - | - | D(0.81) | - | - | 22.4 | Christina M. Sloan-Heggen, 2015 [13] | ||
| small deletion | ||||||||||||
| c.827delT | p.(Ile276fs) | 9 | 0.000025 | N/A | - | - | - | N/A | - | - | 35 | Shearer, 2014 [9]; Christina M. Sloan-Heggen, 2016 [16]; Sommen, 2016 [14] |
| c.1765delC | p.(Gln589fs) | 17 | 0.000025 | N/A | - | - | - | D | - | - | 28.5 | Bong Jik Kim, 2019 [12] |
| c.2960_2961delAT | p.(His987fs) | 25 | 0.000094 | N/A | - | - | - | N/A | - | - | 25.3 | Sommen, 2016 [14] |
| Hereditary | Onset | Average hearing level | Zygosity | Allele #1 | Allele #2 | Reference | ||
|---|---|---|---|---|---|---|---|---|
| AR/Spo | 3y | moderate | homo | whole gene deletion | whole gene deletion | this study | ||
| AR/Spo | childhood | severe | homo | whole gene deletion | whole gene deletion | this study | ||
| AR | prelingual | N/A | homo | whole gene deletion | whole gene deletion | Shahin, 2010 [23] | ||
| AR | N/A | mild to moderate | homo | whole gene deletion | whole gene deletion | Bademci, 2014 [24] | ||
| AR | 0−10y | moderate to severe | homo | Whole gene deletion | whole gene deletion | Shearer, 2014 [9] | ||
| N/A | 21−30y | N/A | homo | whole gene deletion | whole gene deletion | Shearer, 2014 [9] | ||
| AR | prelingual | moderate to severe | homo | whole gene deletion | whole gene deletion | Christina M. Sloan-Heggen, 2015 [13] | ||
| N/A | N/A | N/A | homo | whole gene deletion | whole gene deletion | Christina M. Sloan-Heggen, 2016 [16] | ||
| AD | adult | severe to profound | homo | whole gene deletion | whole gene deletion | Christina M. Sloan-Heggen, 2016 [16] | ||
| Spo | congenital | severe to profound | homo | whole gene deletion | whole gene deletion | Christina M. Sloan-Heggen, 2016 [16] | ||
| AR | 1−13y | severe | homo | 58000bp deletion | 58000bp deletion | Alkowari, 2017 [25] | ||
| AR | prelingual | severe | homo | c.151+1G>A | c.151+1G>A | Christina M. Sloan-Heggen, 2015 [13] | ||
| AR/Spo | 0m | moderate | homo | c.647T>C | p.(Phe216Ser) | c.647T>C | p.(Phe216Ser) | this study |
| AR | prelingual | moderate to severe | homo | c.1025A>T | p.(Asp342val) | c.1025A>T | p.(Asp342val) | Walsh, 2006 [18] |
| AR | prelingual | moderate to severe | homo | c1320+2T>C | c.1320+2T>C | Zwaenepoel, 2002 [10] | ||
| AR | prelingual | severe | homo | c.1352G>A | p.(Gly451Asp) | c.1352G>A | p.(Gly451Asp) | K Lee, 2013 [21] |
| AR | prelingual | severe to profound | homo | c.1807G>T | p.(Val603Phe) | c.1807G>T | p.(Val603Phe) | Ammar-Khodja, 2015 [22] |
| AR | prelingual | severe | homo | c.1879C>T | p.(Pro627Ser) | c.1879C>T | p.(Pro627Ser) | K Lee, 2013 [21] |
| AR | prelingual | moderate to severe | homo | c.1879C>T | p.(Pro627Ser) | c.1879C>T | p.(Pro627Ser) | Christina M. Sloan-Heggen, 2015 [13] |
| AR | prelingual | moderate to severe | homo | c.1939G C | p.(Gly647Arg) | c.1939G>C | p.(Gly647Arg) | Christina M. Sloan-Heggen, 2015 [13] |
| AR | prelingual | moderately severe to profound | homo | c.2201A>G | p.(Gln734Arg) | c.2201A>G | p.(Gln734Arg) | Christina M. Sloan-Heggen, 2015 [13] |
| AR/Spo | 7y | moderate | compound hetero | whole gene deletion | c.235C>T | p.(Arg79Trp) | this study | |
| N/A | 0−10y | N/A | compound hetero | whole gene deletion | c.446C>A | p.(Ala149Asp) | Shearer, 2014 [9] | |
| AR/Spo | 5y | moderate | compound hetero | whole gene deletion | c.469C>T | p.(Arg157Cys) | this study | |
| N/A | 0−10y | N/A | compound hetero | whole gene deletion | c.827delT | p.(Ile276fs) | Shearer, 2014 [9] | |
| Spo | congenital | N/A | compound hetero | whole gene deletion | c.827delT | p.(Ile276fs) | Christina M. Sloan-Heggen, 2016 [16] | |
| AR | childhood | N/A | compound hetero | whole gene deletion | c.1282G>T | p.(Val428Phe) | Cabanillas, 2018 [20] | |
| AD | congenital | N/A | compound hetero | whole gene deletion | c.1728T>G | p.(Ile576Met) | Christina M. Sloan-Heggen, 2016 [16] | |
| AR/Spo | congenital | moderate | compound hetero | whole gene deletion | c.1705A>G | p.(Lys569Glu) | this study | |
| Spo | childhood | severe to profound | compound hetero | whole gene deletion | c.1807G>T | p.(Val603Phe) | Christina M. Sloan-Heggen, 2016 [16] | |
| Spo | congenital | mild to moderate | compound hetero | whole gene deletion | c.1814G>C | p.(Cys605Ser) | Christina M. Sloan-Heggen, 2016 [16] | |
| AR | prelingual | severe | compound hetero | whole gene deletion | c.1865T>A | p.(Leu622His) | P Fontana, 2017 [15] | |
| N/A | N/A | N/A | compound hetero | multi exon deletion | c.1249C>T | p.(Leu417Phe) | Tsai, 2013 [19]t | |
| AR/Spo | 0m | moderate | compound hetero | deletion | c.442C>T | p.(Arg148*) | this study | |
| AR | prelingual | N/A | compound hetero | deletion | c.2960_2961delAT | p.His987fs | Sommen, 2016 [14] | |
| Spo | before 6 years | moderate | compound hetero | micro deletion | c.878A>G | p.(Gln293Arg) | L. He, 2018 [17] | |
| Spo | congenital | mild to moderate | compound hetero | c.131T>C | p.(Ile44Thr) | c.313A>T | p.(Lys105*) | Christina M. Sloan-Heggen, 2016 [16] |
| AR | prelingual | N/A | compound hetero | c.827delT | p.(Ile276fs) | c.2960_2961delAT | p.(His987fs) | Sommen, 2016 [14] |
| AR | congenital | moderate | compound hetero | c.1320+5G>C | c.1765delC | p.(Gln589fs) | Bong Jik Kim, 2019 [12] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugiyama, K.; Moteki, H.; Kitajiri, S.-i.; Kitano, T.; Nishio, S.-y.; Yamaguchi, T.; Wakui, K.; Abe, S.; Ozaki, A.; Motegi, R.; et al. Mid-Frequency Hearing Loss Is Characteristic Clinical Feature of OTOA-Associated Hearing Loss. Genes 2019, 10, 715. https://doi.org/10.3390/genes10090715
Sugiyama K, Moteki H, Kitajiri S-i, Kitano T, Nishio S-y, Yamaguchi T, Wakui K, Abe S, Ozaki A, Motegi R, et al. Mid-Frequency Hearing Loss Is Characteristic Clinical Feature of OTOA-Associated Hearing Loss. Genes. 2019; 10(9):715. https://doi.org/10.3390/genes10090715
Chicago/Turabian StyleSugiyama, Kenjiro, Hideaki Moteki, Shin-ichiro Kitajiri, Tomohiro Kitano, Shin-ya Nishio, Tomomi Yamaguchi, Keiko Wakui, Satoko Abe, Akiko Ozaki, Remi Motegi, and et al. 2019. "Mid-Frequency Hearing Loss Is Characteristic Clinical Feature of OTOA-Associated Hearing Loss" Genes 10, no. 9: 715. https://doi.org/10.3390/genes10090715
APA StyleSugiyama, K., Moteki, H., Kitajiri, S.-i., Kitano, T., Nishio, S.-y., Yamaguchi, T., Wakui, K., Abe, S., Ozaki, A., Motegi, R., Matsui, H., Teraoka, M., Kobayashi, Y., Kosho, T., & Usami, S.-i. (2019). Mid-Frequency Hearing Loss Is Characteristic Clinical Feature of OTOA-Associated Hearing Loss. Genes, 10(9), 715. https://doi.org/10.3390/genes10090715